
Abstract
Primary infection of humans with varicella zoster virus (VZV) causes varicella (chickenpox), after which the virus becomes latent in cranial nerve ganglia, dorsal root ganglia and autonomic ganglia along the entire neuraxis. As VZV-specific cell-mediated immunity declines in elderly and immunocompromised individuals, VZV reactivates from one or more ganglia and typically causes herpes zoster (shingles). Zoster may also be complicated by VZV vasculopathy due to productive virus infection of the cerebral arteries. In recent decades, the clinical spectrum of VZV vasculopathy has expanded to include not only transient ischemic attacks and ischemic and hemorrhagic stroke, but also multifocal VZV vasculopathy, with temporal artery infection mimicking giant cell arteritis, extracranial vasculopathy, aneurysm with and without subarachnoid hemorrhage, arterial dissection and dolichoectasia, ischemic cranial neuropathies, cerebral venous sinus thrombosis, spinal cord infarction and peripheral thrombotic disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Varicella zoster virus (VZV) is an exclusively human, neurotropic double-stranded DNA alphaherpesvirus. Primary infection causes varicella (chickenpox), after which virus becomes latent in cranial nerve ganglia, dorsal root ganglia and autonomic ganglia along the entire neuraxis. With a decline in VZV-specific cell-mediated immunity in elderly individuals, as well as in AIDS and other immunocompromised patients, VZV reactivates from one or more ganglia to cause herpes zoster (shingles). Zoster may also be complicated by VZV vasculopathy due to productive viral infection of cerebral arteries.
Historically, VZV vasculopathy presented as acute hemiplegia after contralateral herpes zoster ophthalmicus or as a postvaricella arteriopathy in children. However, in recent years, the clinical spectrum of VZV vasculopathy has been expanded to include transient ischemic attacks (TIAs), ischemic and hemorrhagic stroke often involving both large and small vessels (Fig. 1), multifocal VZV vasculopathy with temporal artery (TA) infection mimicking giant cell arteritis (GCA), extracranial vasculopathy, aneurysm with and without subarachnoid hemorrhage, arterial dissection and dolichoectasia, ischemic cranial neuropathy, cerebral venous sinus thrombosis, spinal cord infarction and peripheral thrombotic disease.

Protean manifestations of VZV vasculopathy
Epidemiology
The exact frequency of VZV vasculopathy is unknown. However, given that >95 % of the world population is latently infected with VZV and that 50 % will experience virus reactivation and develop zoster by 85 years of age, it is probably not uncommon. In Taiwan, adults with zoster have an attendant 31 % increased risk of stroke in the following year [1], and when zoster is in the ophthalmic distribution of the trigeminal nerve, the stroke risk increases 4.5-fold [2]. In Denmark, the risk of stroke after zoster is reportedly increased by 27 % during the first 2 weeks, by 17 % from 2 weeks to 1 year, and by 5 % after 1 year. The association was found in both young and older groups, with the highest increased risk of stroke seen in patients <40 years of age [3••]. Most recently, a study based in the UK showed that the risk of TIAs and myocardial infarctions (MIs) after zoster was increased 1.15-fold and 1.10-fold, respectively, and in subjects <40 years of age, the risk of stroke, TIAs and MIs was increased 2.42-fold,1.49-fold and 1.74-fold, respectively [4••]. In children, 31 % of all arterial ischemic strokes and 44 % of transient cerebral arteriopathy are associated with varicella [5, 6].
Pathogenesis
Upon reactivation from ganglia, VZV travels transaxonally to arteries where productive infection is established, followed by pathological vascular remodeling and stroke. Evidence for productive VZV infection of affected arteries was first provided by virological analysis of a patient who died after VZV vasculopathy. The infected arteries contained Cowdry A inclusion bodies, multinucleated giant cells, herpes virions and both VZV DNA and antigen [7]. The notion that virus spreads transaxonally after reactivation from trigeminal and other cranial nerve ganglia is supported by the demonstration of afferent fibers from trigeminal ganglia to intracranial blood vessels, venous sinuses and dural structures [8, 9].
