
Abstract
Purpose of Review
The intricate interplay between inflammatory and reparative responses in the context of heart injury is central to the pathogenesis of heart failure. Recent clinical studies have shown the therapeutic benefits of anti-inflammatory strategies in the treatment of cardiovascular diseases. This review provides a comprehensive overview of the cross-talk between immune cells and fibroblasts in the diseased heart.
Recent Findings
The role of inflammatory cells in fibroblast activation after cardiac injury is well-documented, but recent single-cell transcriptomics studies have identified putative pro-inflammatory fibroblasts in the infarcted heart, suggesting that fibroblasts, in turn, can modify inflammatory cell behavior. Furthermore, anti-inflammatory immune cells and fibroblasts have been described. The use of spatial and temporal-omics analyses may provide additional insights toward a better understanding of disease-specific microenvironments, where activated fibroblasts and inflammatory cells are in proximity.
Summary
Recent studies focused on the interplay between fibroblasts and immune cells have brought us closer to the identification of cell type–specific targets for intervention. Further exploration of these intercellular communications will provide deeper insights toward the development of novel therapeutics.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Heart failure is a clinical syndrome defined as the inability of the heart to pump an adequate amount of blood to meet metabolic demands. It is a leading cause of mortality and morbidity throughout the world [1]. Multiple cardiovascular diseases, including myocardial infarction, hypertension, valve disease, and cardiomyopathies, can cause heart failure. Extracellular matrix (ECM) proteins are normal components of the myocardium that provide stability for dynamic contractions and insulate electrical activity, and some expansion of ECM is part of myocardial healing, for example, to prevent life-threatening complications such as cardiac rupture. Accumulation of ECM proteins in the heart interstitium results in fibrosis, a requisite component of cardiac remodeling. This excess ECM deposition leads to reduced compliance of the ventricular wall, leading to diastolic dysfunction. Furthermore, progressive remodeling of fibrotic tissue changes the geometry of the heart, producing less efficient contraction and reduced cardiac output. It can also disrupt normal conduction, leading to arrhythmias [2]. Thus, it would be useful to control fibrosis, obtaining its benefits while limiting its less desirable effects [3].
Cardiac fibroblasts that reside in the heart interstitium play central roles in cardiac remodeling because of their capacity to produce ECM proteins. They balance the secretion and degradation of ECM proteins [4]. Once tissue injury occurs, cardiomyocyte stress and inflammatory processes activate fibroblasts, promoting the reparative program that includes proliferation, migration, and increased ECM-related production [5, 6]. At the same time, tissue injury triggers inflammatory cascades, which induce the infiltration of immune cells [7]. Because these cellular components are also key players in fibrosis, it is imperative to elucidate the interactions between fibroblasts and immune cells and to develop a complete understanding of cardiac fibrosis with a goal of therapeutic manipulation.
Here, we provide an overview of the pathophysiology of inflammation and fibrotic heart disease by describing the immune cell populations associated with cardiac inflammation and the fibrotic process. We then outline how fibroblasts participate in the inflammatory process, highlighting that future research is needed to discover potential avenues for intervention.
Inflammation in Cardiac Diseases
Inflammation is an indispensable response to injury and is needed for the clearance of harmful stimuli and damaged tissue. In the heart, resident immune cells serve as first responders. Both innate and adaptive immune components contribute to the cardiac inflammatory response [7, 8].
Resident macrophages and mast cells initiate innate inflammation via the NOD-like receptor family pyrin domain–containing 3 (NLRP3) inflammasome pathway. Under pathological conditions, stressed or dying cells release cytoplasmic components such as ATP, mitochondrial DNA, sarcomeric proteins, and heat shock proteins [9,10,11,12]. Such molecules termed damage-associated molecular patterns (DAMPs) are recognized by the pattern recognition receptors (PRRs), which are expressed on resident cells, including fibroblasts and immune cells. Engagement of DAMPs and PRRs promotes the nuclear factor-κB (NF-κB) pathway, followed by the recruitment of the NLRP3 inflammasome complex composed of the apoptotic speck protein containing a caspase recruitment domain (ASC) and pro-caspase 1. The activated inflammasome is responsible for the production of inflammatory molecules including interleukins, IL1 and IL18, and Gasdermin D (GSDMD) [13]. IL1β and IL18 rapidly recruit neutrophils to the site of injury [14, 15]. Additionally, the p38-mitogen activated protein kinase pathway is activated, leading to the production of inflammatory mediators such as tumor necrosis factor-α (TNF-α) and IL6 [16, 17]. Multiple heart conditions can trigger these inflammatory cascades. We describe a few below.
Ischemia
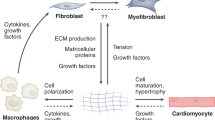
Ischemia is one of the most common and well-studied cardiac injuries in humans. Hypoxia and accumulation of toxic metabolites induce cardiomyocyte stress and death, initiating DAMP-related pathways (Fig. 1). In animal injury models, the first 3 days after insult is referred to as the inflammatory phase, during which neutrophils, monocytes, and macrophages play prominent roles. These immune cells produce not only pro-inflammatory cytokines/chemokines but proteolytic enzymes that degrade ECM structures. Inflammatory mediators such as IL1β and IL6 also stimulate the proliferation and migration of resident fibroblasts [18, 19]. The following 2 weeks is called the proliferative phase. During this period, phagocytosis by neutrophils and macrophages removes cell and ECM debris. Lymphocytes are recruited and activate the adaptive immunity [20]. Neutrophils rapidly decrease by apoptosis and are largely gone by day 7 [21]. Macrophages shift from pro-inflammatory to anti-inflammatory [22]. Activated fibroblasts and myofibroblasts display a peak of proliferation and secrete ECM to stabilize the tissue. As the proliferative phase gives way to the maturation phase, ECM cross-linking and inflammatory cell apoptosis occur [23]. Some fibroblasts return to a quiescent state, while some in the scar produce ECM-modifying proteins [24].

Potential cross-talk between fibroblasts and immune cells after the heart injury. The cellular response to tissue damage is initiated by resident cells that serve as first responders to damage-associated molecular patterns (DAMPs) and reactive oxidative species (ROS). Inflammasome pathway cytokines activate fibroblasts and recruit neutrophils and monocytes, which facilitate inflammation by the secretion of pro-inflammatory cytokines and proteolytic enzymes. Activated fibroblasts initially exhibit a pro-inflammatory phenotype characterized by the expression of multiple chemotactic mediators. CD4+ T cells secrete cytokines that enhance fibroblast functions such as proliferation and migration. Subsequently, neutrophils and macrophages undergo a transition to anti-inflammatory populations in response to TGF-β and concomitant with increased phagocytic actions. Regulatory T cells, eosinophils, and basophils also contribute to the resolution of inflammation and scar formation. Two subsets of fibroblasts, Wif1.+ and IFN-stimulated, are not depicted, but they may also interact with immune cells [101••, 102]
In addition to responses to acute injury, inflammation associated with fibrosis is linked to the pathogenesis of chronic heart failure, even in the absence of obvious tissue damage or infection. Low-grade inflammation occurs in the diseased heart regardless of the etiology [25]. Indeed, inflammatory cytokines including IL1β, IL6, and TNF-α are upregulated in the serum of patients with heart failure [26]. Histologically, the end-stage failing heart of transplant recipients contains infiltrating macrophages, lymphocytes, and mast cells [27]. The cause of persistent inflammation is not always clear, but one finding implicates angiotensin II. Angiotensin II is commonly elevated in the serum of patients with chronic heart failure, and not only activates fibroblast directly but also induces the NLRP3 inflammasome, contributing to sustained inflammation and fibrosis [28]. Oxidative stress induced by reactive oxygen species (ROS) is another contributor to inflammation in failing hearts [29].
Aging
Aging is associated with the progression of heart failure with diastolic dysfunction. The aging heart exhibits structural alterations including cardiomyocyte hypertrophy and interstitial fibrosis, and increased monocyte-derived macrophages and T cells are observed in the fibrotic area [30]. Single-cell RNA-sequencing of the aged, murine heart has shown that fibroblasts in the aged heart have upregulated inflammatory and osteogenic genes [31]. The osteogenic program, defined by the expression of bone and cartilage genes, is implicated as a pro-fibrotic response [32]. Somatic mutations also accumulate with aging. Aging-related mutations in the transcriptional regulators, DMNT3A, ASXL1, or TET2, are associated with clonal expansion of pathogenic immune cells and activated inflammasome pathway [33]. Patients with heart failure carrying these mutations have higher all-cause mortality [34•], which may reflect extensive chronic inflammation. Similarly, epigenetic alterations such as DNA methylation and histone modifications also accumulate with aging. The gene dysregulation associated with aging-related genetic change disturbs homeostasis, leading to mitochondrial stress [35].
Diabetes and Obesity
Diabetes mellitus and obesity are risk factors for heart failure. Although the risk of heart failure can be partially attributed to the prevalence of ischemic heart disease and hypertension, diabetes can independently contribute to the pathogenesis of heart failure, leading to diabetic cardiomyopathy [36]. Diabetic cardiomyopathy is characterized by cardiomyocyte hypertrophy, interstitial fibrosis, and infiltration of inflammatory cells. Hyperglycemia leads to ROS generation, subsequently triggering the NLRP3 inflammasome [37, 38]. One animal study has suggested that hyperglycemia stimulates ROS production in T cells, followed by transforming growth factor (TGF)-β activation. Oxidants also contribute to inflammation in patients with obesity. Oxidized fatty acid by-products can activate apoptosis and inflammation [39]. Adipocytes and progenitor cells can also be a source of pro-inflammatory cytokines. A recent cohort study has shown that higher levels of IL6, IL18, CC motif chemokine ligand 2 (CCL2), and CCL7 were observed in patients with a higher body mass index [40]. Local or systemic hypoxia due to impaired angiogenesis, or sleep apnea syndrome in obesity may trigger oxidative stress driving an inflammatory cascade.
HIV
Infection with the human immunodeficiency virus (HIV) increases the risk for cardiovascular disease and patients often develop heart failure in the absence of ischemic heart disease [41]. Several factors may elicit inflammation in the myocardium, aside from the response to the virus. For example, HIV-infected patients have dysfunctional mucus in the intestine, resulting in microbial translocation, and systemic inflammation even when on anti-viral therapy. Indeed, monocytes infiltrate HIV-infected human hearts with diastolic dysfunction [42]. Monocyte and monocyte-derived macrophages persistently contribute to fibroblast activation and fibrosis as discussed below. Importantly, platelets in HIV patients are activated by the viral envelope proteins. TGF-β is also upregulated in platelets, contributing to the exacerbation of fibrosis [43, 44].
How Do Immune Cells Modulate Fibroblasts?
Neutrophils
Neutrophils and other granulocytes are rare populations in the healthy heart. In the setting of injury, the activated inflammasome signaling from resident cells stimulates the mobilization of neutrophils from bone marrow. IL1, granulocyte colony stimulating factor (G-CSF), and complement proteins act as attractants of neutrophils [45•]. Neutrophils are essential during the early inflammatory phase of cardiac injury. They serve to remove cell and ECM debris, can be stimulated by DAMPs, and express pro-inflammatory cytokines such as TNF-α and IL6 [45•].
Pro-inflammatory cytokines from neutrophils have important roles in cardiac fibrosis, although the evidence regarding direct effects on cardiac fibroblasts in vivo is still limited. Several in vitro studies have demonstrated cytokine activation of fibroblasts. For example, TNF-α stimulated the proliferation of fibroblasts, although ECM synthesis was unaffected [46, 47]. Also, IL-6 activates fibroblast proliferation and ECM synthesis [19, 48]. In addition to inflammatory cytokines, neutrophil granules contain anti-microbial agents (myeloperoxidase: MPO, lactoferrin) and matrix metalloproteinases MMP8 and MMP9. Recruited neutrophils release these components when stimulated by IL1β. MPO catalyzes ROS production [49], which subsequently stimulates fibroblast proliferation, activation, and ECM remodeling [50]. Some ECM fragments released during matrix proteolysis can further enhance fibroblast activation. Taken together, neutrophils act on fibroblasts through multiple mechanisms early in fibrosis.
