
Abstract
Varicella zoster virus (VZV) infects >95 % of the world population. Typically, varicella (chickenpox) results from primary infection. The virus then becomes latent in ganglionic neurons along the entire neuraxis. In immunocompromised individuals, VZV reactivates and causes herpes zoster (shingles), pain, and rash in 1–2 dermatomes. Multiple case reports showed a link between stroke and zoster, and recent studies have emerged which reveal that VZV infection of the cerebral arteries directly causes pathological vascular remodeling and stroke (VZV vasculopathy). In the past few years, several large epidemiological studies in Taiwan, Denmark, and the U.K. demonstrated that zoster is a risk factor for stroke and that antiviral therapy may reduce this risk. Herein, the history, clinical features, and putative mechanisms of VZV vasculopathy, as well as recent epidemiological studies demonstrating that zoster increases the risk of stroke, are discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The onset of stroke after herpes zoster due to reactivation of varicella zoster virus (VZV) was described as early as 1896 by Dumary and published by Baudouin and Lantuejoul [1], when a patient developed right-sided ophthalmic zoster followed 4 weeks later by left-sided hemiplegia without any signs of generalized encephalitis. This was followed by case reports of contralateral hemiplegia that developed 4 and 3 weeks after ophthalmic-distribution zoster [2–4], as well as after zoster in other dermatomes. While VZV infection can produce stroke without inducing coagulopathy, evidence is emerging that VZV reactivation with or without zoster rash directly causes pathological vascular remodeling resulting in stroke (VZV vasculopathy). Herein, we review VZV vasculopathy, potential mechanisms by which VZV causes stroke, and epidemiological studies which reveal that the incidence of stroke is significantly increased after zoster.
VZV Vasculopathy
Varicella zoster virus (VZV) is a highly neurotropic DNA alphaherpesvirus that infects >95 % of the world population. Varicella (chickenpox) is the usual outcome of infection, most often in children. Virus latency is then established in the cranial nerve, dorsal root, and autonomic ganglionic neurons. In latently infected ganglia, VZV transcription is limited without production of virions. The VZV-specific cell-mediated immune response decreases with advancing age or when humans are immunocompromised by immunosuppressive or anti-inflammatory drugs or in HIV+ individuals. After reactivation, VZV moves transaxonally to the skin resulting in zoster (shingles). Zoster rash typically resolves over a 2-week period but zoster is often complicated by persistent pain (postherpetic neuralgia). In addition, patients with zoster develop cranial neuropathies, meningitis, and encephalitis, sometimes isolated to the cerebellum, transient ischemic attacks and stroke. Multiple ocular disorders, particularly ischemic optic neuropathy and retinal necrosis also occur. Importantly, the neurologic and ocular diseases noted above may develop without zoster rash.
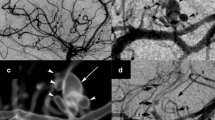
Ischemic and hemorrhagic stroke after zoster results from cerebral arterial infection by VZV (VZV vasculopathy). The average time between zoster and onset of focal neurological deficits is 4 months. Brain MRI is abnormal in 97 % of cases, frequently revealing lesions at the junction of the gray and white matter, and angiography demonstrates involvement of large and small arteries in nearly 70 % of cases [5]. Definitive diagnosis of VZV vasculopathy is made by detection in the CSF of anti-VZV IgG antibody, which is more often positive than the presence of amplifiable VZV DNA in the CSF [6]; also, if the serum/CSF ratio of anti-VZV IgG antibody is reduced, the diagnosis of VZV vasculopathy is further confirmed. Interestingly, about approximately 30 % of patients with VZV vasculopathy have no history of earlier zoster, indicating that VZV can reactivate from the ganglia and travel centrally to infect the cerebral arteries without peripheral spread to the skin.
