
Abstract
Purpose of Review
The pathological remodeling of cardiac tissue after injury or disease leads to scar formation. Our knowledge of the role of nonmyocytes, especially fibroblasts, in cardiac injury and repair continues to increase with technological advances in both experimental and clinical studies. Here, we aim to elaborate on cardiac fibroblasts by describing their origins, dynamic cellular states after injury, and heterogeneity in order to understand their role in cardiac injury and repair.
Recent Findings
With the improvement in genetic lineage tracing technologies and the capability to profile gene expression at the single-cell level, we are beginning to learn that manipulating a specific population of fibroblasts could mitigate severe cardiac fibrosis and promote cardiac repair after injury.
Summary
Cardiac fibroblasts play an indispensable role in tissue homeostasis and in repair after injury. Activated fibroblasts or myofibroblasts have time-dependent impacts on cardiac fibrosis. Multiple signaling pathways are involved in modulating fibroblast states, resulting in the alteration of fibrosis. Modulating a specific population of cardiac fibroblasts may provide new opportunities for identifying novel treatment options for cardiac fibrosis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Heart failure is becoming the leading cause of morbidity and mortality worldwide [1]. In the course of cardiomyocyte death, the cardiac tissue is replaced by a reparative fibrotic scar, which progressively remodels the tissue. A healthy heart consists of cardiomyocytes and nonmyocytes. Cardiomyocytes occupy only 30% of the cell numbers and are responsible for contraction and relaxation [2]. Nonmyocytes consist of epicardial cells, endothelial cells, fibroblasts, pericytes, smooth muscle cells, lymphocytes, macrophages, and some other cell types. Of these, cardiac fibroblasts play an irreplaceable role in regulating the extracellular environment by increasing or decreasing the extracellular matrix (ECM) [3]. Cardiac injuries, including pressure overload-induced heart failure and ischemic injury, usually result in cardiac fibrosis because of the excessive deposition of ECM, which is produced by myofibroblasts differentiated from resident fibroblasts in the injured heart [4, 5]. Given the significant role of cardiac fibroblasts in pathophysiological conditions, it is important to learn more about fibroblasts and myofibroblasts, including their origin, state, regulating genes, heterogeneity, and signaling pathways, in order to develop novel therapeutic targets for treating cardiac fibrosis. Recent advances in technology, such as genetic lineage tracing and single-cell RNA sequencing, have provided knowledge on the role of cardiac fibroblasts in cardiovascular disease [5,6,7, 8•, 9,10,11]. This review aims to provide updates on the recent advancements in fibroblast research and on the roles of fibroblasts after cardiac injuries.
Origins of Cardiac Fibroblasts
As cardiac fibroblasts play a significant role in maintaining the structural integrity of the injured heart by differentiating into myofibroblasts and modulating the ECM, it is necessary to clarify their origin. However, the identification of fibroblasts remains difficult because of a lack of clear markers that can specifically label fibroblasts [12, 13]. According to recent findings based on flow cytometric and histological analyses, fibroblasts account for approximately 13% of cells in the healthy mouse heart [14, 15]. Fibroblasts could also be divided into subpopulations based on gene expression files during tissue homeostasis and after injury. In addition, resident cardiac fibroblasts have distinct developmental origins, which may indicate their unique gene profiles and functions in cardiac diseases.
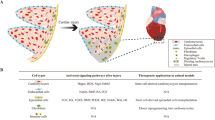
For understanding the developmental origin, genetic lineage-tracing based on the Cre–loxP recombination system is used to permanently trace labeled cells and follow their fate over time during tissue repair and regeneration [16]. Using the mouse lines that express Cre, including Tbx18Cre [17, 18], Gata5Cre [19], Sema3DCre [20], and Wt1Cre [21,22,23], even the inducible CreER, such as Wt1CreERT2 [21], Tcf21mCrem [24, 25], and Tbx18CreERT2 [26], previous studies have demonstrated that a large population of cardiac fibroblasts is derived from the epicardium through epithelial-to-mesenchymal transition (EMT) [27] (Fig. 1a). In the developing heart, some epicardial progenitor cells delaminate from the epicardial layer and invade the myocardium on embryonic day (E) 12.5. Fibroblasts with distinct molecular markers are formed and distributed throughout the ventricle at later embryonic stages [25, 28,29,30,31]. Moreover, single-cell RNA sequencing (scRNA-seq) data have suggested that epicardium-derived fibroblasts express genes associated with cell migration and cell metabolism and account for a major population of cardiac fibroblasts [32].

Origins of fibroblasts. a, Epicardium and endocardium to fibroblasts by EMT. b, Epi-Fb and Endo-Fb occupy distinct regions of hearts. Epi-Fb, epicardium-derived fibroblasts; Endo-Fb, endocardium-derived fibroblasts
In addition to the epicardium being a major source of fibroblasts, recent genetic lineage tracing studies have suggested that the embryonic endothelium represents an indispensable developmental source of fibroblasts. By tracing the endothelial lineage with Tie2Cre, Ali and Moore-Morris reported that around 10% of fibroblasts in the left ventricle and around 65% of fibroblasts in the interventricular septum are derived from endothelial progenitor cells [5, 33] (Fig. 1b). By exploring the fate map of these endothelium-derived fibroblasts, Moore-Morris clarified that an endothelial-to-mesenchymal transition (EndoMT) process by E9.5 was responsible for this fibroblast lineage [5] (Fig. 1a). Furthermore, according to scRNA-seq data, endothelium-derived fibroblasts express genes associated with valve leaflets and account for a small population of cardiac fibroblasts [32]. With the development of the lineage tracing strategy, dual lineage tracing tools have been used to specifically trace endocardium-derived cells and have revealed that endothelial cells have the potential to produce intermediate mesenchymal cells that can subsequently differentiate into fibroblasts, smooth muscle cells, pericytes, and adipocytes [34]. In addition to epicardium- and endothelium-derived cells, neural crest cells are reported to contribute to a small population of cardiac fibroblasts [35].
Cardiac fibroblasts have multiple developmental origins. Whether these different origins affect their behaviors and functions under pathophysiological conditions remains unknown. The mouse model for myocardial infarction (MI) has recently been used to study the functions of fibroblasts having different origins. After MI, adult epicardial cells are reactivated and generate a thickened epicardial layer consisting of fibroblasts in response to cardiac injury [36, 37]. In previous studies using the thoracic aortic constriction (TAC) model, few fibroblasts originating from the adult epicardium were found [5, 33], indicating that fibroblasts derived from the epicardium are injury type-dependent in adults. To analyze the gene expression profiles in distinct fibroblast populations based on their origin, epicardium-derived fibroblasts from Tbx18Cre;Rosa26mTmG mice and endothelium-derived fibroblasts from Tie2Cre;Rosa26mTmG mice were sorted by flow cytometry. Little difference was noted between their RNAs after pressure overload-induced heart failure [5, 38, 39]. The proliferative activity of the two fibroblast populations was found to be similar as the proportion of fibroblasts of each origin did not change significantly after pressure overload-induced heart failure in comparison with the sham control [33, 40]. The aforementioned data indicated that the origins of fibroblasts have rare effects on their function and performance in pressure overload-induced cardiac injury. However, the epicardium or endothelium contributes to not only cardiac fibroblasts but also pericytes, adipocytes, and other cell types. In addition, there is preference in terms of the location where the two fibroblast populations reside. Immunostaining for fibroblast markers and lineage tracing markers may obscure the true difference between the two fibroblast populations in response to injury. In the future, more advanced lineage tracing strategies could be developed in order to re-assess whether fibroblasts of different origins have different functions and gene expression profiles during heart injury and repair.
State of Cardiac Fibroblasts After Cardiac Injury
As a plastic cell type, fibroblasts exhibit variable differentiated states to respond to wound healing and to remodel for scar formation. Recently, Fu et al. identified four different states of fibroblasts, namely resident fibroblasts, active fibroblasts, myofibroblasts, and matrifibrocytes, after MI using genetic lineage tracing to monitor the fate of fibroblasts [41••]. They used Tcf21MCM/+;R26EGFP mice to trace tissue-resident cardiac fibroblasts and calculated the proliferation rate of fibroblasts through 5-ethynyl-2′-deoxyuridine (EdU) and Ki67 staining of heart sections collected from mice during different periods after MI. They reported that the activation and proliferation of fibroblasts are the highest within 2–4 days after MI [41••]. After the activation of fibroblasts, the activated fibroblasts get converted to myofibroblasts that express smooth muscle α-actin (αSMA), contain extensive endoplasmic reticulum [42], and secrete adequate extracellular matrix proteins to fill the injured area within 3–7 days [41••]. Furthermore, the activated fibroblasts and myofibroblasts reach the exhausted proliferation rate by 7 days after MI; this finding is consistent with that of another study conducted by an independent group [43]. By 7–10 days, the myofibroblasts enter an alternate differentiated state without proliferation ability and αSMA expression, while the scar fully matures with increased extracellular matrix deposition [41••, 44]. The final stage for fibroblasts is matrifibrocytes. They exhibit weak contractile and secretory abilities but express universal and specific extracellular matrix and tendon genes. Specifically, they express Cilp2 and Comp, which are related to bone and fiber remodeling, and may play an important role in maintaining the mature scar [41••] (Fig. 2).

Different states of fibroblasts respond to wounds healing in a time-dependent way
A subset of cardiac fibroblasts undergoes senescence in response to injury and aging. These senescent fibroblasts modulate innate immunity by secreting inflammatory molecules [45]. The p53 signaling pathway is involved in promoting the occurrence of senescent fibroblasts and in modulating the inflammatory domains and ECM deposition [46]. As matrifibrocytes have a poor proliferation rate in the final stage of injury, they are considered to be a fibroblast cell type with some senescent characteristics [47]. Although senescent fibroblasts have been reported to express differential genes in comparison with other fibroblast subtypes, further studies should be conducted to assess the impact of senescent fibroblasts on tissue fibrosis and cardiac remodeling after injury.
Using scRNA-seq, many studies have started to unravel the diversity and heterogeneity of fibroblast populations [48]. Ren et al. analyzed the transcriptomes of 11,492 single cells in a pressure overload-induced cardiac injury model in order to unravel the heterogeneity of fibroblasts by grouping them into six clusters (FB1–FB6) [8•]. Skelly et al. defined a new cluster of fibroblasts that could express both fibroblast and immune cell markers through scRNA-seq of cells isolated from uninjured hearts; however, their functional role in the heart has not yet been studied [32]. Another study reported the emergence of two new fibroblast subpopulations, fibroblast-Cilp and fibroblast-Thbs4, after tissue stress and accelerated fibrosis [49]. As the transcriptional profile of fibroblasts is variably modified in response to external stimulation, it will be helpful to reveal their indispensable phenotypic plasticity using single-cell transcriptomic technologies.
The aforementioned data demonstrate the different states of fibroblasts in different injury models and generalize the function of each fibroblast state during fibrosis; the correlations between fibroblasts in each state remain unknown. Further research will be needed to reveal their functions and contributions to cardiac injury and repair.
Table 1 Marker genes expression in different states of fibroblasts (summarized from [41••]). Col1a1, collagen type I alpha 1; Col13a1, collagen type III alpha 1; PAGFRa, platelet-derived growth factor receptor alpha; Tcf21, transcription factor 21; Acta2, actin alpha 2, smooth muscle; Lox, lysyl oxidase; Cilp2, cartilage intermediate layer protein 2; Comp, cartilage oligomeric matrix protein. +, represents marker genes expression, more“+” indicates higher expression.
Pathways Implicated in Cardiac Fibroblasts After Cardiac Injury
Transforming growth factor beta (TGF-β) is one of the most well-studied factors involved in the pathogenesis of cardiac fibrosis, including tissue inflammation, myofibroblast differentiation, ECM synthesis, and gene expression [50,51,52,53,54,55,56]. TGF-β1, TGF-β2, and TGF-β3 are the three distinct isoforms of TGF-β [57]. Once TGF-βs bind to the heteromeric serine–threonine kinase receptors TGFBRs on the cell surface, downstream signaling cascades, including the Smad tri-complex (Smad2–Smad3–Smad4) [58–60] or Smad-independent pathways, are activated to execute important functions in tissue fibrosis [59]. TGF-β–Smad2/3 signaling is considered to be the canonical signaling pathway for inducing the differentiation of fibroblasts to myofibroblasts [52]. Accordingly, Khalil et al. provided direct genetic evidence that fibroblast-specific TGF-β–Smad2/3 signaling plays a role in cardiac fibrosis using a tissue-specific knockout model in vivo [61]. Specifically, they used lineage tracing strategies to explore cardiac fibrosis in pressure overload-induced heart failure by using fibroblast-specific and myofibroblast-specific inducible Cre mouse lines simultaneously with the conditional deletion of Tgfbr1/2, Smad2, or Smad3 [61]. They found that the specific deletion of Tgfbr1/2 or Smad3, instead of Smad2, in fibroblasts remarkably reduced the fibrotic response after TAC. The deletion of TGF-β receptor 1/2 in fibroblasts could relieve pressure overload-induced cardiac hypertrophy by adjusting abundant regulatory genes engaged in cardiomyocyte homeostasis and disease compensation [61]. This finding was consistent with that of Li et al., who proved that Smad2 plays a redundant role in ovarian granulosa cells in vivo [62]. Furthermore, mitogen-activated protein kinase (MAPK) signaling pathways, including p38 MAPK[62], ERK1/2[63, 64], PI3K[65, 66], and JNK[67], are considered to be the noncanonical signaling pathways in response to TGF-β stimulation; they form a complicated network of signaling pathways for modulating cardiac fibrosis.
Bone morphogenetic proteins (BMPs) are believed to play an indispensable role in the TGF-β–Smad signaling pathway [68,69,70,71]. TGF-β1 and BMPs directly bind to type II receptors, which dimerize with and stimulate type I receptors for signal transduction through Smads [72]. TGF-β1 triggers counterregulatory pathways to reduce its own activity. For instance, ALK5/Smad3 and ALK1/Smad1 stimulated by TGF-β1 counterregulate each other to modulate the TGF-β1 state in cardiac fibrosis [68–71]. BMP7 has been found to reduce cardiac fibrosis by activating Smad1 in endothelial cells [73]. In order to assess the effect of BMPs on cardiac fibroblasts, Kevin et al. subjected BMP9−/− mice to TAC and observed that the deletion of BMP9 aggregated cardiac fibrosis by increasing phosphorylated Smad3 (pSmad3) levels in the left ventricle [72]. Their findings suggest that the manipulation of the BMP signaling pathway could be a potential therapeutic target for reducing cardiac fibrosis and improving cardiac repair.
There is extensive evidence that Wnt/β-catenin signaling is involved in inflammation, immune response, and scar formation in multiple mouse models [74, 75]. The Wnt signaling system consists of 19 lipophilic proteins [76]. Wnts play an indispensable role in the induction of cardiogenesis [77]. Recent research has revealed that Wnt1 could promote the proliferation of cardiac fibroblasts and the expression of specific genes that are associated with the profibrotic process in injured regions of the infarcted heart [74]. Wnt5a expression is increased in regions of immune cell infiltration during cardiac fibrosis caused by autoimmune myocarditis [75]. To assess the role of β-catenin in pressure overload-induced heart failure, the β-catenin gene in resident cardiac fibroblasts was manipulated and cardiac fibroblasts were activated in Tcf21MerCreMer and Periostin (Postn)MerCreMer mouse lines, respectively. The loss of function study revealed that the inactivation of β-catenin in cardiac fibroblasts was extremely beneficial in alleviating cardiac hypertrophy by reducing interstitial fibrosis without changing the number of activated fibroblasts in vivo [78]. Notably, cardiomyocyte hypertrophy was found to be reduced with the loss of β-catenin after TAC [78], indicating some non-cell autonomous effect. Additional research should be conducted to study the interaction of the Wnt/β-catenin signaling pathway with other pathways in cardiac fibroblasts in order to provide new insights into cardiac injury and repair.
The Hippo pathway is known for its key role in cardiomyocyte proliferation [79, 80]. Large tumor suppressor kinase1 (Lats1) and Lats2 are two negative regulators of Yap, which is the core transcriptional coactivator in the Hippo pathway [81]. The results of scRNA-seq have revealed that uninjured Lats1/2-mutant cardiac fibroblasts could automatically transition to the myofibroblast state [82]; this is consistent with the finding of another study [83]. Jamie et al. used Yap F/F;Tcf21 MCM mouse line to knock down Yap specifically in cardiac fibroblasts and found that Yap induced myofibroblast differentiation and gene expression involved in ECM through the activation of the TEAD domain transcription factor 1 and the subsequent de novo expression of myocardin-related transcription factor A [79]. These findings suggest that the manipulation of the Hippo signaling pathway is a prospective therapeutic strategy for improving cardiac remodeling in heart failure. As Wnt/β-catenin and Hippo pathways interact to regulate cardiomyocyte proliferation in the developing heart [84], it would be interesting to understand whether these two pathways converge or interact with each other to regulate fibroblast activation and proliferation in cardiac fibrosis.
Manipulation of Cardiac Fibroblasts for Cardiac Repair
As cardiac fibrosis is an important predictor of sudden cardiac death [85], manipulating the number and activity of cardiac fibroblasts to alleviate severe fibrosis could be a promising therapeutic target for treating cardiac fibrosis. In terms of gene regulation, the selective knockdown of Tgfbr1/2 or Smad3 in cardiac fibroblasts could significantly inhibit the fibrotic response and reduce fibrosis [61, 86]. The deletion of Yap in fibroblasts could prominently attenuate cardiac fibrosis and ameliorate cardiac dysfunction after MI or other injuries [79]. Another study reported that the activation of TGF-β1 could be inhibited by decreasing nuclear Yap levels [87]. In addition, recombinant BMP9 acts as a potential candidate to reduce cardiac fibrosis and improve cardiac function in heart failure patients [72]. The deletion of Mapk14 from fibroblasts can also arrest fibrosis by blocking the process of myofibroblast differentiation [88].
With the exploration of the underlying mechanism, many therapeutic approaches for potentially targeting tissue fibrosis have been reported in patients. Angiotensin-converting enzyme inhibitors (ACEIs) and angiotensin II receptor blockers (ARBs) have already shown considerable potency in alleviating cardiac fibrosis in human and animal models of heart failure; however, they have not been approved for treating cardiac fibrosis [89]. TβRI, an inhibitor of ALK5, reduces TGF-β activity by restraining collagen synthesis and slowing fibrosis progression; it can alleviate cardiac dysfunction and improve cardiac remodeling in heart injury patients [90, 91]. Evodiamine reduces the expression of α-SMA in rat cardiac fibroblasts to enhance cardiac function after injury [92]. Moreover, tranilast and pirfenidone are two antifibrotic agents that could limit TGF-β activation; however, the underlying mechanisms remain unknown [93]. The profibrotic Wnt/β-catenin signaling pathway has been reported to be controlled by angiotensin II receptor 1 in experimental autoimmune myocarditis [94]. XAV939, an inhibitor of the Wnt/β-catenin pathway, reduces cardiac fibroblast activation induced by TGFβ1 and promotes cardiac repair [78]. With regard to Smad-dependent and Smad-independent pathways, halofuginone and the c-abl kinase inhibitor imatinib are proposed to inhibit fibrosis [95]. Halofuginone resists fibrosis by inhibiting the activity of Smad3 and rapidly increasing the expression of Smad7 [96]. Imatinib alleviates renal [97] and pulmonary [98] fibrosis by affecting the Smad-independent TGF-β signaling pathway. Osthole treatment decreases the expression of α-SMA, pSmad2/3, Smad4, and TβRI and the deposition of collagen I and III but increases the expression of Smad7 [99]. Verteporfin inhibits the YAP–TEAD association and decreases the expression of MRTF-A and α-SMA to attenuate cardiac fibrosis [79]. CCG-203971, the inhibitor of MRTF-A, also reduces collagen lattice contraction [79, 100]. Moreover, cilengitide reduces the scar size in collagen V-deficient mice by inhibiting specific integrins [101]. JQ1, a small-molecule BET bromodomain inhibitor, has been verified to reduce fibrosis in TAC models [102].
Through scRNA-seq analysis, collagen triple helix repeat containing 1 (CTHRC1) has been found to be a novel regulator that plays a crucial role in cardiac fibroblasts to heal scars after MI [103]. Moreover, scRNA-seq and transposase-accessible chromatin sequencing (scATAC-seq) have revealed that Meox1 is highly upregulated in myofibroblasts after TAC; it is considered to be a central mediator of fibroblast activation that could be targeted to regulate cardiac dysfunction [104•]. Recently, chimeric antigen receptor (CAR) T cells have been engineered to recognize and prompt the ablation of myofibroblasts [105]. These advances in the modulation of cardiac fibroblasts provide us with new approaches for treating cardiac fibrosis and promoting tissue repair after injury.
Conclusion
With technological advancements, such as lineage tracing and scRNA-seq, the origins of fibroblasts, the relationships between different fibroblast states, and fibroblast heterogeneity have begun to be unraveled in recent years. The coordination between tremendous complex and diverse signaling pathways that regulate the fate of fibroblasts and modulate their transformation during diseases has also been elucidated in recent studies. While some existing treatments targeting cardiac fibroblasts could alleviate fibrosis after cardiac injury, their specific mechanisms and pharmacokinetics remain unknown. As fibroblasts are a type of nonmyocytes in the heart that play an indispensable role in cardiac injury and repair, efforts will also be needed to understand the interaction of fibroblasts with other cell lineages during the pathological process of cardiac fibrosis. It is necessary to conduct more basic research using state-of-the-art technology to understand fibroblast biology in depth and explore more therapeutic targets for treating cardiac fibrosis.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Virani SS, Alonso A, Aparicio HJ, Benjamin EJ, Bittencourt MS, Callaway CW, et al. Heart Disease and Stroke Statistics-2021 Update: A Report From the American Heart Association. Circulation. 2021;143(8):e254–e743. https://doi.org/10.1161/CIR.0000000000000950.
Tallquist MD. Developmental Pathways of Cardiac Fibroblasts. Cold Spring Harb Perspect Biol. 2020;12(4). https://doi.org/10.1101/cshperspect.a037184.
Eghbali M, Weber KT. Collagen and the Myocardium - Fibrillar Structure, Biosynthesis and Degradation in Relation to Hypertrophy and Its Regression. Mol Cell Biochem. 1990;96(1):1–14.
Rockey DC, Bell PD, Hill JA. Fibrosis - A Common Pathway to Organ Injury and Failure. N Engl JMed. 2015;372(12):1138–49. https://doi.org/10.1056/NEJMra1300575.
Moore-Morris T, Guimaraes-Camboa N, Banerjee I, Zambon AC, Kisseleva T, Velayoudon A, et al. Resident fibroblast lineages mediate pressure overload-induced cardiac fibrosis. J Clin Invest. 2014;124(7):2921–34. https://doi.org/10.1172/JCI74783.
Paik DT, Cho S, Tian L, Chang HY, Wu JC. Single-cell RNA sequencing in cardiovascular development, disease and medicine. Nat Rev Cardiol. 2020;17(8):457–73. https://doi.org/10.1038/s41569-020-0359-y.
Muhl L, Genove G, Leptidis S, Liu J, He L, Mocci G, et al. Single-cell analysis uncovers fibroblast heterogeneity and criteria for fibroblast and mural cell identification and discrimination. Nat Commun. 2020;11(1):3953. https://doi.org/10.1038/s41467-020-17740-1.
• Ren Z, Yu P, Li D, Li Z, Liao Y, Wang Y, et al. Single-Cell Reconstruction of Progression Trajectory Reveals Intervention Principles in Pathological Cardiac Hypertrophy. Circulation. 2020;141(21):1704–19. https://doi.org/10.1161/circulationaha.119.043053. Findings from this study unravel six clusters of fibroblasts in a pressure overload-induced cardiac injury model by single-cell sequencing.
Wang L, Yu P, Zhou B, Song J, Li Z, Zhang M, et al. Single-cell reconstruction of the adult human heart during heart failure and recovery reveals the cellular landscape underlying cardiac function. Nat Cell Biol. 2020;22(1):108-+. https://doi.org/10.1038/s41556-019-0446-7.
Wang Y, Yao F, Wang L, Li Z, Ren Z, Li D, et al. Single-cell analysis of murine fibroblasts identifies neonatal to adult switching that regulates cardiomyocyte maturation. Nat Commun. 2020;11(1):2585. https://doi.org/10.1038/s41467-020-16204-w.
Hesse J, Owenier C, Lautwein T, Zalfen R, Weber JF, Ding Z, et al. Single-cell transcriptomics defines heterogeneity of epicardial cells and fibroblasts within the infarcted murine heart. Elife. 2021;10. https://doi.org/10.7554/eLife.65921.
Borthwick LA, Wynn TA, Fisher AJ. Cytokine mediated tissue fibrosis. Biochimica Et Biophysica Acta-Molecular Basis of Disease. 2013;1832(7):1049-60. https://doi.org/10.1016/j.bbadis.2012.09.014.
Davis J, Molkentin JD. Myofibroblasts: Trust your heart and let fate decide. Journal of Molecular and Cellular Cardiology. 2014;70:9–18. https://doi.org/10.1016/j.yjmcc.2013.10.019.
Zhang H, Pu W, Li G, Huang X, He L, Tian X, et al. Endocardium Minimally Contributes to Coronary Endothelium in the Embryonic Ventricular Free Walls. Circ Res. 2016;118(12):1880–93. https://doi.org/10.1161/CIRCRESAHA.116.308749.
Pinto AR, Ilinykh A, Ivey MJ, Kuwabara JT, D'Antoni ML, Debuque R, et al. Revisiting Cardiac Cellular Composition. Circ Res. 2016;118(3):400–9. https://doi.org/10.1161/CIRCRESAHA.115.307778
Kebschull JM, Zador AM. Cellular barcoding: lineage tracing, screening and beyond. Nat Methods. 2018;15(11):871–9. https://doi.org/10.1038/s41592-018-0185-x.
Cai CL, Martin JC, Sun Y, Cui L, Wang L, Ouyang K, et al. A myocardial lineage derives from Tbx18 epicardial cells. Nature. 2008;454(7200):104–8. https://doi.org/10.1038/nature06969.
Trowe MO, Shah S, Petry M, Airik R, Schuster-Gossler K, Kist R, et al. Loss of Sox9 in the periotic mesenchyme affects mesenchymal expansion and differentiation, and epithelial morphogenesis during cochlea development in the mouse. Dev Biol. 2010;342(1):51–62. https://doi.org/10.1016/j.ydbio.2010.03.014.
Merki E, Zamora L, Raya A, Kawakami Y, Wang JM, Zhang XX, et al. Epicardial retinoid X receptor alpha is required for myocardial growth and coronary artery formation. Proc Nat Acad Sci U S A. 2005;102(51):18455–60. https://doi.org/10.1073/pnas.0504343102.
Katz TC, Singh MK, Degenhardt K, Rivera-Feliciano J, Johnson RL, Epstein JA, et al. Distinct compartments of the proepicardial organ give rise to coronary vascular endothelial cells. Dev Cell. 2012;22(3):639–50. https://doi.org/10.1016/j.devcel.2012.01.012.
Zhou B, Ma Q, Rajagopal S, Wu SM, Domian I, Rivera-Feliciano J, et al. Epicardial progenitors contribute to the cardiomyocyte lineage in the developing heart. Nature. 2008;454(7200):109–13. https://doi.org/10.1038/nature07060.
Wessels A, van den Hoff MJ, Adamo RF, Phelps AL, Lockhart MM, Sauls K, et al. Epicardially derived fibroblasts preferentially contribute to the parietal leaflets of the atrioventricular valves in the murine heart. Dev Biol. 2012;366(2):111–24. https://doi.org/10.1016/j.ydbio.2012.04.020.
Peralta M, Gonzalez-Rosa JM, Marques IJ, Mercader N. The Epicardium in the Embryonic and Adult Zebrafish. J Dev Biol. 2014;2(2):101–16. https://doi.org/10.3390/jdb2020101.
Acharya A, Baek ST, Banfi S, Eskiocak B, Tallquist MD. Efficient inducible Cre-mediated recombination in Tcf21 cell lineages in the heart and kidney. Genesis. 2011;49(11):870–7. https://doi.org/10.1002/dvg.20750.
Acharya A, Baek ST, Huang G, Eskiocak B, Goetsch S, Sung CY, et al. The bHLH transcription factor Tcf21 is required for lineage-specific EMT of cardiac fibroblast progenitors. Development. 2012;139(12):2139–49. https://doi.org/10.1242/dev.079970.
Guimaraes-Camboa N, Cattaneo P, Sun Y, Moore-Morris T, Gu Y, Dalton ND, et al. Pericytes of Multiple Organs Do Not Behave as Mesenchymal Stem Cells In Vivo. Cell Stem Cell. 2017;20(3):345-+. https://doi.org/10.1016/j.stem.2016.12.006.
Doppler SA, Carvalho C, Lahm H, Deutsch M-A, Dressen M, Puluca N, et al. Cardiac fibroblasts: more than mechanical support. J Thorac Dis. 2017;9:S36–51. https://doi.org/10.21037/jtd.2017.03.122.
Smith CL, Baek ST, Sung CY, Tallquist MD. Epicardial-Derived Cell Epithelial-to-Mesenchymal Transition and Fate Specification Require PDGF Receptor Signaling. Circ Res. 2011;108(12):E15–U28. https://doi.org/10.1161/circresaha.110.235531.
Gittenberger-de Groot AC, Peeters M, Mentink MMT, Gourdie RG, Poelmann RE. