
Abstract
Although both genetics and environmental factors play important roles in the etiology of type 2 diabetes (T2D), the extent to which genetics influence the environment (or vice versa) is still an open question. In this chapter, we first motivate the study of gene-environment (G × E) interaction with T2D, present statistical models of interaction, and then share illustrating examples of G × E interaction in the literature: PPGARG with T2D, COBLL1/GRB14 with fasting insulin levels and FTO with T2D, obesity, and physical activity.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Body Mass Index Group
- Fasting Insulin Level
- Body Mass Index Level
- High Body Mass Index Group
- High Body Mass Index Level
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Debates on the causes of type 2 diabetes (T2D) often center on the question of “nature vs nurture”—to what extent is the disease caused by genetics or exposure to environmental risk factors? In this chapter, we explore the idea of incorporating these external, non-genetic variables in genetic association studies of diabetes and diabetes-related traits with the goals of identifying novel genetic associations which might be modulated by environmental factors (or vice versa) and thus better understand disease mechanisms. We will present the methodology and current best practices of gene-environment interaction studies.
It is important to distinguish between biological and statistical interaction. Biological interaction is a term used to describe dependent biological systems. The simplest example relevant to diabetes physiology concerns the canonical relationship between insulin secretion and insulin action, captured by the molecular interaction between circulating insulin and its receptor.
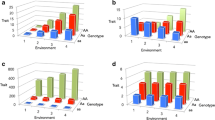
The statistical study of gene-environment interaction is concerned with finding genetic variants for which the association effect between an outcome and the variant is modified by an additional covariate, the environmental variable. In the presence of statistical interaction, the association between the genetic variant and the outcome is different for the various levels of the environmental variable (see Fig. 12.1 ). Note that the relationship can be viewed in a reciprocal manner, that is, the effect of the environmental variable on the outcome differs by genotype (or degree of genetic exposure).

Interpreting gene-environment interaction: For each panel, two plots are presented. In the top plot, the outcome (Y) value is displayed for the three genotypes under an additive genetic model. The association in the exposed group (blue) is compared to the genetic association in the unexposed subgroup (red). In the bottom plot, Y values are displayed for the two exposure groups with the three lines showing the values in the three genotype groups (0/0, purple; 0/1, red; and 1/1, blue). Three different interaction models are presented: (a) positive interaction occurs when the genetic effect and the interaction effect are in the same direction. Here, there is a genetic association in the absence of the exposure variable (when E = 0, βSNP = 0.1). In the exposed group, the association is strengthened (when E = 1, βSNP = 0.3). (b) Masking can occur when the genetic association is not present in one exposure group. Here, there is no genetic association in the non-exposed group (when E = 0, βSNP = 0). In a test of only the genetic main effect (black line), the association may still be detectable (βSNP = 0.09) but it is much stronger in the exposed group (when E = 1, βSNP = 0.2). (c) The third example illustrates a situation where the genetic effect in the exposed group (βSNP = 0.1) is in the opposite direction as the genetic effect in the unexposed group (βSNP = −0.1)
Questions related to gene-environment interaction include:
-
Are there individuals with particular genetic profiles who are more likely to develop T2D when exposed to sedentary lifestyles and/or poor nutrition? (See Chaps. 27 and 28.)
-
Can we personalize treatment knowing that certain medications work best in individuals with particular genetic profiles? (See Chaps. 24 and 25.)
1.1 Type 2 Diabetes, Glycemic Traits, and Gene-Environment Interactions
Type 2 diabetes is a disease of deteriorating beta-cell function and increasing insulin resistance with lifestyle risk factors such as lack of physical activity and obesity (McCarthy 2010). Candidate gene studies of genes in known diabetes pathways or previously implicated in neonatal diabetes or maturity-onset diabetes of the young (MODY) (Altshuler et al. 2000; Gloyn et al. 2004; Winckler et al. 2007; Sandhu et al. 2007) and genome-wide association studies (GWAS) of common genetic variants (Voight et al. 2010; Saxena et al. 2012; Morris et al. 2012) point to two mechanistic pathways of T2D disease progression: (1) variants near genes such as CDKAL1, CDKN2A, and CDKN2B reduce beta-cell mass and variants near genes such as MTNR1B, TCF7L2, and KCNJ11 influence beta-cell dysfunction, both of which result in reduced insulin secretion to lower glucose levels, and (2) insulin resistance, where cells and tissues become resistant to the effects of insulin, with association in or near genes such as FTO (related to obesity), IRS1, and PPARG (McCarthy 2010). Genetic variants associated with fasting glucose levels (related to beta-cell dysfunction) and fasting insulin levels (related to insulin resistance) have also been published (Prokopenko et al. 2008; Dupuis et al. 2010; Manning et al. 2012; Scott et al. 2012) (see Chaps. 2 and 3).