Mechanisms of VZV Vasculopathy
No other human virus is known that infects the cerebral arteries and produces pathological vascular changes resulting in stroke. Initial studies leading to the notion that VZV infection causes pathological vascular remodeling began in 1959 with a description of “a non-infectious granulomatous angiitis with a predilection for the nervous system, characterized by thrombosis in the cerebral arteries and distinguished from other vasculitides by the nature of the inflammatory response, which consisted predominantly of histiocytes, mononuclear cells and multinucleated giant cells” [7]. Thereafter, patients with lymphoma and zoster who developed granulomatous angiitis in the brain were described, characterized by mononuclear giant cell infiltration in the cerebral arteries [8]. In 1974, angiography of the large cerebral arteries in a patient with ophthalmic zoster followed by hemiparesis on the opposite side of the body revealed focal narrowing in the carotid siphon [9]. Definitive evidence of VZV infection of the cerebral arteries came from analysis of a patient with a waxing-waning course of vasculitis [10]; postmortem examination of the affected cerebral arteries revealed Cowdry A inclusions, multinucleated giant cells, VZV DNA, VZV antigen, and herpesvirus particles [11••].
Since there are no animal models for VZV vasculopathy, further mechanistic studies used virus-infected cerebral and temporal arteries from subjects who died of VZV vasculopathy. Immunohistochemistry with antibodies directed against VZV gene 63 protein revealed VZV antigen in the outermost layer of the artery (adventitia) within months of the disease and in the media and intima later in the disease [12•], a finding expected from spread of virus transaxonally after reactivation from the cranial or cervical ganglia to the arterial adventitia followed by virus extension transmurally. Additional studies using antibodies directed against endothelial CD31, smooth muscle actin, and myosin heavy chains demonstrated that VZV-infected arteries contain a thickened intima composed of cells expressing all these markers except for the endothelial cell marker CD31; often, the internal elastic lamina was fractured and smooth muscle cells in the media were reduced [12•]. While the origin of cells in the thickened intima is unknown, the cells expressing myosin suggest that they are derived from smooth muscle cells in the media.
A later study of VZV-infected arteries at 3 days after disease onset (early) and when disease was prolonged at 10 months (late) revealed T cells that were CD4+ and CD8+, CD68+ macrophages, and rarely CD20+ B cells mostly in the adventitia and intima, with sparing of the media [13]. Only when VZV vasculopathy was early were abundant neutrophils present in the adventitia. Later, VZV was found in the media; however, no inflammatory cells were present, indicating that the media may be immunoprivileged [14]. No significant inflammation was seen in control arteries. Importantly, in a vaso vasorum vessel in early VZV vasculopathy, adventitial inflammation was associated with an overlying thickened intima, whereas another region of the same artery did not contain inflammatory cells or show a thickened intima [15, 16], indicating that the inflammatory response after virus infection lead to remodeling of the vessel wall.
Overall, the studies to date suggest that VZV infection of the cerebral arteries with subsequent inflammation leads to pathological vascular remodeling with development of a thickened intima that contributes to vascular occlusion and ischemia, and with a disruption of the media contributing to the aneurysm formation and hemorrhage seen in patients with virologically confirmed VZV vasculopathy.
Epidemiological Studies
Given that VZV reactivates to cause zoster in about one half of individuals by 85 years of age, with approximately 1 million zoster cases yearly in the USA alone, and that recent studies from Asia, Europe, and the UK indicate that zoster is a risk factor for stroke, VZV vasculopathy is not uncommon. A retrospective study of records from >7000 adults who had zoster and >23,000 control subjects with no history of zoster from Taiwan revealed that for a year from the time of zoster, the stroke risk was increased by 30 %, and was 4-fold greater with ophthalmic-distribution zoster [17]; further analysis of >600 persons who had zoster in the ophthalmic distribution of the trigeminal nerve and nearly 2000 control subjects also showed 4.5-fold greater stroke risk for the first 12 month, and treatment of zoster with antivirals had no effect on the incidence of subsequent cerebral vascular disease [18]. Unfortunately, the two studies from Taiwan were confounded by atrial fibrillation, body mass index, and the fact that the risk of stroke was determined at one time-point (12 months following zoster).
Analysis of 4.6 million people adults from Denmark in which >117,926 patients received antiviral therapy showed that stroke increased 126 % in the first 2 weeks from the onset of zoster, 17 % from 2 weeks to the first year after zoster, and 5 % overall in contrast to patients with zoster who did not receive any treatment [19]. Unfortunately the Danish study included treatment with antiviral agents as an indication that the patient had zoster, this could have led to false-positives because anyone with HSV infection would have been treated. Other controls were also insufficient for confounders.
The most recent analysis of >100,000 patients with zoster and >200,000 control subjects showed a statistically significant increase in both cerebral and cardiovascular disease after zoster; an important finding was that the risk of a warning of a stroke and of myocardial infarction was greatest in individuals whose zoster occurred before the age of 40 [20]. Unfortunately, the increased risk of stroke was not determined at multiple times following zoster. Subsequently, records were analyzed from >6000 people from the UK who had a stroke prior to or after rash were studied from the UK General Practice Research Database [21••]. For each person, the occurrence was studied from zoster onset up to a year and was compared with (1) the time of enrollment up to a month prior to zoster, and (2) from a year after zoster until the close of the study. Calculation of age-adjusted incidence ratios and 95 % confidence intervals compared to control time periods showed that stroke risk decreased with time after zoster. There was more stroke one to 4 weeks (1.63), 5 to 12 weeks (1.42), and 13 to 26 weeks after zoster (1.23), but not after those periods. Once again, zoster in the ophthalmic distribution of the trigeminal nerve was shown to be associated with 3-fold increased risk of stroke from 5 to 12 weeks following zoster. Finally, analysis of the 55 % of subjects with zoster who were given oral antivirals revealed a reduced stroke risk that was not found in untreated zoster patients.
Together, epidemiological studies from three countries in Asia and Europe revealed that zoster is a significant risk for subsequent stroke, especially ophthalmic-distribution zoster, and studies from the UK indicate that treatment with antiviral agents reduces this risk.
Conclusion
Herpes zoster due to VZV reactivation has been historically perceived as a benign disease, with resolution of rash after several weeks and only rare complications. However, recent studies demonstrating VZV-induced pathological vascular remodeling, together with epidemiological studies, indicate that zoster is clearly an important risk factor for stroke and that stroke is likely a direct effect of virus infection in the arteries. In fact, the frequency of stroke after zoster is probably underestimated since VZV can reactivate in the absence of rash. An increased recognition that VZV infection is associated with stroke will lead to further studies to determine the optimal treatment with antiviral agents, as well as zoster vaccination, in reducing zoster-associated stroke risk.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Baudouin E, Lantué joul P. Les troublecas moteurs dans le zona. Gazette des Hopitaux. 1919.
Gordon IR, Tucker JF. Lesions of the central nervous sytem in herpes zoster. J Neurol Neurosurg Psychiatry. 1945;8:40–66.
Hughes WN. Herpes zoster of the right trigeminal nerve with left hemiplegia. Neurology. 1951;1:167–9.
Cope S, Jones AT. Hemiplegia complicating ophthalmic zoster. Lancet. 1954;267:898–9.
Nagel MA, Cohrs RJ, Mahalingam R, et al. The varicella zoster virus vasculopathies: clinical, CSF, imaging, and virologic features. Neurology. 2008;70:853–60.
Nagel MA, Forghani B, Mahalingam R, et al. The value of detecting anti-VZV IgG antibody in CSF to diagnose VZV vasculopathy. Neurology. 2007;68:1069–73.
Cravioto H, Feigin I. Noninfectious granulomatous angiitis with a predilection for the nervous system. Neurology. 1959;9:599–609.
Rosenblum WI, Hadfield MG. Granulomatous angiitis of the nervous system in cases of herpes zoster and lymphosarcoma. Neurology. 1972;22:348–54.
Gilbert GJ. Herpes zoster ophthalmicus and delayed contralateral hemiparesis. Relationship of the syndrome to central nervous system granulomatous angiitis. JAMA. 1974;229:302–4.
Case records of the Massachusetts General Hospital. Weekly clinicopathological exercises. Case 5–1995. A 73-year-old man with focal brain lesions and peripheral-nerve disease. N Engl J Med. 1995;332:452–9.
Gilden DH, Kleinschmidt-DeMasters BK, Wellish M, et al. Varicella zoster virus, a cause of waxing and waning vasculitis: the New England Journal of Medicine case 5–1995 revisited. Neurology. 1996;47:1441–6. This article was the first to demonstrate VZV DNA, antigen and herpesvirus particles, as well as Cowdry A inclusions and giant cells, in the cerebral arteries of a patient who died of VZV vasculopathy.
Nagel MA, Traktinskiy I, Azarkh Y, et al. Varicella zoster virus vasculopathy: analysis of virus-infected arteries. Neurology. 2011;77:364–70. Nagel et al. examined cerebral and temporal arteries from 3 patients with VZV vasculopathy and characterized changes in the intima, media and internal elastic lamina associated with infection.
Nagel MA, Traktinskiy I, Stenmark KR, et al. Varicella-zoster virus vasculopathy: immune characteristics of virus-infected arteries. Neurology. 2013;80:62–8.
Dal Canto AJ, Swanson PE, O’Guin AK, et al. IFN-gamma action in the media of the great elastic arteries, a novel immunoprivileged site. J Clin Invest. 2001;107:15–22.
Frid MG, Brunetti JA, Burke DL, et al. Hypoxia-induced pulmonary vascular remodeling requires recruitment of circulating mesenchymal precursors of a monocyte/macrophage lineage. Am J Pathol. 2006;168:659–69.
Stenmark KR, Yeager ME, El Kasmi KC, et al. The adventitia: essential regulator of vascular wall structure and function. Annu Rev Physiol. 2013;75:23–47.
Kang JH, Ho JD, Chen YH, et al. Increased risk of stroke after a herpes zoster attack: a population-based follow-up study. Stroke. 2009;40:3443–8.
Lin HC, Chien CW, Ho JD. Herpes zoster ophthalmicus and the risk of stroke: a population-based follow-up study. Neurology. 2010;74:792–7.
Sreenivasan N, Basit S, Wohlfahrt J, et al. The short- and long-term risk of stroke after herpes zoster—a nationwide population-based cohort study. PLoS One. 2013;8:e69156.
Breuer J, Pacou M, Gauthier A, Brown MM. Herpes zoster as a risk factor for stroke and TIA: a retrospective cohort study in the UK. Neurology. 2014;82:206–12.
Langan SM, Minassian C, Smeeth L, Thomas SL. Risk of stroke following herpes zoster: a self-controlled case-series study. Clin Infect Dis. 2014;58:1497–503. Langan et al. studied 6584 individuals with zoster and stroke using a self-controlled case series method and showed that there was an increased stroke rate within 6 months following zoster and that antiviral therapy may reduce this risk.
Acknowledgment
The study was supported by grants from the National Institutes of Health (AG032958 and AG006127 to Dr. Gilden and NS06070 to Dr. Nagel). We thank Marina Hoffman for editorial review and Cathy Allen for manuscript preparation.
Compliance with Ethics Guidelines
ᅟ
Conflict of Interest
Maria A. Nagel has received the following grant: NIH/NIA PPG, Molecular Pathogenesis of VZV Infection.
Don Gilden has received the following grants: NIH/NIA PPG, Molecular Pathogenesis of VZV Infection and NIH/NINDS R01, Neurobiology of VZV.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Stroke
Rights and permissions
About this article

Cite this article
Nagel, M.A., Gilden, D. The Relationship Between Herpes Zoster and Stroke. Curr Neurol Neurosci Rep 15, 16 (2015). https://doi.org/10.1007/s11910-015-0534-4
Published:
DOI: https://doi.org/10.1007/s11910-015-0534-4