Traditionally, neutrophils are regarded as pro-inflammatory. Indeed, excessive and persistent infiltration of neutrophils exacerbates tissue injury by the combined effect of the inflammatory cytokines, ROS, and proteolytic enzymes [51]. However, in the mouse, ablation of neutrophils resulted in decreased systolic function after MI, suggesting that neutrophils may have protective roles in the maintenance of cardiac function [52]. Novel subsets of Ly6G+/CD206− neutrophils with a pro-inflammatory phenotype have been described after MI [53]. The same study described a Ly6G+/CD206+ anti-inflammatory population that secretes the pro-fibrotic cytokine, IL-10. These subsets were referred to as N1 and N2 populations similar to the M1-M2 nomenclature in macrophages described below. The polarization from N1 to N2 can be induced by IL4, and N2 subsets were more abundant 7 days post-MI. Taken together, these data suggest that neutrophils actively stimulate fibroblast migration and proliferation.
Macrophages and Monocytes
Tissue-resident macrophages, characterized by the absence of CC motif receptor 2 (CCR2) expression, maintain heart homeostasis by regulating angiogenesis and tissue repair and removing dysfunctional mitochondria [54,55,56]. Ablation of this population results in loss of regenerative capacity in neonatal mice [57]. Recent studies in the mouse have shown that this resident population can be subdivided into two groups: a TIMD4+, LYVE1+, FOLR2+ subset and an MHCII+ subset, which may be conserved in humans [58].
In the setting of injury, resident macrophages detect ROS and DAMPs, triggering inflammatory cascades [59]. CCL2 secreted by cardiomyocytes, fibroblasts, resident macrophages, and B cells recruits CCR2-positive monocytes [60, 61]. CCR2+/Ly6c+ monocytes infiltrate at the site of injury and differentiate into CCR2+ macrophages. These monocyte-derived macrophages secrete IL1β, IL6, and TNF-α, recruiting immune cells and stimulating fibroblasts [62]. Like neutrophils, CCR2+ macrophages secrete proteolytic enzymes such as cathepsins and MMPs to yield bioactive ECM degradation products, promoting fibroblast proliferation and ECM secretion.
Although current standards suggest that M1/M2 nomenclature for macrophages is an oversimplification, CCR2+ macrophages, generally categorized as M1, are considered pro-inflammatory. Phagocytosis can shift macrophages to an anti-inflammatory mode that is broadly categorized as M2 [52, 63]. These phagocytic macrophages are characterized by the MER proto-oncogene tyrosine kinase (MERTK) and contribute to the clearance of dead neutrophils and cardiomyocyte debris; they are also considered pro-fibrotic. The M2 macrophages promote ECM synthesis in fibroblasts by secreting IL10 and TGF-β. IL10 treatment in the mouse MI model also drives macrophages toward the M2 phenotype, leading to increased fibroblast proliferation, migration, and ECM synthesis [64]. Matricellular proteins, such as osteopontin produced by reparative macrophages, can also stimulate ECM production by fibroblasts [65, 66]. Indeed, a spatial multi-omics study has suggested that SPP1-expressing macrophages are in proximity to activated fibroblasts in the human infarcted heart [67••]. Several studies have attempted to examine whether the ablation of macrophages is beneficial after cardiac injury; these results remain inconclusive [65, 68,69,70,71]. Based on the complicated roles of macrophages during tissue repair, it may be more beneficial to target fibroblast/macrophage interactions than macrophages directly.
Lymphocytes
T cells are primarily involved in the adaptive immune response and activated by antigen-presenting cells. Triggered by chemotactic signals, from neutrophils and macrophages, T cells infiltrate in the later stages of heart injury. The NLRP3 inflammasome and GM-CSF, CCL2, and CXC motif ligand (CXCL) families all serve as attractants for T cells [8]. CD4+ T cells secrete interferon (IFN)-γ and activate additional cells at the site of injury. Ablation of CD4+ T cells attenuates fibrosis and adverse remodeling in mouse disease models, suggesting that they play a pro-inflammatory role [20, 72]. Additionally, a recent study has demonstrated that IFN-γ-stimulated fibroblasts may increase antigen presentation to CD4+ cells via Class II MHC molecules during mouse pressure overload [73]. Th17 cells are a minor but not insignificant population of CD4+ T cells, characterized by a pro-inflammatory profile and abundant IL17A secretion. Studies have shown that IL17A stimulates fibroblast-derived GM-CSF, which activates monocyte recruitment and differentiation into inflammatory macrophages [74, 75]. Fibroblast-specific deficiency of the IL17 receptor is associated with a better prognosis and a decrease in GM-CSF in a murine MI model. Regulatory T cells (Tregs) are also a subset of CD4+ T cells, identified by the expression of FOXP3. In contrast to Th17 cells, Tregs play reparative roles by secreting IL10 [76]. Interestingly, ST2, the receptor of IL33 on Tregs, has been implicated in the expansion of Tregs, and fibroblasts are the main producers of IL33 in the infarcted heart. In vitro experiments from this same study demonstrated that SPARC-expressing Tregs stimulated collagen synthesis in fibroblasts [77]. The limited data on the roles of T cells later in fibrosis illustrate the need for further studies.
B cells, a second arm of the adaptive immune response, can contribute to cardiac hypertrophy and tissue remodeling. A percentage of B cells can be found in the normal heart, but their roles in homeostasis are unclear. B cells are recruited to areas of tissue injury, producing antibodies and cytokines. B cells recruit monocytes by releasing chemokines such as CCL2 and CCL7, contributing to an expansion of pro-inflammatory macrophages [61]. Overall, it is likely that B cells are a pro-inflammatory cell type, and several studies have shown that B cell ablation ameliorates cardiac function and attenuates fibrosis [78, 79]. In humans, the involvement of auto-antibodies against cardiomyocyte components has been implicated in the pathogenesis of dilated cardiomyopathy [80]. Although B cells may contribute to the sustained activation of fibroblasts through chronic inflammation, the details of interactions between B cells and fibroblasts are still unknown.