Studies show that both dietary fats and free fatty acids impact insulin resistance, possibly through mediating genetic factors such as PPARG variation (Roden et al. 1996; Kubota et al. 1999; Haag and Dippenaar 2005). External environmental variables (lifestyle factors such as diet or exercise) that impact T2D disease progression, beta-cell deterioration, and/or insulin sensitivity have been proposed as environmental exposures that may interact with genetics in the etiology of T2D. Furthermore, obesity contributes to insulin resistance by creating an “obesogenic environment,” making continuous body mass index (BMI) or categorical obesity (as defined by BMI ≥ 30 kg/m2) attractive candidate variables for interaction studies.
The measurement of lifestyle variables (physical activity, diet, and smoking status) can differ between studies, making meta-analysis and replication of genetic associations more difficult. For example, physical activity is a measure of an individual’s energy expenditure that can be summarized through questionnaires or more direct means such as continuous heart rate monitoring. Crude categories (sedentary, active, and/or very active) are often used in order to reach concordance between the various measures of physical activity used across the study designs, resulting in a loss of information in the subset of studies using sophisticated measurement tools. Other exposures such as smoking status and diet may be inconsistently measured across studies.
The InterAct project was designed to investigate lifestyle interactions in the development of T2D (The InterAct Consortium 2011) using a case-cohort sample of 10,901 incident diabetes cases from eight EPIC countries and a control cohort of 15,352 participants including 736 cases of incident T2D. Findings include an increased incidence of T2D with high total protein intake (HR = 1.13, 95 % confidence interval [CI]: 1.08–1.19) and high animal protein intake (HR = 1.12 95 % CI: 1.07–1.17), where effect modification was observed by sex (P < 0.001) and BMI among women (P < 0.001).
2 Statistical Model, Study Designs, and Interpretation
Numerous reviews of the design, implementation, and interpretation of GWAS of gene-environment interaction are available (Ottman 1996; Thomas 2010; Ober and Vercelli 2011; Aschard et al. 2012; Gauderman et al. 2013). Here, we first present the basic methodology of gene-environment interaction analyses and then describe several popular extensions.
2.1 Type 2 Diabetes as the Outcome
Although other models might be appropriate for the scientific question at hand, and complex diseases can be studied with a variety of models (Clayton 2012), the association between T2D and genetic factors is often assessed using logistic regression models in appropriate samples.
The term “main-effects model ” refers to a test of the marginal association between a genetic variant and the outcome (without interaction). Here, disease status is dichotomous and coded as T2D = 1 for individuals with T2D and T2D = 0 otherwise. Along with the independent genetic variable G, coded for an appropriate genetic model, additional covariates such as age and sex are usually included in the main-effects model:
Using the regression estimates, an estimate of the odds ratio for the association between G and T2D can be obtained: \( O{R}_G={e}^{{\widehat{\beta}}_3} \).
Statistical interaction by an independent variable E is defined as a departure from the multiplicative odds ratio model for the joint effect of G and E. Using the odds ratio for the association between T2D and E, OR E , a relationship between OR G and OR E can be defined: if there is no interaction, and the association between G and T2D is the same for all levels of E, then the two variables are independent and \( O{R}_{G,E}=O{R}_G\times O{R}_E \). When \( O{R}_{G,E}\ne O{R}_G\times O{R}_E \), or \( \frac{O{R}_{G,E}}{O{R}_GO{R}_E}\ne 1, \) statistical interaction is present.
One statistical test for interaction can be performed by including term for G, E, and the product of the two variables in the regression model:
In this model, the odds ratio estimate for the increased or decreased risk of T2D must be derived using both \( {\widehat{\beta}}_3 \) and \( {\widehat{\beta}}_5 \) (see Fig. 12.1).
2.2 Quantitative Outcomes
With quantitative outcomes such as glucose or insulin levels, linear regression models can be used to assess G × E interaction effects. There are several assumptions of linear regression that should be considered and are discussed in many statistical texts.
As with T2D, covariates such as age and BMI are commonly included in the genetic association tests of diabetes-related quantitative traits.
The main-effects regression model describes the relationship between Y and G:
A test for gene-environment interaction can be performed by adding an interaction term to the main-effects regression model:
When fit to data, there is evidence for statistical interaction if the estimate of the regression estimate \( {\widehat{\beta}}_5 \) is significantly different from zero.
2.3 Dichotomous Environmental Variable
Alternatively, if the E is dichotomous, the sample can be split into two strata, one with E = 1 and another with E = 0. The main-effects regression models, in which only the association between G and the outcome is considered, are assessed within strata of the environmental variable.
For logistic regression:
For linear regression:
Here, statistical interaction is present if the estimate \( {\hat{\beta}}_3^{E=1} \) differs significantly from the estimate \( {\hat{\beta}}_3^{E=0} \) (Aschard et al. 2010).