The mechanism(s) by which VZV causes pathological vascular remodeling can be surmised from studies of VZV-infected arteries from patients with virologically confirmed VZV vasculopathy. For example, in VZV-infected cerebral and TAs from three patients with VZV vasculopathy, histological and immunohistochemical analyses using antibodies directed against VZV, endothelium and smooth muscle actin and myosin revealed the presence of VZV antigen in the outermost arterial layer (adventitia) early in infection and in the media and intima layers, consistent with both transaxonal spread of reactivated VZV to the arterial adventitia and with transmural spread of virus [10]. Moreover, virus-infected arteries revealed a disrupted internal elastic lamina, a thickened intima composed of cells expressing alpha-smooth muscle actin and smooth muscle myosin heavy chain but not endothelial cells expressing CD31 and decreased numbers of medial smooth muscle cells [10]. The loss of medial smooth muscle cells and the presence of cells expressing myosin in the thickened intima suggest that some of these latter cells are of medial smooth muscle origin.
In a follow-up study of inflammatory cells and their distribution in six normal cerebral arteries and in two VZV-infected arteries at 3 days after onset of disease (early) and 10 months after protracted neurological disease (late) [11•], immunohistochemical analysis revealed VZV antigen in the adventitia in early disease and in the media during late disease. In addition, arteries contained CD4+ and CD8+ T cells, CD68+ macrophages, and rare CD20+ B cells found throughout the adventitia and intima, but not in the media. Early VZV vasculopathy was distinguished by the presence of abundant neutrophils in the adventitia that were absent in late VZV vasculopathy. In late VZV vasculopathy, viral antigen was seen in the media in the absence of leukocytes, supporting the notion that the media is an immunoprivileged site [12]. Inflammatory cells were absent in control arteries. Finally, a thickened intima was associated with inflammation in vasa vasorum vessels in early VZV vasculopathy, consistent with a role for virus-induced inflammation in vessel wall remodeling [13, 14].
Together, the findings point to the role of altered arterial caliber and contractility, produced in part by abnormal accumulation of smooth muscle cells and myofibroblasts in the thickened neointima and by disruption of the media associated with the presence of viral antigen and inflammatory cells in VZV-associated stroke.
Ischemic and Hemorrhagic Stroke
Productive VZV infection of cerebral arteries causes ischemic and hemorrhagic stroke. VZV vasculopathy should be suspected in a patient with a recent history of zoster or varicella who presents with a TIA, stroke, chronic headache or altered mental status, and should also be considered in patients with vasculopathy in which a specific cause has not been determined, particularly among immunocompromised and HIV+ subjects. Importantly, the absence of a history of rash should not deter the clinician from pursuing a diagnostic evaluation for VZV, since one-third of patients with virologically verified VZV vasculopathy have no preceding rash [15].
Supportive data to aid in diagnosis include a mononuclear pleocytosis in cerebrospinal fluid (CSF) and magnetic resonance imaging (MRI) findings consistent with an ischemic or hemorrhagic lesion, particularly at gray–white matter junctions (Fig. 2a). A study of 30 subjects with virologically confirmed VZV vasculopathy [15] revealed rash in 63 %, CSF pleocytosis in 67 % and imaging abnormalities in 97 %. Angiography revealed abnormalities in 70 % of subjects, with large and small arteries involved in 50 %, small arteries only in 37 % and large arteries only in 13 %. The CSF of 30 % of subjects contained VZV DNA, whereas 93 % had anti-VZV IgG antibody in CSF with a reduced serum/CSF ratio of anti-VZV IgG that confirmed intrathecal synthesis of anti-VZV IgG. Detection of anti-VZV IgG antibody is the best diagnostic test (Fig. 3) [16], since although viral DNA can be detected by polymerase chain reaction in early disease, VZV vasculopathy is often chronic and protracted. Importantly, diagnosis of this treatable cause of stroke is often missed because one-third of subjects have no history of zoster rash, one-third of subjects have normal CSF, and there is an average 4.2-month delay from zoster to neurological symptoms and signs, with VZV DNA often absent in CSF [15].