Other Immune Populations
Although eosinophils and basophils are less abundant than other immune cells, recent studies have suggested that they contribute to tissue repair. The removal of eosinophils or basophils exacerbates adverse remodeling in the MI mouse model. They both produce the pro-fibrotic cytokine, IL4, that has a direct effect on fibroblast ECM production [81]. Mast cells reside in the baseline heart and can rapidly respond to tissue damage. The expansion and degranulation of mast cells release pro-inflammatory mediators such as TNF-α, histamine, and renin activating other immune cells and fibroblasts [7, 8]. The relative contribution of mast cells to the process of fibrosis is unknown because the secreted mediators are also produced by other cells.
The Immune Modulatory Potential of Fibroblasts in Tissue Injury
Fibroblasts in the quiescent state are characterized by the expression of PDGFRα, Tcf21, and continuous basal production of ECM proteins [5, 6]. During tissue injury, fibroblasts undergo phenotypic alterations and contribute to the repair process. One classical characteristic of an activated fibroblast is expression of actin intermediate filament proteins such as α-smooth muscle actin (αSMA), but more recent studies suggest that activated fibroblasts can exist in multiple gene expression states dependent on spatial and temporal influences. One intermediate state that has been described predominantly expresses inflammatory cytokines and ECM proteins in the absence of αSMA [5, 6]. A population of late-injury stage fibroblasts, termed matrifibrocytes, is characterized by the expression of abundant matricellular proteins related to cartilage [24].
A wide variety of factors can trigger fibroblast activation. For example, the renin-angiotensin system directly activates fibroblasts [82, 83]. Engagement with the type 1 angiotensin receptor (AT1) stimulates fibroblast increases in proliferation, migration, and ECM synthesis. DAMPs, released by damaged cardiomyocytes, can directly trigger fibroblast activation [13]. TGF-β, appreciated for its pro-fibrotic activities, is a central regulator of myofibroblast conversion. The receptor complex transduces signaling by phosphorylation of SMAD3, leading to the upregulation of αSMA and ECM proteins [84]. As mentioned above, inflammation, hypoxia, and metabolic perturbations stimulate ROS production, which promotes fibroblast ECM synthesis [50, 85]. Several studies have suggested that the pro-fibrotic effect of TGF-β may be partially attributed to ROS generation [86, 87]. Additionally, changes in ECM composition and matrix stiffness can lead to pro-fibrotic phenotypes by activation of mechanosensing receptors [88].
Recent single-cell RNA-seq analyses have provided additional information regarding the diversity of fibroblast gene expression. An examination of interstitial cell transcriptomes after MI classified fibroblasts into several populations, including four general subsets: homeostatic, activated (injury-response), myofibroblasts, and matrifibrocytes [89]. Interestingly, this study noted that pro-inflammatory cytokines and chemokines such as CCL7, CCL2, and CXCL1 were extensively enriched in the activated fibroblasts in the early inflammatory phase, suggesting a role in orchestrating immune cell recruitment. CCL2 is a chemokine that attracts CCR2-positive monocytes [90]. CCL7 binds to multiple receptors including CCR2, serving as an attractant for monocytes and macrophages [91]. CXCL1 has been reported to recruit neutrophils, monocytes, and T cells via its receptor, CXCR2 [92]. A recent in vitro study has suggested that fibroblasts may attract macrophages by deformation of the ECM, thereby providing mechanical cues for macrophages [93]. Thus, activated fibroblasts may contribute to the recruitment of inflammatory cells early in heart injury.
The NLRP3 inflammasome pathway has also been implicated in the pro-inflammatory actions of fibroblasts [94]. Cardiac fibroblasts also express PRRs such as TLRs, and activate the inflammasome pathways, potentially contributing to the initiation of inflammation [95]. Fibroblast-derived IL1β and IL18 can recruit neutrophils and monocytes, leading to further activation of fibroblasts [14, 96]. Because the NLRP3 inflammasome pathways can be activated in other immune cells which solicit the pro-inflammatory program, the relative contribution of the fibroblast-derived inflammasome is unclear. Given the beneficial effect of IL1β neutralizing antibodies in patients with heart failure, a better understanding of IL1β production and signaling is warranted [97].
Activated fibroblasts and myofibroblasts also secrete anti-inflammatory cytokines. TGF-β is a central regulator for inflammatory and pro-fibrotic programs. Although the canonical role of TGF-β is the conversion of fibroblasts to myofibroblasts, TGF-β exerts pleiotropic effects on immune cells at the site of injury. In vitro studies have shown that TGF-β can activate the migration and degranulation of neutrophils, promoting inflammation. In contrast, TGF-β can be anti-inflammatory by suppressing the NF-κB pathway and cytokines such as CCL2 and IL1β in macrophages [98]. Consistent with these actions, TGF-β treatment attenuates inflammatory cytokines. Given that the upregulation of TGF-β occurs at the end of the inflammatory phase, TGF-β may comprehensively orchestrate inflammation and tissue repair. Relevant to TGF-β signaling, IL11 is another cytokine produced by cardiac fibroblasts. In human fibroblasts, IL11 is secreted in response to TGF-β stimulation, and recombinant IL11 induces ECM synthesis, independent of TGF-β. Deletion of the IL11 receptor, IL11RA1, in an MI mouse model resulted in attenuation of fibrosis, suggesting a pro-fibrotic role of fibroblasts [99]. However, the effect on immune cells is unclear, as IL11RA1 is exclusively expressed by fibroblasts. In vitro evidence has implicated that IL11 may suppress inflammatory cytokines such as TNF-α and IL1β of macrophages [100].
Conclusion
Although the matrix producing capability of fibroblasts has been long appreciated, an understanding of the complex interactions between fibroblasts and inflammatory cells is currently less understood. A growing body of evidence supporting the therapeutic potential of anti-inflammatory strategies for treating cardiovascular disease highlights the significance of regulatory mechanisms by fibroblasts. Here, we have described the intricate interactions between immune cells and fibroblasts in the setting of heart injury. The process of inflammation and fibrosis involves diverse participants including multiple cell types and the extracellular microenvironment. In addition, many of the mediators exert pleiotropic effects, making it particularly challenging to elucidate pivotal interactions. Recent advances in transcriptomic technologies have enabled researchers to analyze not only gene expression but also the profiles of chromatin accessibility, at the single-cell resolution, helping to illuminate different cell states and transcriptional activities. Also, spatial information on transcriptomes in vivo is becoming available, although the resolution is not yet at a single-cell level. Accumulating spatial information with higher resolution and an understanding of the spectrum of fibroblast roles will provide novel insights into the cell–cell communication orchestrating inflammation and tissue remodeling processes.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Tsao CW, Aday AW, Almarzooq ZI, et al. Heart Disease and Stroke Statistics—2022 update: a report from the American Heart Association. Circulation. 2022;145:e153–639.
Frangogiannis NG. Cardiac fibrosis. Cardiovasc Res. 2020;117:1450–88.
Heidenreich PA, Bozkurt B, Aguilar D, et al. 2022 AHA/ACC/HFSA guideline for the management of heart failure: a report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 2022;145:e895–1032.
Kuwabara JT, Hara A, Bhutada S, et al. Consequences of PDGFRa+ fibroblast reduction in adult murine hearts. Elife. 2022. https://doi.org/10.7554/elife.69854.
Tallquist MD, Molkentin JD. Redefining the identity of cardiac fibroblasts. Nat Rev Cardiol. 2017;14:484–91.
Tallquist MD. Cardiac fibroblast diversity. Annu Rev Physiol. 2020;82:63–78.
Adamo L, Rocha-Resende C, Prabhu SD, Mann DL. Reappraising the role of inflammation in heart failure. Nat Rev Cardiol. 2020;17:269–85.
Rurik JG, Aghajanian H, Epstein JA. Immune cells and immunotherapy for cardiac injury and repair. Circ Res. 2021;128:1766–79.
Bliksøen M, Mariero LH, Torp MK, Baysa A, Ytrehus K, Haugen F, Seljeflot I, Vaage J, Valen G, Stensløkken K-O. Extracellular mtDNA activates NF-κB via toll-like receptor 9 and induces cell death in cardiomyocytes. Basic Res Cardiol. 2016;111:42.
Lipps C, Nguyen JH, Pyttel L, et al. N-terminal fragment of cardiac myosin binding protein-C triggers pro-inflammatory responses in vitro. J Mol Cell Cardiol. 2016;99:47–56.
McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CCM, Beck PL, Muruve DA, Kubes P. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science. 2010;330:362–6.
Martine P, Rébé C. Heat shock proteins and inflammasomes. Int J Mol Sci. 2019;20:4508.
Fan J, Ren M, Adhikari BK, Wang H, He Y. The NLRP3 inflammasome as a novel therapeutic target for cardiac fibrosis. J Inflamm Res. 2022;15:3847–58.
Jorgensen I, Lopez JP, Laufer SA, Miao EA. IL-1β, IL-18, and eicosanoids promote neutrophil recruitment to pore-induced intracellular traps following pyroptosis. Eur J Immunol. 2016;46:2761–6.
Jia C, Chen H, Zhang J, et al. Role of pyroptosis in cardiovascular diseases. Int Immunopharmacol. 2019;67:311–8.
Bageghni SA, Hemmings KE, Zava N, Denton CP, Porter KE, Ainscough JFX, Drinkhill MJ, Turner NA. Cardiac fibroblast-specific p38α MAP kinase promotes cardiac hypertrophy via a putative paracrine interleukin-6 signaling mechanism. Faseb J. 2018;32:4941–54.
Craig R, Larkin A, Mingo AM, Thuerauf DJ, Andrews C, McDonough PM, Glembotski CC. p38 MAPK and NF-kappa B collaborate to induce interleukin-6 gene expression and release. Evidence for a cytoprotective autocrine signaling pathway in a cardiac myocyte model system. J Biol Chem. 2000;275:23814–24.
Saxena A, Chen W, Su Y, Rai V, Uche OU, Li N, Frangogiannis NG. IL-1 induces proinflammatory leukocyte infiltration and regulates fibroblast phenotype in the infarcted myocardium. J Immunol. 2013;191:4838–48.
Leicht M, Briest W, Zimmer H-G. Regulation of norepinephrine-induced proliferation in cardiac fibroblasts by interleukin-6 and p42/p44 mitogen activated protein kinase. Mol Cell Biochem. 2003;243:65–72.
Bansal SS, Ismahil MA, Goel M, Patel B, Hamid T, Rokosh G, Prabhu SD. Activated T lymphocytes are essential drivers of pathological remodeling in ischemic heart failure. Circulation Hear Fail. 2017;10:e003688.
Yan X, Anzai A, Katsumata Y, et al. Temporal dynamics of cardiac immune cell accumulation following acute myocardial infarction. J Mol Cell Cardiol. 2013;62:24–35.
Hilgendorf I, Gerhardt LMS, Tan TC, et al. Ly-6Chigh monocytes depend on Nr4a1 to balance both inflammatory and reparative phases in the infarcted myocardium. Circ Res. 2014;114:1611–22.
Prabhu SD, Frangogiannis NG. The biological basis for cardiac repair after myocardial infarction. Circ Res. 2016;119:91–112.
Fu X, Khalil H, Kanisicak O, et al. Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart. J Clin Invest. 2018;128:2127–43.
Dick SA, Epelman S. Chronic heart failure and inflammation. Circ Res. 2016;119:159–76.
Frantz S, Falcao-Pires I, Balligand J, et al. The innate immune system in chronic cardiomyopathy: a European Society of Cardiology (ESC) scientific statement from the Working Group on Myocardial Function of the ESC. Eur J Heart Fail. 2018;20:445–59.
Medzhitov R. The spectrum of inflammatory responses. Science. 2021;374:1070–5.
Gan W, Ren J, Li T, Lv S, Li C, Liu Z, Yang M. The SGK1 inhibitor EMD638683, prevents angiotensin II–induced cardiac inflammation and fibrosis by blocking NLRP3 inflammasome activation. Biochim Biophys Acta Mol Basis Dis. 2018;1864:1–10.
Peters A, Nawrot TS, Baccarelli AA. Hallmarks of environmental insults. Cell. 2021;184:1455–68.
Ramos GC, van den Berg A, Nunes-Silva V, et al. Myocardial aging as a T-cell–mediated phenomenon. Proc National Acad Sci. 2017;114:E2420–9.
Vidal R, Wagner JUG, Braeuning C, et al. Transcriptional heterogeneity of fibroblasts is a hallmark of the aging heart. JCI Insight. 2019;4:e131092.
Pillai ICL, Li S, Romay M, et al. Cardiac fibroblasts adopt osteogenic fates and can be targeted to attenuate pathological heart calcification. Cell Stem Cell. 2017;20:218-232.e5.
Yura Y, Sano S, Walsh K. Clonal hematopoiesis: a new step linking inflammation to heart failure. JACC Basic Transl Sci. 2020;5:196–207.
• Dorsheimer L, Assmus B, Rasper T, et al. Association of mutations contributing to clonal hematopoiesis with prognosis in chronic ischemic heart failure. JAMA Cardiol. 2019;4:25–33. This study identified causal genes of clonal hematopoiesis, which were associated with an increase in mortality and hospitalization of elderly patients with heart failure.
Wagner JUG, Dimmeler S. Cellular cross-talks in the diseased and aging heart. J Mol Cell Cardiol. 2019;138:136–46.
Dillmann WH. Diabetic cardiomyopathy. Circ Res. 2019;124:1160–2.
Nishikawa T, Edelstein D, Du XL, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404:787–90.
Iyer SS, He Q, Janczy JR, et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity. 2013;39:311–23.
Sies H, Jones DP. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat Rev Mol Cell Bio. 2020;21:363–83.
Pang Y, Kartsonaki C, Lv J, et al. Associations of adiposity, circulating protein biomarkers, and risk of major vascular diseases. JAMA Cardiol. 2021;6:276–86.
Deeks SG, Tracy R, Douek DC. Systemic effects of inflammation on health during chronic HIV infection. Immunity. 2013;39:633–45.
Zanni MV, Awadalla M, Toribio M, et al. Immune correlates of diffuse myocardial fibrosis and diastolic dysfunction among aging women with human immunodeficiency virus. J Infect Dis. 2019;221:1315–20.
Meyer A, Wang W, Qu J, Croft L, Degen JL, Coller BS, Ahamed J. Platelet TGF-β1 contributions to plasma TGF-β1, cardiac fibrosis, and systolic dysfunction in a mouse model of pressure overload. Blood. 2012;119:1064–74.
Ahamed J, Terry H, Choi ME, Laurence J. Transforming growth factor-&bgr;1-mediated cardiac fibrosis. AIDS. 2016;30:535–42.
• Daseke MJ, Chalise U, Becirovic-Agic M, Salomon JD, Cook LM, Case AJ, Lindsey ML. Neutrophil signaling during myocardial infarction wound repair. Cell Signal. 2020;77:109816. This review comprehensively describes the roles of neutrophils during myocardial infarction, encompassing both molecular biology and pathophysiology.
Venkatachalam K, Venkatesan B, Valente AJ, Melby PC, Nandish S, Reusch JEB, Clark RA, Chandrasekar B. WISP1, a pro-mitogenic, pro-survival factor, mediates tumor necrosis factor-α (TNF-α)-stimulated cardiac fibroblast proliferation but inhibits TNF-α-induced cardiomyocyte death*. J Biol Chem. 2009;284:14414–27.
Siwik DA, Chang DL-F, Colucci WS. Interleukin-1β and tumor necrosis factor-α decrease collagen synthesis and increase matrix metalloproteinase activity in cardiac fibroblasts in vitro. Circ Res. 2000;86:1259–65.
Mir SA, Chatterjee A, Mitra A, Pathak K, Mahata SK, Sarkar S. Inhibition of signal transducer and activator of transcription 3 (STAT3) attenuates interleukin-6 (IL-6)-induced collagen synthesis and resultant hypertrophy in rat heart. J Biol Chem. 2012;287:2666–77.
Aminjan HH, Abtahi SR, Hazrati E, Chamanara M, Jalili M, Paknejad B. Targeting of oxidative stress and inflammation through ROS/NF-kappaB pathway in phosphine-induced hepatotoxicity mitigation. Life Sci. 2019;232:116607.
Siwik DA, Pagano PJ, Colucci WS. Oxidative stress regulates collagen synthesis and matrix metalloproteinase activity in cardiac fibroblasts. Am J Physiol Cell Physiol. 2001;280:C53–60.
Bratton DL, Henson PM. Neutrophil clearance: when the party is over, clean-up begins. Trends Immunol. 2011;32:350–7.
Horckmans M, Ring L, Duchene J, Santovito D, Schloss MJ, Drechsler M, Weber C, Soehnlein O, Steffens S. Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur Heart J. 2017;38:187–97.
Ma Y, Yabluchanskiy A, Iyer RP, Cannon PL, Flynn ER, Jung M, Henry J, Cates CA, Deleon-Pennell KY, Lindsey ML. Temporal neutrophil polarization following myocardial infarction. Cardiovasc Res. 2016;110:51–61.
Nicolás-Ávila JA, Lechuga-Vieco AV, Esteban-Martínez L, et al. A network of macrophages supports mitochondrial homeostasis in the heart. Cell. 2020;183:94-109.e23.
Epelman S, Lavine KJ, Beaudin AE, et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity. 2014;40:91–104.
Lavine KJ, Epelman S, Uchida K, Weber KJ, Nichols CG, Schilling JD, Ornitz DM, Randolph GJ, Mann DL. Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc National Acad Sci. 2014;111:16029–34.
Aurora AB, Porrello ER, Tan W, Mahmoud AI, Hill JA, Bassel-Duby R, Sadek HA, Olson EN. Macrophages are required for neonatal heart regeneration. J Clin Invest. 2014;124:1382–92.
Dick SA, Macklin JA, Nejat S, et al. Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat Immunol. 2019;20:29–39.
Lavine KJ, Pinto AR, Epelman S, Kopecky BJ, Clemente-Casares X, Godwin J, Rosenthal N, Kovacic JC. The macrophage in cardiac homeostasis and disease JACC macrophage in CVD series (part 4). J Am Coll Cardiol. 2018;72:2213–30.
Frangogiannis NG, Dewald O, Xia Y, Ren G, Haudek S, Leucker T, Kraemer D, Taffet G, Rollins BJ, Entman ML. Critical role of monocyte chemoattractant protein-1/CC chemokine ligand 2 in the pathogenesis of ischemic cardiomyopathy. Circulation. 2007;115:584–92.
Zouggari Y, Ait-Oufella H, Bonnin P, et al. B lymphocytes trigger monocyte mobilization and impair heart function after acute myocardial infarction. Nat Med. 2013;19:1273–80.
Bajpai G, Bredemeyer A, Li W, et al. Tissue resident CCR2− and CCR2+ cardiac macrophages differentially orchestrate monocyte recruitment and fate specification following myocardial injury. Circ Res. 2019;124:263–78.
Zhang S, Weinberg S, DeBerge M, et al. Efferocytosis fuels requirements of fatty acid oxidation and the electron transport chain to polarize macrophages for tissue repair. Cell Metab. 2019;29:443-456.e5.
Jung M, Ma Y, Iyer RP, DeLeon-Pennell KY, Yabluchanskiy A, Garrett MR, Lindsey ML. IL-10 improves cardiac remodeling after myocardial infarction by stimulating M2 macrophage polarization and fibroblast activation. Basic Res Cardiol. 2017;112:33.
Shiraishi M, Shintani Y, Shintani Y, Ishida H, Saba R, Yamaguchi A, Adachi H, Yashiro K, Suzuki K. Alternatively activated macrophages determine repair of the infarcted adult murine heart. J Clin Invest. 2016;126:2151–66.
Shirakawa K, Endo J, Kataoka M, et al. MerTK expression and ERK activation are essential for the functional maturation of osteopontin-producing reparative macrophages after myocardial infarction. J Am Heart Assoc. 2020;9:e017071.
•• Kuppe C, Flores ROR, Li Z, et al. Spatial multi-omic map of human myocardial infarction. Nature. 2022;608:766–77. This study identified several transcription factors involved in the myofibroblast activation in the human MI using multi-omics approaches. The spatial transcriptomics analysis has suggested the presence of a special milieu consisting of distinct macrophage subsets and myofibroblasts in the infarction.
Patel B, Bansal SS, Ismahil MA, Hamid T, Rokosh G, Mack M, Prabhu SD. CCR2+ monocyte-derived infiltrating macrophages are required for adverse cardiac remodeling during pressure overload. JACC Basic Transl Sci. 2018;3:230–44.
Kubota A, Frangogiannis NG. Macrophages in myocardial infarction. Am J Physiol Cell Physiol. 2022;323:C1304–24.
van Amerongen MJ, Harmsen MC, van Rooijen N, Petersen AH, van Luyn MJA. Macrophage depletion impairs wound healing and increases left ventricular remodeling after myocardial injury in mice. Am J Pathology. 2007;170:818–29.
Bevan L, Lim ZW, Venkatesh B, Riley PR, Martin P, Richardson RJ. Specific macrophage populations promote both cardiac scar deposition and subsequent resolution in adult zebrafish. Cardiovasc Res. 2019;116:1357–71.
Laroumanie F, Douin-Echinard V, Pozzo J, et al. CD4+ T cells promote the transition from hypertrophy to heart failure during chronic pressure overload. Circulation. 2014;129:2111–24.
Ngwenyama N, Kaur K, Bugg D, et al. Antigen presentation by cardiac fibroblasts promotes cardiac dysfunction. Nat Cardiovasc Res. 2022;1:761–74.
Zhang Y, Zhang Y-Y, Li T-T, et al. Ablation of interleukin-17 alleviated cardiac interstitial fibrosis and improved cardiac function via inhibiting long non-coding RNA-AK081284 in diabetic mice. J Mol Cell Cardiol. 2018;115:64–72.
Wu L, Ong S, Talor MV, et al. Cardiac fibroblasts mediate IL-17A–driven inflammatory dilated cardiomyopathy. J Exp Med. 2014;211:1449–64.
Saxena A, Dobaczewski M, Rai V, Haque Z, Chen W, Li N, Frangogiannis NG. Regulatory T cells are recruited in the infarcted mouse myocardium and may modulate fibroblast phenotype and function. Am J Physiol-heart C. 2014;307:H1233–42.
Xia N, Lu Y, Gu M, et al. A unique population of regulatory T cells in heart potentiates cardiac protection from myocardial infarction. Circulation. 2020;142:1956–73.
Cordero-Reyes AM, Youker KA, Trevino AR, Celis R, Hamilton DJ, Flores-Arredondo JH, Orrego CM, Bhimaraj A, Estep JD, Torre-Amione G. Full expression of cardiomyopathy is partly dependent on B-cells: a pathway that involves cytokine activation, immunoglobulin deposition, and activation of apoptosis. J Am Heart Assoc. 2016;5:e002484.
Adamo L, Rocha-Resende C, Lin C-Y, et al. Myocardial B cells are a subset of circulating lymphocytes with delayed transit through the heart. JCI Insight. 2020. https://doi.org/10.1172/jci.insight.134700.
Lazzerini PE, Capecchi PL, Laghi-Pasini F, Boutjdir M. Autoimmune channelopathies as a novel mechanism in cardiac arrhythmias. Nat Rev Cardiol. 2017;14:521–35.
Peng H, Sarwar Z, Yang X-P, Peterson EL, Xu J, Janic B, Rhaleb N, Carretero OA, Rhaleb N-E. Profibrotic role for interleukin-4 in cardiac remodeling and dysfunction. Hypertension. 2018;66:582–9.
Kawano H, Do YS, Kawano Y, Starnes V, Barr M, Law RE, Hsueh WA. Angiotensin II has multiple profibrotic effects in human cardiac fibroblasts. Circulation. 2000;101:1130–7.
Swaney JS, Roth DM, Olson ER, Naugle JE, Meszaros JG, Insel PA. Inhibition of cardiac myofibroblast formation and collagen synthesis by activation and overexpression of adenylyl cyclase. Proc National Acad Sci. 2005;102:437–42.
Khalil H, Kanisicak O, Prasad V, et al. Fibroblast-specific TGF-β–Smad2/3 signaling underlies cardiac fibrosis. J Clin Invest. 2017;127:3770–83.
Sanchez MC, Lancel S, Boulanger E, Neviere R. Targeting oxidative stress and mitochondrial dysfunction in the treatment of impaired wound healing: a systematic review. Antioxidants. 2018;7:98.
Jain M, Rivera S, Monclus EA, Synenki L, Zirk A, Eisenbart J, Feghali-Bostwick C, Mutlu GM, Budinger GRS, Chandel NS. Mitochondrial reactive oxygen species regulate transforming growth factor-β signaling. J Biol Chem. 2013;288:770–7.
Cucoranu I, Clempus R, Dikalova A, Phelan PJ, Ariyan S, Dikalov S, Sorescu D. NAD(P)H oxidase 4 mediates transforming growth factor-β1–induced differentiation of cardiac fibroblasts into myofibroblasts. Circ Res. 2005;97:900–7.
Saucerman JJ, Tan PM, Buchholz KS, McCulloch AD, Omens JH. Mechanical regulation of gene expression in cardiac myocytes and fibroblasts. Nat Rev Cardiol. 2019;16:361–78.
Forte E, Skelly DA, Chen M, Daigle S, Morelli KA, Hon O, Philip VM, Costa MW, Rosenthal NA, Furtado MB. Dynamic interstitial cell response during myocardial infarction predicts resilience to rupture in genetically diverse mice. Cell Rep. 2020;30:3149-3163.e6.
Dewald O, Zymek P, Winkelmann K, Koerting A, Ren G, Abou-Khamis T, Michael LH, Rollins BJ, Entman ML, Frangogiannis NG. CCL2/monocyte chemoattractant protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circ Res. 2005;96:881–9.
Cheng JW, Sadeghi Z, Levine AD, Penn MS, von Recum HA, Caplan AI, Hijaz A. The role of CXCL12 and CCL7 chemokines in immune regulation, embryonic development, and tissue regeneration. Cytokine. 2014;69:277–83.
Wu CL, Yin R, Wang S-N, Ying R. A review of CXCL1 in cardiac fibrosis. Frontiers Cardiovasc Medicine. 2021;8:674498.
Pakshir P, Alizadehgiashi M, Wong B, Coelho NM, Chen X, Gong Z, Shenoy VB, McCulloch CA, Hinz B. Dynamic fibroblast contractions attract remote macrophages in fibrillar collagen matrix. Nat Commun. 2019;10:1850.
Kawaguchi M, Takahashi M, Hata T, et al. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation. 2011;123:594–604.
Sandanger Ø, Ranheim T, Vinge LE, et al. The NLRP3 inflammasome is up-regulated in cardiac fibroblasts and mediates myocardial ischaemia–reperfusion injury. Cardiovasc Res. 2013;99:164–74.
Bageghni SA, Hemmings KE, Yuldasheva NY, et al. Fibroblast-specific deletion of interleukin-1 receptor-1 reduces adverse cardiac remodeling following myocardial infarction. JCI Insight. 2019;4:e125074.
Everett BM, Cornel J, Lainscak M, Anker SD, Abbate A, Thuren T, Libby P, Glynn RJ, Ridker PM. Anti-inflammatory therapy with canakinumab for the prevention of hospitalization for heart failure. Circulation. 2019;139:1289–99.
Frangogiannis NG. Transforming growth factor-β in myocardial disease. Nat Rev Cardiol. 2022:1–21.
Schafer S, Viswanathan S, Widjaja AA, et al. IL-11 is a crucial determinant of cardiovascular fibrosis. Nature. 2017;552:110–5.
Trepicchio WL, Bozza M, Pedneault G. Dorner AJ (1996) Recombinant human IL-11 attenuates the inflammatory response through down-regulation of proinflammatory cytokine release and nitric oxide production. J Immunol Baltim Md. 1950;157:3627–34.
•• Farbehi N, Patrick R, Dorison A, Xaymardan M, Janbandhu V, Wystub-Lis K, Ho JW, Nordon RE, Harvey RP. Single-cell expression profiling reveals dynamic flux of cardiac stromal, vascular and immune cells in health and injury. Elife. 2019;8:e43882. This pioneering research presents the utilization of single-cell RNA-sequencing to analyze mouse interstitial heart cells after MI, deciphering the heterogeneity among fibroblasts and macrophages, and provided valuable data for exploring the cellular interactions and processes of differentiation in silico.
McLellan MA, Skelly DA, Dona MSI, et al. High-resolution transcriptomic profiling of the heart during chronic stress reveals cellular drivers of cardiac fibrosis and hypertrophy. Circulation. 2020;142:1448–63.
Funding
Akitoshi Hara reports a Naito Foundation Grant for Studying Overseas and an Overseas Research Fellowship from the Japan Society for the Promotion of Science. Michelle D. Tallquist reports funding from the National Institutes of Health (R01HL144067).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Hara, A., Tallquist, M.D. Fibroblast and Immune Cell Cross-Talk in Cardiac Fibrosis. Curr Cardiol Rep 25, 485–493 (2023). https://doi.org/10.1007/s11886-023-01877-8
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11886-023-01877-8