The interpretation of interaction for linear regression is in terms of the different slopes of the relationship between G and Y for different values of E, when E is a continuous measure, or different strata of E, when it is dichotomous (see Fig. 12.1).
3 Genome-Wide Interaction Tests
3.1 Screening for Interaction and “Case-Only” Tests
Often genome-wide main-effects analyses are performed prior to interaction studies being undertaken. This approach runs the risk of type II error (the failure to reject a false null hypothesis), as genetic variants with strong environmental interactions may have weak overall main effects and greater heterogeneity in main-effects testing, hindering their detection by main-effects screens. One proposed method studying gene-environment interaction while reducing the number of interaction tests performed was to choose a threshold P 0 (e.g., 0.05 or 0.01) and only investigate the interaction model (testing only the interaction term) on those genome-wide variants with a main-effects P-value less than P 0 (Kooperberg and LeBlanc 2008).
Another statistical test for interaction is the case-only test for gene-environment interaction (Piegorsch et al. 1994), which tests for an association between the environmental exposure (E ) and the genetic variant (G ) in a sample of “cases,” people with T2D, for example. Under the assumption that the genetic variable is not associated with the environmental variable, the estimated odds ratio, OR E , from this model is mathematically identical to the interaction odds ratio \( O{R}_{G=1,E=1} \) from formula (12.2). Although a large increase in statistical power is observed with the case-only test (Yang et al. 1997), there is a strong assumption that G is independent of E in the overall population. The case-only method and other methods that leverage the G and E independence assumption can exhibit both an increase in type I error (the false-positive rate) and a decrease in statistical power (Wu et al. 2013; Gauderman et al. 2013). Several methods that use the case-only test, but retain statistical power when the G-E independence assumption is violated, have been proposed. These include screening methods (Murcray et al. 2009, 2011) and “cocktail” methods (Mukherjee et al. 2012; Hsu et al. 2012) that combine the case–control test for interaction with case-only methods by using screening or model averaging (Mukherjee et al. 2008; Li and Conti 2009) approaches. Another method proposed for testing for interactions include a screening method that jointly assess the significance of the environment-gene association (as in the case-only test) and the disease-gene association in the screening step (Gauderman et al. 2013).
3.2 Joint Test of Main and Interaction Effects
The joint test of marginal G and interaction effects was introduced as a flexible test for genome-wide discovery of genetic associations when the underlying interaction model is suspected but unknown (Kraft et al. 2007). A statistical test is constructed to test if either or both of the genetic terms in the interaction model are significantly different from zero (\( {H}_0:{\beta}_3={\beta}_5=0 \)). The statistic can be constructed using a likelihood ratio test or Wald test; it follows a 2 degree-of-freedom chi-square distribution and remains valid when the gene-environment independence assumption is violated. Over a range of models, the joint test has comparable or better power than the interaction or case-only test, making it an attractive approach for genome-wide analysis, as only one statistical model needs to be applied to the genetic data.
3.3 Meta-Analysis Methods
Meta-analysis has become the de facto standard for performing genetic discovery analyses when the genetic effects are too small for detection with individual cohorts. Most common genetic discoveries were possible only when consortia were formed to conduct these meta-analysis (Prokopenko et al. 2008; Dupuis et al. 2010; Voight et al. 2010). In order to detect genetic interactions, much larger samples are required than that needed to detect comparable main effects (Aschard et al. 2010). One recent efficient and powerful meta-analysis method for testing the interaction effect across multiple studies has been proposed (Li et al. 2014). This method uses summary statistics from the individual studies (as in other meta-analysis methods) and a meta-regression approach to adaptively estimate the gene-environment interaction effect.
The joint test has been extended to a meta-analysis framework (Aschard et al. 2010; Manning et al. 2011). The joint meta-analysis, or JMA, is a meta-analysis method that allows individual cohorts to submit regression statistics from the interaction model: \( Y={\beta}_0+{\beta}_1\mathrm{SEX}+{\beta}_2\mathrm{AGE}+{\beta}_3G+{\beta}_4E+{\beta}_5G\times E+\varepsilon \). The statistics that need to be submitted for meta-analysis are the estimates of \( {\widehat{\beta}}_4 \) and \( {\widehat{\beta}}_5 \), the robust standard error and robust covariance of these estimates. The method is implemented in a modified version of the METAL software (Willer et al. 2010), available from the corresponding authors of the JMA paper (Manning et al. 2011), which produces summarized regression estimates of β 3 and β 5 and a 2 degree-of-freedom chi-square test of significance.