MRI in VZV vasculopathy and myelopathy. a Multifocal VZV vasculopathy is characterized by superficial and deep-seated lesions, typically at gray–white matter junctions (arrows). b VZV myelopathy is characterized by longitudinal serpiginous lesions in the spinal cord (arrow). (Permission granted from Neurology Clinical Practice)

Diagnosis and treatment of VZV vasculopathy
Immunocompetent patients with VZV vasculopathy should be treated with a full 14-day course of intravenous acyclovir, 10 – 15 mg/kg given three times daily (Fig. 3). Immunocompromised patients or those with recurrent VZV vasculopathy may need a longer course. Since virus-infected arteries typically contain inflammatory cells, we give oral prednisone 1 mg/kg daily for 5 days without taper, in conjunction with intravenous acyclovir. Patients with renal disease must be monitored closely when treated with intravenous acyclovir.
Multifocal VZV Vasculopathy with Temporal Artery Infection and Giant Cell Arteritis
Recently, three subjects with multifocal VZV vasculopathy and TA infection were described [17–19]. All three presented with ischemic optic neuropathy, with one patient subsequently developing acute retinal necrosis; VZV infection of the ipsilateral TA was confirmed in all three patients. Importantly, these patients experienced symptoms, signs and laboratory abnormalities characteristic of GCA, a vasculitis of unclear etiology that is treated with corticosteroids. However, histopathological examination of their TAs was negative for GCA. These cases raise the possibility that patients with suspected GCA but whose arteries are pathologically negative for GCA have multifocal VZV vasculopathy with TA infection. It is essential to differentiate GCA from multifocal VZV vasculopathy because treatment with corticosteroids for presumed GCA may potentiate VZV infection and lead to loss of vision. In contrast, patients with multifocal VZV vasculopathy require immediate antiviral (intravenous acyclovir) treatment.
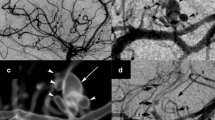
To further address the incidence of VZV infection in GCA biopsy-negative patients, 24 TAs from patients with clinical suspicion of GCA, but biopsy-negative, were examined by immunohistochemistry for the presence of VZV antigen [20]. Remarkably, the TAs from five of the patients (21 %) contained VZV. All five subjects whose TAs contained VZV antigen had presented with clinical and laboratory features of GCA and early visual disturbances. Thirteen normal TAs did not contain VZV antigen. In another GCA-negative TA, detection of VZV antigen and VZV DNA in multiple regions (skip areas) as well as in skeletal muscle adjacent to the infected artery led to additional pathological analysis of sections contiguous with those containing VZV antigen (Fig. 4). Remarkably, inflammation involving the arterial media and abundant multinucleated giant cells was seen, resulting in a change in pathological diagnosis from GCA-negative to GCA-positive [21••]. Overall, multifocal VZV vasculopathy with TA infection can present with the full spectrum of clinical features and laboratory abnormalities characteristically seen in GCA. VZV is considered a major cause of GCA and its role is under intense study.

VZV antigen in the temporal artery of a patient with giant cell arteritis. Immunohistochemistry revealed VZV antigen (a red) in the adventitia (solid arrow) and its nerve bundles (dashed arrow), as well as in the media and intima (not shown) after staining with anti-VZV antibody; No staining is seen using anti-HSV-1 antibody (b) or normal rabbit serum (not shown) (×600). Hematoxylin & eosin staining of cross-section of the temporal artery (c) reveals extensive inflammation in the adventitia (long black arrow), as well as in the media (short black arrow) and vaso vasorum (dashed black arrow). The white arrow spans the thickened intima and points to an occluded lumen. Numerous multinucleated giant cells were seen throughout the artery (insets) (×200). (Permission granted from Journal of Neurological Sciences)
Extracranial Vasculopathy
VZV vasculopathy with widespread intracranial and extracranial arterial disease has recently been described [22]. A 30-year-old immunocompetent man developed aphasia and a right hemiplegia with multiple ischemic lesions in the left basal ganglia, external capsule and left temporal region, as well as loss of vision in the left eye from a central retinal artery occlusion. Vascular studies revealed complete occlusion of the left common carotid artery and most of the external and internal carotid arteries, thrombosis and vasospasm of the left middle cerebral artery and thrombosis of the A1 segment of the anterior cerebral artery. There was no history of skin rash preceding stroke. The diagnosis of VZV vasculopathy was confirmed by the presence of anti-VZV IgG antibodies in CSF. After treatment with intravenous acyclovir, motor and speech deficits improved, although visual acuity in the left eye was limited to light perception. Importantly, involvement of extracranial arteries in VZV vasculopathy provides a source of virus to the superior TA, a terminal branch of the external carotid circulation.