If the environmental variable is dichotomous, a simplified version of the joint test can be applied using a score test approach (Aschard et al. 2010). Regressions are performed in the stratum-specific main-effects models and the regression estimates of \( {\beta}_3^{E=1} \) and \( {\beta}_3^{E=0} \) are meta-analyzed using a standard inverse v ariance approach (de Bakker et al. 2008; Zeggini and Ioannidis 2009). The joint test, constructed with a sum of the fixed-effects tests for \( {\beta}_3^{E=1}=0 \) and \( {\beta}_3^{E=0}=0 \), follows a chi-square distribution with 2 degrees-of-freedom.
3.4 Statistical Issues and Best Practices
Statistical tests for interaction can be dependent on the trait scale—the interactions can be present in one scale (after a log-transformation, for example) and undetectable if modeled on another scale. Issues of environmental exposure and departures from the gene-environment independence assumption have been recently discussed (Lindström et al. 2009; Cornelis et al. 2012). Generally, the joint test performs well in the presence of environmental misspecification, a problem that can be somewhat controlled for through the use of robust standard error estimates (Cornelis et al. 2012). Furthermore, the use of robust standard error estimates corrects an issue of apparent QQ-plot inflation, from violations of assumptions such as linearity and homoscedasticity between Y and E, observed when comparing the P-values for a test of β 5, to those from the expected P-value distribution (Voorman et al. 2011).
Finally, a recent paper discusses the implication of confounding on the interaction term in the interaction model (12.2) (Keller 2014). The G × E term will be biased if either (a) a covariate (C) is associated with the SNP and the relationship between E and Y differs according to C (β C×E ≠ 0) or (b) C is associated with E and the relationship between G and Y differs according to C (β C×G ≠ 0). This implies that if either of these relationships holds, then the interaction terms β C × E or β C × SNP should be included in the model for each covariate considered. These models should be considered on a case-by-case basis, depending on the outcome, environmental variable, and whether or not the additional covariates could be independently associated with the SNP or be candidates for G × E interaction.
4 Illustrating Examples
4.1 PPARG
Of the candidate gene associations previously described, one of the early confirmed genetic associations with T2D was the Pro12Ala polymorphism in the PPARG gene (Altshuler et al. 2000; McCarthy 2010). Replication of this association was not universal, with several studies confirming the association and other studies failing to replicate it (see Ludovico et al. for a comprehensive citation list of the PPARG studies and Gouda et al. for a comprehensive review). Among the first attempts to explain this heterogeneity of effects, some groups found that along with increasing the risk for diabetes, the Pro121 allele also decreased insulin sensitivity, possibly lowered BMI, and was associated with increased adipose tissue formation (Roden et al. 1996; Kubota et al. 1999; Haag and Dippenaar 2005; Cecil et al. 2006).
In an analysis of “time to onset of diabetes” in the Diabetes Prevention Program, a significant gene-environment interaction was found between the Pro12Ala variant and obesity traits (Florez et al. 2007). Self-reported ethnicity was considered as an additional variable in a test for potential interaction but was not found to be significant. The Pro121 carriers progressed more quickly to diabetes (HR, 1.24; 95 % CI, 0.99–1.57; P = 0.07), and in models with P121Q-adiposity interactions with BMI (interaction P = 0.03) and waist circumference (interaction P = 0.002), the incidence of diabetes increased for higher mean BMI levels, showing that the protective effects of the alanine allele were attenuated at higher BMI levels.
A large meta-analysis (N = 42,910) was conducted based on 41 published studies and 2 unpublished studies to determine possible sources of the effect heterogeneity in the association of PPARG Pro121Ala with T2D (Ludovico et al. 2007). The association was confirmed (Ala12 OR = 0.81, P = 0.005), and population-specific differences in the reduced risk of T2D due to the Ala12 variant were also reported: the odds ratio was 0.65 in the Asian subgroup, 0.82 in the North American subgroup, and 0.85 in the European subgroup. Although the authors describe that the difference in the Asian subgroup could be due to BMI (48 % of the heterogeneity was explained by the BMI in the control groups), different population-specific genetic backgrounds were stated as a more likely cause for the heterogeneity observed in the European and North American studies.
In a subsequent meta-analysis, of 60 studies with up to 32,849 type 2 diabetes cases and 47,456 controls, the estimated odds ratio for the 121Ala allele was 0.86 (95 % CI: 0.91–0.90) and 0.85 (CI: 0.82–0.88) for random-effects and fixed-effects meta-analyses, respectively (Gouda et al. 2010). The authors report a moderate degree of inconsistency among the studies contributing to this meta-analysis (I 2 = 37 %, 95 % CI 9–54; P = 0.003). Ethnicity accounted for some of the heterogeneity (14 % of the between-study variance), but mean BMI levels among the T2D cases in the studies varied widely: although not significant, a trend was observed such that the protective effect of the variant was strongest (the odds ratio was lowest) for studies with mean case BMI < 25 kg/m2, and the protective effect was attenuated (the odds ratio increased toward the null) as the mean BMI in cases increased.