Aneurysm/Hemorrhage
The protean complications of VZV vasculopathy include cerebral aneurysm and subarachnoid and intracerebral hemorrhage. The first report of a CNS aneurysm produced by VZV infection was in a 24-year-old healthy woman who developed ophthalmic-distribution zoster with peripheral facial palsy and contralateral hemiplegia [23]. Although an initial angiogram was normal, she developed decreased hearing and a left-sided Horner’s syndrome 4 weeks later. Repeat angiography revealed an aneurysm in the intrapetrosal region of the left internal carotid artery. Three further cases of aneurysm caused by VZV infection in patients with AIDS were recently reported [24]. In another case of VZV vasculopathy, a 42-year-old renal transplant recipient with concurrent aneurysm in the vertebral artery and dissection was successfully treated with embolization and acyclovir therapy [25]. Most remarkable is the recent report of a patient with zoster in multiple dermatomes, severe headache and a normal magnetic resonance angiogram (MRA) and four-vessel digital subtraction angiogram who developed nine anterior circulation aneurysms 2 months later. Antiviral treatment resulted in clinical improvement, size reduction of most aneurysms and complete resolution of the two largest aneurysms [26].
Arterial Dissection and Dolichoectasia
Two cases of dissection after VZV infection have been described [27]. The first was a 15-year-old boy, who at 4 weeks after ophthalmic-distribution zoster, developed a left hemiparesis while jogging. Angiography revealed carotid artery dissection. His clinical symptoms resolved and the angiogram normalized in 3 months. The other case was a 4-year-old boy, who at 2 weeks after recovery from varicella developed left-sided weakness while playfully wrestling with another child. Cerebral angiogram revealed a right internal carotid artery dissection. Clinical examination and radiological improvement revealed full recovery after 2 months. Although trauma and exercise are frequently associated with arterial dissection, VZV infection in the wall of an artery may predispose the artery to dissection if it is subjected to further trauma.
VZV has also been associated with dolichoectasia of the anterior and posterior intracranial circulation with multiple small deep-seated infarcts [28]. A comprehensive study of VZV vasculopathy revealed the occurrence of stenosis, ectasia and aneurysm in several patients [24]. Elble [29] described intracerebral hemorrhage after ophthalmic-distribution zoster. Another patient with VZV vasculitis presented with intracranial hemorrhage and concomitant zoster on the back; cerebral angiography revealed vasculitic changes involving small and medium-sized vessels [30]. Because VZV infection occurs primarily in the media of arteries with disruption of the internal elastic membrane, it is surprising that arterial ectasia and aneurysm with subarachnoid and intracerebral hemorrhage is not more frequent.
Cranial Neuropathies
Cranial neuropathies after zoster are not uncommon. Although the mechanism of disease is unknown, their development days to weeks after rash parallels the development of VZV vasculopathy in the brain weeks after zoster, suggesting that cranial neuropathies might be secondary to microvascular infarction. Such a notion is supported by the demonstration of three arterial systems that each supply multiple cranial nerves [31]. The first is the inferolateral trunk, which arises from the internal carotid artery and supplies cranial nerves III, IV, V1 and VI. The second is the middle meningeal artery, which is derived from the external carotid artery and supplies cranial nerves V2, V3 and VII. The intrapetrous portion of nerve VII has two defined vascular territories: the stylomastoid artery, which is limited to nerve VII, and the middle meningeal artery, which supplies nerves VII and V. The third is the ascending pharyngeal artery, also derived from the external carotid artery, which supplies nerves IX, X, XI and XII; cranial nerve XI has a dual vascularization, which explains why it can either be spared or involved along with other cranial nerves in zoster. VZV vasculopathy should be considered when cranial nerve lesions occur in mono- or polyneuritis cranialis syndromes with or without rash.
Venous Sinus Thrombosis
Three cases of VZV vasculopathy with associated cerebral venous sinus thrombosis have been reported. Chan and colleagues [32] described a 55-year-old woman who presented with a 3-day history of left temporal and postauricular pain, nausea, vomiting and photophobia. Examination was remarkable for hyperesthesia in the left C2 nerve root distribution without rash. Based on elevated serum anti-VZV IgG, a diagnosis of zoster sine herpete was made. Hypercoagulable studies were normal. Computed tomography revealed minimal fluid in the left mastoid and thrombosis in the left lateral and sigmoid sinuses, consistent with VZV reactivation from the left C2 dorsal root ganglia and transaxonal spread to the ipsilateral cerebral venous sinuses in anatomical pathways previously described [33]. The patient improved after treatment with intravenous acyclovir, gabapentin and anticoagulation. Siddiqi and colleagues [34] described two additional cases of cerebral venous sinus thrombosis associated with varicella and zoster. The first case was a 15-year-old boy who developed varicella associated with fever and diffuse headaches; 3 weeks later he had grand mal seizures and was drowsy, was disoriented and developed a right hemiparesis. Protein C and S levels were deficient. MRA and magnetic resonance venography (MRV) revealed filling defects in the superior sagittal, straight, bilateral transverse and sigmoid sinuses, extending into both jugular veins. There was focal narrowing in the left distal middle cerebral artery just before trifurcation, with a paucity of the left Sylvian branches, suggesting vasculitis. The patient improved with intravenous acyclovir, adequate hydration and oral levetiracetam. The second case was a 20-year-old diabetic man who developed disseminated rash, fever, vertigo and headache followed 1 week later by generalized tonic–clonic seizures. Hypercoagulable laboratory results were negative. MRI brain scanning revealed bilateral thalamic and left parieto-occipital hemorrhagic infarcts with surrounding edema; MRV revealed a filling defect in the left transverse sinus extending into the left sigmoid sinus and straight sinus involving the vein of Galen and extending into the left jugular vein, consistent with cerebral venous sinus thrombosis. The patient recovered after treatment with intravenous acyclovir, levetiracetam and subcutaneous enoxaparin.
Spinal Cord Infarction
Although putative spinal cord infarctions produced by VZV vasculopathy have been diagnosed based on acute-onset myelopathy associated with zoster or based on virological evidence of VZV infection in patients with acute myelopathy without rash (Fig. 2b), a few cases have been verified pathologically [35]. Before development of diffusion-weighted MRI, the only way to establish the diagnosis of VZV vasculopathy in the spinal cord was post-mortem spinal cord necrosis secondary to VZV vasculitis with evidence of productive VZV infection [35, 36]. Diffusion-weighted MRI, which is superior to conventional MRI in detecting spinal cord ischemia and infarction [37], was used in conjunction with virological analysis of CSF to diagnose VZV infarction of the spinal cord in a patient who presented with abrupt-onset myelopathy followed by zoster [38]. Diffusion-weighted MRI revealed spinal cord infarction, while virological analysis detected anti-VZV IgG antibody, with decreased serum/CSF ratios of anti-VZV IgG antibody indicative of intrathecal synthesis of anti-VZV IgG.
Peripheral Thrombotic Disease
Massano et al. [39] reported a case of multifocal VZV vasculopathy with peripheral thrombotic disease associated with transient hypercoagulation abnormalities. One week after varicella, a 39-year-old man developed central retinal artery occlusion, ischemic stroke with a small hemorrhage and proximal right middle cerebral artery occlusion, along with occlusive left femoral artery and two thrombi in the left common iliac and right femoral arteries. Diagnosis of VZV vasculopathy was confirmed by the presence of anti-VZV IgM and IgG antibodies in CSF. Interestingly, the patient also had increased anti-cardiolipin antibodies and low protein S, which normalized after antiviral treatment. Additional cases of VZV vasculopathy with arterial thrombotic complications, typically in the lower extremities, have been reported and associated with C3 deposits [40] and deficiencies in protein S [41].
Several cases of postvaricella thrombosis in children have also been reported, often with transient hypercoagulation abnormalities [42]. Of two of these cases described by Rabah and colleagues [43], case 1 was a 10-year-old girl who developed varicella followed 2 weeks later by left leg pain and swelling. Doppler ultrasonography revealed a thrombus extending along the left iliac, left superficial femoral vein and popliteal vein. In addition, a thrombus was found in the left common iliac vein extending into the left side of the lower vena cava for 2 – 3 cm. Abnormal coagulation studies included prolonged prothrombin time and activated partial thromboplastin time, in addition to protein S deficiency. After anticoagulation and treatment antibiotics for complicating cellulitis (no antivirals were given), protein S normalized at 4 months. Case 2 was a 3-year-old girl who developed right limb swelling 9 days after varicella. Doppler ultrasonography revealed a thrombus in the right distal superficial femoral vein and popliteal vein. She had elevated fibrinogen, anticardiolipin IgG and anti-β2-glycoprotein I IgG antibodies and elevated C4, IgA, IgG, and IgM. She was treated with anticoagulants and with intravenous acyclovir. Although no follow-up laboratory studies were performed, she had partial recanalization of thrombosed vessels 5 months later.
While the above cases were associated with coagulation abnormalities, evidence for VZV infection of vessels in the same dermatome as the rash is provided by a case reported by Tezcan and colleagues [44]. A 71-year-old man developed left C6-distribution zoster. Two weeks later, he developed numbness in the left hand with an absent left radial artery pulse. Double-subtraction angiography showed occlusion of the proximal left radial artery. No thrombophilic factors were found. The presence of arterial thrombosis at the site of zoster raises the possibility that a peripheral VZV vasculopathy developed in the radial artery.
Conclusions
Initially, VZV vasculopathy was characterized as a large-vessel stroke resulting in hemiplegia after zoster ophthalmicus or varicella. However, an increasing number of cases have revealed that both large and small intracranial vessels, dural sinuses and extracranial vessels can be involved in VZV vasculopathy with or without rash. In fact, VZV vasculopathy can present as ischemic or hemorrhagic stroke, GCA, aneurysm with and without hemorrhage, cranial neuropathies, venous sinus thrombosis, spinal cord infarction and peripheral thrombotic disease. It is important to recognize that all of these variants can present without rash and that the best test for diagnosis is determination of intrathecal synthesis of anti-VZV antibodies. Treatment of VZV vasculopathy requires intravenous acyclovir therapy, which may be prolonged in immunosuppressed individuals in whom recurrence is common. Overall, VZV is emerging as an important cause of vascular disease with protean manifestations.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Kang JH, Ho JD, Chen YH, Lin HC. Increased risk of stroke after a herpes zoster attack: a population-based follow-up study. Stroke. 2009;40:3443–8.
Lin HC, Chien CW, Ho JD. Herpes zoster ophthalmicus and the risk of stroke: a population-based follow-up study. Neurology. 2010;74:792–7.
Sreenivasan N, Basit S, Wohlfahrt J, et al. The short- and long-term risk of stroke after herpes zoster – a nationwide population-based cohort study. PLoS One. 2013;8:e69156. This large nationwide cohort study found an increased risk of stroke after treatment for herpes zoster: individuals who had received medication had a 127 % increased risk in the first 2 weeks, 17 % between 2 weeks and 1 year, and 5 % after the first year. The increased risk was greatest in the youngest age group (<40 years of age).
Breuer J, Pacou M, Gauthier A, Brown MM. Herpes zoster as a risk factor for stroke and TIA: a retrospective cohort study in the UK. Neurology. 2014;82:206–12. This is the largest study showing that herpes zoster is an independent risk factor for vascular disease in the UK population, particularly for stroke, TIA, and MI in subjects affected before the age of 40 years (adjusted hazard ratios of 1.74, 2.42, and 1.49, respectively).
Askalan R, Laughlin S, Mayank S, et al. Chickenpox and stroke in childhood: a study of frequency and causation. Stroke. 2001;32:1257–62.
Braun KP, Bulder MM, Chabrier S, et al. The course and outcome of unilateral intracranial arteriopathy in 79 children with ischaemic stroke. Brain. 2009;132:544–57.
Gilden DH, Kleinschmidt-DeMasters BK, Wellish M, Hedley-Whyte ET, Rentier B, Mahalingam R. Varicella zoster virus, a cause of waxing and waning vasculitis: the New England Journal of Medicine case 5-1995 revisited. Neurology. 1996;47:1441–46.
Mayberg M, Langer RS, Zervas NT, Moskowitz MA. Perivascular meningeal projections from cat trigeminal ganglia: possible pathway for vascular headaches in man. Science. 1981;213:228–30.
Mayberg MR, Zervas NT, Moskowitz MA. Trigeminal projections to supratentorial pial and dural blood vessels in cats demonstrated by horseradish peroxidase histochemistry. J Comp Neurol. 1984;223:46–56.
Nagel MA, Traktinskiy I, Azarkh Y, et al. Varicella zoster virus vasculopathy: analysis of virus-infected arteries. Neurology. 2011;77:364–70.
Nagel MA, Traktinskiy I, Stenmark KR, Frid MG, Choe A, Gilden D. Varicella-zoster virus vasculopathy: immune characteristics of virus-infected arteries. Neurology. 2013;80:62–8. This study of VZV-infected arteries demonstrates inflammation associated with pathological vascular remodeling and the presence of neutrophils in early VZV vasculopathy.
Dal Canto AJ, Swanson PE, O’Guin AK, Speck SH, Virgin HW. IFN-gamma action in the media of the great elastic arteries, a novel immunoprivileged site. J Clin Invest. 2001;107:15–22.
Frid MG, Brunetti JA, Burke DL, et al. Hypoxia-induced pulmonary vascular remodeling requires recruitment of circulating mesenchymal precursors of a monocyte/macrophage lineage. Am J Pathol. 2006;168:659–69.
Stenmark KR, Yeager ME, El Kasmi KC, et al. The adventitia: essential regulator of vascular wall structure and function. Annu Rev Physiol. 2013;75:23–47.
Nagel MA, Cohrs RJ, Mahalingam R, et al. The varicella zoster virus vasculopathies: clinical, CSF, imaging, and virologic features. Neurology. 2008;70:853–60.
Nagel MA, Forghani B, Mahalingam R, et al. The value of detecting anti-VZV IgG antibody in CSF to diagnose VZV vasculopathy. Neurology. 2007;68:1069–73.
Salazar R, Russman AN, Nagel MA, et al. Varicella zoster virus ischemic optic neuropathy and subclinical temporal artery involvement. Arch Neurol. 2011;68:517–20.
Nagel MA, Russman AN, Feit H, et al. VZV ischemic optic neuropathy and subclinical temporal artery infection without rash. Neurology. 2013;80:220–2.
Mathias M, Nagel MA, Khmeleva N, et al. VZV multifocal vasculopathy with ischemic optic neuropathy, acute retinal necrosis and temporal artery infection in the absence of zoster rash. J Neurol Sci. 2013;325:180–2.
Nagel MA, Bennett JL, Khmeleva N, et al. Multifocal VZV vasculopathy with temporal artery infection mimics giant cell arteritis. Neurology. 2013;80:2017–21.
Nagel MA, Khmeleva N, Boyer PJ, Choe A, Bert R, Gilden D. Varicella zoster virus in the temporal artery of a patient with giant cell arteritis. J Neurol Sci. 2013;335:228–30. This report shows that the presence of VZV antigen in the temporal artery is associated with the histopathological changes of giant cell arteritis.
Verma R, Lalla R, Patil TB. Extensive extracranial and intracranial varicella zoster vasculopathy. BMJ Case Rep. 2013. doi:10.1136/bcr-2013-009222.
Gursoy G, Aktin E, Bahar S, Tolun R, Ozden B. Post-herpetic aneurysm in the intrapetrosal portion of the internal carotid artery. Neuroradiology. 1980;19:279–82.
de Broucker T, Verollet D, Schoindre Y, et al. Cerebral vasculitis with aneurysms caused by varicella-zoster virus infection during AIDS: a new clinicoangiographical syndrome. Rev Neurol (Paris). 2008;164:61–71.
Bhayani N, Ranade P, Clark NM, McGuinn M. Varicella-zoster virus and cerebral aneurysm: case report and review of the literature. Clin Infect Dis. 2008;47:e1–3.
Liberman AL, Nagel MA, Hurley MC, Caprio FZ, Bernstein RA, Gilden D. Rapid development of nine cerebral aneurysms in varicella zoster virus vasculopathy. Neurology. 2014, in press.
Constantinescu CS. Association of varicella-zoster virus with cervical artery dissection in 2 cases. Arch Neurol. 2000;57:427.
Dalton CM, Jager HR, Losseff NA, Greenwood RJ. Neurological picture. Varicella zoster virus and intracranial dolichoectasia in a late adult cancer survivor. J Neurol Neurosurg Psychiatry. 2008;79:573.
Elble RJ. Intracerebral hemorrhage with herpes zoster ophthalmicus. Ann Neurol. 1983;14:591–2.
Jain R, Deveikis J, Hickenbottom S, Mukherji SK. Varicella-zoster vasculitis presenting with intracranial hemorrhage. AJNR Am J Neuroradiol. 2003;24:971–4.
Lapresle J, Lasjaunias P. Cranial nerve ischaemic arterial syndromes. A review. Brain. 1986;109:207–16.
Chan J, Bergstrom RT, Lanza DC, Oas JG. Lateral sinus thrombosis associated with zoster sine herpete. Am J Otolaryngol. 2004;25:357–60.
Saito K, Moskowitz MA. Contributions from the upper cervical dorsal roots and trigeminal ganglia to the feline circle of Willis. Stroke. 1989;20:524–26.
Siddiqi SA, Nishat S, Kanwar D, Ali F, Azeemuddin M, Wasay M. Cerebral venous sinus thrombosis: association with primary varicella zoster virus infection. J Stroke Cerebrovasc Dis. 2012;8:917.
Devinsky O, Cho ES, Petito CK, Price RW. Herpes zoster myelitis. Brain. 1991;114:1181–96.
Kenyon LC, Dulaney E, Montone KT, Goldberg HI, Liu GT, Lavi E. Varicella-zoster ventriculo-encephalitis and spinal cord infarction in a patient with AIDS. Acta Neuropathol. 1996;92:202–5.
Thurnher MM, Bammer R. Diffusion-weighted MR imaging (DWI) in spinal cord ischemia. Neuroradiology. 2006;48:795–801.
Orme HT, Smith AG, Nagel MA, Bert RJ, Mickelson TS, Gilden DH. VZV spinal cord infarction identified by diffusion-weighted MRI (DWI). Neurology. 2007;69:398–400.
Massano J, Ferreira D, Toledo T, Mansilha A, Azevedo E, Carvalho M. Stroke and multiple peripheral thrombotic events in an adult with varicella. Eur J Neurol. 2008;15:e90–1.
Farge D, Ribaud P, Boulay I, Scrobohaci ML, Rouffy J, Gluckman E. Tibial artery thrombosis due to varicella zoster virus in a transplant recipient under cyclosporine. Eur J Med. 1993;2:123–24.
Peyton BD, Cutler BS, Stewart FM. Spontaneous tibial artery thrombosis associated with varicella pneumonia and free protein S deficiency. J Vasc Surg. 1998;27:563–67.
Levin M, Eley BS, Louis J, Cohen H, Young L, Heyderman RS. Postinfectious purpura fulminans caused by an autoantibody directed against protein S. J Pediatr. 1995;127:355–63.
Rabah F, El-Banna N, Abdel-Baki M, et al. Post varicella thrombosis – report of two cases and literature review. Pediatr Infect Dis J. 2012;31:985–7.
Tezcan ME, Teksut TK, Onal AB, Ozturk MA. Reactivated varicella zoster virus may cause peripheral arterial thrombosis. J Rheumatol. 2010;37:1785–86.
Acknowledgments
This work was supported in part by NIH grants AG006127 and AG032958 to D.G. and NS067070 to M.A.N. We thank Marina Hoffman for editorial assistance and Lori DePriest for manuscript preparation.
Compliance with Ethics Guidelines
ᅟ
Conflict of Interest
Maria A. Nagel received a grant from the NIH. Don Gilden has received grants from the NIH.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by the author.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Central Nervous System Infections.
Rights and permissions
About this article

Cite this article
Nagel, M.A., Gilden, D. Update on Varicella Zoster Virus Vasculopathy. Curr Infect Dis Rep 16, 407 (2014). https://doi.org/10.1007/s11908-014-0407-z
Published:
DOI: https://doi.org/10.1007/s11908-014-0407-z