4.2 BMI Interactions with Fasting Insulin
Initial publications from the Meta-Analysis of Glucose and Insulin-related traits Consortium (MAGIC) described 16 loci associated with fasting glucose levels compared to two loci associated with fasting insulin levels (Prokopenko et al. 2008; Dupuis et al. 2010), indicating differences in the genetic architectures for beta-cell dysfunction and insulin resistance. The marginal models investigated by MAGIC included minimal adjustments for age and sex. Subsequently, two efforts were undertaken to investigate the role of obesity in the variation of quantitative glycemic traits: (1) interaction models on a subset of MAGIC cohorts for which obesity measures were available (Manning et al. 2012) and (2) meta-analyses of main-effects models adjusting for obesity measures including larger sample sizes with the inclusion of Metabochip genotype data (Scott et al. 2012) (see Chap. 3).
For the first analysis, two terms were added to models for fasting glucose and log-transformed fasting insulin: the adjustment for body mass index (BMI) and the interaction between a genetic variant and BMI.
The joint meta-analysis was applied in a genome-wide analysis of 2.4 million single nucleotide polymorphisms (SNPs) and six and seven additional loci were found to be associated with fasting insulin and fasting glucose, respectively (Manning et al. 2012), with one locus, PPP1R3B, showing association with both fasting insulin and fasting glucose levels. Of these loci, one fasting insulin association (rs7607980 in the COBLL1/GRB14 locus, joint P = 4.3 × 10−20) and three fasting glucose loci displayed a greater degree of significance in the joint test compared to a model that only included an adjustment for BMI. All loci were reported as significant in the second analysis, demonstrating that either larger sample sizes or adjustment for BMI was necessary for their discovery (Scott et al. 2012). Although a number of the loci reported in Manning et al. showed differential evidence for significance and effect sizes in the high BMI group compared to the low BMI group (as defined by a BMI cutoff of 28 kg/m2), only rs7607980 showed evidence for an interaction effect when a meta-analysis of the interaction term was performed (P = 0.0002). The additive main effect of rs7607980 on log-transformed fasting insulin levels was 0.02 (with standard error 0.0033), similar to the BMI-adjusted main effect of 0.028 (0.0033). In the subset with high BMI, the effect was 0.041 (0.0064) with P = 3.0 × 10−10, while in the lower BMI stratum, the effect was weaker and less significant at 0.0175 (0.0041) with P = 1.8 × 10−5. The stratum-specific effect was consistent with the jointly estimated effect from the interaction model (Fig. 12.2). These findings support the sensible assumption that taking adiposity into account can augment discovery of genetic variants that underlie insulin resistance.

The genetic effect estimate of rs7607980 from the COBLL1/GRB14 locus accounting for the interaction with BMI. The additive genetic effect of rs7607980 changes for different BMI levels. (a) For the joint meta-analysis, where BMI is a continuous exposure variable, the estimate (solid black line) and 95 % confidence interval (gray curves), the estimate is \( {\widehat{\beta}}_{\mathrm{SNP}}=-0.06+0.003\times BMI \). (b) The studies were dichotomized into high- and low-BMI groups and the estimate of the genetic effect was obtained within each subgroup. The additive genetic effect is displayed with the circles, with the 95 % confidence interval of the estimate shown by the vertical lines. In the subset with high BMI, \( {\widehat{\beta}}_{\mathrm{SNP}}=0.04 \), and in the subset with the lower BMI, \( {\widehat{\beta}}_{\mathrm{SNP}}=0.02 \)
4.3 FTO: Type 2 Diabetes, Body Mass Index, and Interaction with Physical Activity
In 2007, the Wellcome Trust Case Control Consortium (WCCC) performed a genome-wide association study for type 2 diabetes and described a strong increase of risk for T2D associated with SNPs in the first intron of the FTO gene (rs9939609, OR = 1.27; P = 5 × 10−8 in 1,924 T2D cases and 2,938 controls) which was replicated in an independent sample (OR = 1.15; P = 9 × 10−6 in 3,757 T2D cases and 5,346 controls) (Frayling et al. 2007). A strong association with BMI was also observed (P = 3 × 10−35 in 30,081 individuals with BMI values). As a classic demonstration of confounding, the T2D association was abolished in subsequent analyses that adjusted for BMI as a covariate in the regression procedure (OR = 1.03; P = 0.44). FTO is now recognized as a locus harboring strong associations with obesity (Frayling et al. 2007; Scuteri et al. 2007) with associations with T2D appearing because the typical T2D cases are more obese than typical nondiabetic controls.
An analysis was performed in the Danish Inter99 cohort exploring SNP by physical activity interactions at the FTO locus (Andreasen et al. 2008). First the association between the FTO SNP rs9939609 and BMI was established: the AA genotype group had 1.1 kg/m2 higher BMI levels on average compared to the TT genotype group, and those in the AT genotype had BMI levels 0.3 kg/m2 higher than the TT genotype group (additive effect P = 1 × 10−9 with N = 5,722). Physical activity status was assessed by questionnaire and individuals were classified into three groups: physically inactive (N = 1,914), lightly or moderately physically active (N = 3,224), and very physically active (N = 416). A statistically significant interaction was observed (P = 0.007). The genetic effect between rs9939609 and BMI was weaker in the individuals with the highest physical activity: the average BMI in the AA genotype group was 0.47 kg/m2 (not significant) higher compared to the TT group. This association was stronger in the physically inactive group: here, the average BMI in the AA genotype group was 1.95 kg/m2 higher than the TT group, a much larger increase than 0.38 kg/m2 difference observed between the AT group and the TT group.
A careful exploration of this interaction effect was reported in a meta-analysis of 218,166 adults (Kilpeläinen et al. 2011). The physical activity interaction was replicated (P = 0.005), although the effect was not as strong as originally reported, an example of a possible winner’s curse. In all individuals, the additive effect of the BMI-increasing allele (A) of rs9939609 was 0.36 kg/m2 (P = 1.8 × 10−75). In the two physical activity strata applied across the study samples, the additive effect of the BMI-increasing allele was 0.46 kg/m2 in the inactive group (P = 3.7 × 10−23, N = 54,611) and 0.32 kg/m2 in the active group (P = 4.5 × 10−69, N = 163,555). Interestingly, heterogeneity was observed (I 2 = 36 %), mainly from cohorts of European origin. When the North American cohorts were analyzed on their own, the interaction was much stronger (P = 1.6 × 10−9): the additive effects were 0.82 kg/m2 (P = 2.7 × 10−21, N = 9,438) and 0.34 kg/m2 (P = 6.1 × 10−12, N = 38,500) in the inactive and active groups, respectively, with no measurable heterogeneity (I 2 = 0 %). The authors of this study carefully consider sources of bias and confounding in this association, and although they note that this result has importance for public health (being physically active can further alleviate a genetic predisposition toward obesity beyond the obvious health benefits), they further note that the changes in the genetic association due to physical activity could be confounded by correlated lifestyle and environmental factors. The observed interaction does not imply causation—as in other studies of genetic effects, the appropriate epidemiological interpretations apply.
5 Summary
In this chapter, we have introduced the concept of statistical interaction by exposure variables in the study of the genetic determinants of T2D and related traits. The basic methodology of gene-environment interaction studies was presented along with several extensions that have been recently proposed. Finally, three relevant examples of gene-environment interaction in the literature were described.
Of the greatest importance for future studies of gene-lifestyle interaction are the following. First, we suggest a careful consideration of the epidemiological design and hypotheses to be tested in the study—if the harmonization of exposure variables and outcomes increases noise and heterogeneity in your study data, then the potential gain in power from larger sample sizes might be obliterated. Second, appropriate statistical tests must be applied—if there is a reasonable expectation that there is a genetic basis for the exposure variable, then methods that depend upon gene-exposure independence may not be ideal.
Studies that account for differences in genetic effects due to environmental exposures will continue to be important as genetic association studies query low-frequency and rare genetic variants. Testing for interaction, accounting for the variability in the outcome due to the exposure (by using it as a covariate) or looking for genetic associations in distinct subgroups (revealing masking effects), may reveal additional genetic susceptibility loci that could illuminate biological pathways in the pathophysiology of type 2 diabetes.
References
Altshuler D, Hirschhorn JN, Klannemark M (2000) The common PPAR[gamma] Pro12Ala polymorphism is associated with decreased risk of type 2 diabetes. Nat Genet 26(1):76–80
Andreasen CH, Stender-Petersen KL, Mogensen MS et al (2008) Low physical activity accentuates the effect of the FTO rs9939609 polymorphism on body fat accumulation. Diabetes 57:95–101. doi:10.2337/db07-0910
Aschard H, Hancock DB, London SJ, Kraft P (2010) Genome-wide meta-analysis of joint tests for genetic and gene-environment interaction effects. Hum Hered 70:292–300. doi:10.1159/000323318
Aschard H, Lutz S, Maus B et al (2012) Challenges and opportunities in genome-wide environmental interaction (GWEI) studies. Hum Genet 131:1591–1613. doi:10.1007/s00439-012-1192-0
Cecil JE, Watt P, Palmer CN, Hetherington M (2006) Energy balance and food intake: the role of PPARγ gene polymorphisms. Physiol Behav 88(3):227–233
Clayton D (2012) Link functions in multi-locus genetic models: implications for testing, prediction, and interpretation. Genet Epidemiol 36:409–418. doi:10.1002/gepi.21635
Cornelis MC, Tchetgen EJT, Liang L et al (2012) Gene-environment interactions in genome-wide association studies: a comparative study of tests applied to empirical studies of type 2 diabetes. Am J Epidemiol 175:191–202. doi:10.1093/aje/kwr368
de Bakker PIW, Ferreira MAR, Jia X et al (2008) Practical aspects of imputation-driven meta-analysis of genome-wide association studies. Hum Mol Genet 17:R122–R128. doi:10.1093/hmg/ddn288
Dupuis J, Langenberg C, Prokopenko I et al (2010) New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet 42:105–116. doi:10.1038/ng.520
Florez JC, Jablonski KA, Sun MW et al (2007) Effects of the type 2 diabetes-associated PPARG P12A polymorphism on progression to diabetes and response to troglitazone. J Clin Endocrinol Metab 92:1502–1509. doi:10.1210/jc.2006-2275
Frayling TM, Timpson NJ, Weedon MN et al (2007) A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science 316:889–894. doi:10.1126/science.1141634
Gauderman WJ, Zhang P, Morrison JL, Lewinger JP (2013) Finding novel genes by testing G × E interactions in a genome-wide association study. Genet Epidemiol 37:603–613. doi:10.1002/gepi.21748
Gloyn AL, Pearson ER, Antcliff JF et al (2004) Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetes. N Engl J Med 350:1838–1849. doi:10.1056/NEJMoa032922
Gouda HN, Sagoo GS, Harding AH et al (2010) The association between the peroxisome proliferator-activated receptor- 2 (PPARG2) Pro12Ala gene variant and type 2 diabetes mellitus: a HuGE review and meta-analysis. Am J Epidemiol 171:645–655. doi:10.1093/aje/kwp450
Haag M, Dippenaar NG (2005) Dietary fats, fatty acids and insulin resistance: short review of a multifaceted connection. Med Sci Monit 11:RA359–RA367
Hsu L, Jiao S, Dai JY et al (2012) Powerful cocktail methods for detecting genome-wide gene-environment interaction. Genet Epidemiol 36:183–194. doi:10.1002/gepi.21610
Keller MC (2014) Gene × environment interaction studies have not properly controlled for potential confounders: the problem and the (simple) solution. Biol Psychiatry 75:18–24. doi:10.1016/j.biopsych.2013.09.006
Kilpeläinen TO, Qi L, Brage S et al (2011) Physical activity attenuates the influence of FTO variants on obesity risk: a meta-analysis of 218,166 adults and 19,268 children. PLoS Med 8:e1001116–e1001116. doi:10.1371/journal.pmed.1001116
Kooperberg C, LeBlanc M (2008) Increasing the power of identifying gene × gene interactions in genome-wide association studies. Genet Epidemiol 32(3):255–263
Kraft P, Yen Y-C, Stram DO et al (2007) Exploiting gene-environment interaction to detect genetic associations. Hum Hered 63:111–119. doi:10.1159/000099183
Kubota N, Terauchi Y, Miki H et al (1999) PPARγ mediates high-fat diet–induced adipocyte hypertrophy and insulin resistance. Mol Cell 4:597–609. doi:10.1016/S1097-2765(00)80210-5
Li D, Conti DV (2009) Detecting gene-environment interactions using a combined case-only and case–control approach. Am J Epidemiol 169:497–504. doi:10.1093/aje/kwn339
Li S, Mukherjee B, Taylor JMG et al (2014) The role of environmental heterogeneity in meta-analysis of gene-environment interactions with quantitative traits. Genet Epidemiol 38:416–429. doi:10.1002/gepi.21810
Lindström S, Yen Y-C, Spiegelman D, Kraft P (2009) The impact of gene-environment dependence and misclassification in genetic association studies incorporating gene-environment interactions. Hum Hered 68:171–181. doi:10.1159/000224637
Ludovico O, Pellegrini F, Paola R et al (2007) Heterogeneous effect of peroxisome proliferator-activated receptor γ2 Ala12 variant on type 2 diabetes risk. Obesity 15:1076–1081. doi:10.1038/oby.2007.617
Manning AK, LaValley M, Liu C-T et al (2010) Gene–environment-wide association studies: emerging approaches. Nat Publ Group 11:259–272. doi:10.1038/nrg2764
Manning AK, LaValley M, Liu C-T et al (2011) Meta-analysis of gene-environment interaction: joint estimation of SNP and SNP × environment regression coefficients. Genet Epidemiol 35:11–18. doi:10.1002/gepi.20546
Manning AK, Hivert M-F, Scott RA et al (2012) A genome-wide approach accounting for body mass index identifies genetic variants influencing fasting glycemic traits and insulin resistance. Nat Genet 44:659–669. doi:10.1038/ng.2274
McCarthy MI (2010) Genomics, type 2 diabetes, and obesity. N Engl J Med 363:2339–2350. doi:10.1056/NEJMra0906948
Morris AP, Voight BF, Teslovich TM et al (2012) Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet 44:981–990. doi:10.1038/ng.2383
Mukherjee B, Ahn J, Gruber SB et al (2008) Tests for gene-environment interaction from case–control data: a novel study of type I error, power and designs. Genet Epidemiol 32:615–626. doi:10.1002/gepi.20337
Mukherjee B, Ahn J, Gruber SB, Chatterjee N (2012) Testing gene-environment interaction in large-scale case–control association studies: possible choices and comparisons. Am J Epidemiol 175:177–190. doi:10.1093/aje/kwr367
Murcray CE, Lewinger JP, Gauderman WJ (2009) Gene-environment interaction in genome-wide association studies. Am J Epidemiol 169:219–226. doi:10.1093/aje/kwn353
Murcray CE, Lewinger JP, Conti DV et al (2011) Sample size requirements to detect gene-environment interactions in genome-wide association studies. Genet Epidemiol 35:201–210. doi:10.1002/gepi.20569
Ober C, Vercelli D (2011) Gene–environment interactions in human disease: nuisance or opportunity? Trends Genet 27:107–115. doi:10.1016/j.tig.2010.12.004
Ottman R (1996) Gene–environment interaction: definitions and study designs. Prev Med 25:764
Piegorsch WW, Weinberg CR, Taylor JA (1994) Non-hierarchical logistic models and case-only designs for assessing susceptibility in population-based case–control studies. Stat Med 13:153–162
Prokopenko I, Langenberg C, Florez JC et al (2008) Variants in MTNR1B influence fasting glucose levels. Nat Genet 41:77–81. doi:10.1038/ng.290
Roden M, Price TB, Perseghin G et al (1996) Mechanism of free fatty acid-induced insulin resistance in humans. J Clin Invest 97:2859. doi:10.1172/JCI118742
Sandhu MS, Weedon MN, Fawcett KA et al (2007) Common variants in WFS1 confer risk of type 2 diabetes. Nat Genet 39:951–953. doi:10.1038/ng2067
Saxena R, Elbers CC, Guo Y et al (2012) Large-scale gene-centric meta-analysis across 39 studies identifies type 2 diabetes loci. Am J Hum Genet 90:410–425. doi:10.1016/j.ajhg.2011.12.022
Scott RA, Lagou V, Welch RP et al (2012) Large-scale association analyses identify new loci influencing glycemic traits and provide insight into the underlying biological pathways. Nat Genet 44:991–1005. doi:10.1038/ng.2385
Scuteri A, Sanna S, Chen W-M et al (2007) Genome-wide association scan shows genetic variants in the FTO gene are associated with obesity-related traits. PLoS Genet 3, e115. doi:10.1371/journal.pgen.0030115
The InterAct Consortium (2011) Design and cohort description of the InterAct Project: an examination of the interaction of genetic and lifestyle factors on the incidence of type 2 diabetes in the EPIC Study. Diabetologia 54:2272–2282. doi:10.1007/s00125-011-2182-9
Thomas D (2010) Gene–environment-wide association studies: emerging approaches. Nat Publ Group 11:259–272. doi:10.1038/nrg2764
Voight BF, Scott LJ, Steinthorsdottir V et al (2010) Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat Genet 42:579–589. doi:10.1038/ng.609
Voorman A, Lumley T, McKnight B, Rice K (2011) Behavior of QQ-plots and genomic control in studies of gene-environment interaction. PLoS ONE 6, e19416. doi:10.1371/journal.pone.0019416
Willer CJ, Li Y, Abecasis GR (2010) METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26:2190–2191. doi:10.1093/bioinformatics/btq340
Winckler W, Weedon MN, Graham RR et al (2007) Evaluation of common variants in the six known maturity-onset diabetes of the young (MODY) genes for association with type 2 diabetes. Diabetes 56:685–693. doi:10.2337/db06-0202
Wu C, Chang J, Ma B et al (2013) The case-only test for gene-environment interaction is not uniformly powerful: an empirical example. Genet Epidemiol 37:402–407. doi:10.1002/gepi.21713
Yang Q, Khoury MJ, Flanders WD (1997) Sample size requirements in case-only designs to detect gene-environment interaction. Am J Epidemiol 146:713–720
Zeggini E, Ioannidis JPA (2009) Meta-analysis in genome-wide association studies. Pharmacogenomics 10:191–201. doi:10.2217/14622416.10.2.191
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Manning, A.K. (2016). Gene-Environment Interaction: Methods and Examples in Type 2 Diabetes and Obesity. In: Florez, J. (eds) The Genetics of Type 2 Diabetes and Related Traits. Springer, Cham. https://doi.org/10.1007/978-3-319-01574-3_12
Download citation
DOI: https://doi.org/10.1007/978-3-319-01574-3_12
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-01573-6
Online ISBN: 978-3-319-01574-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)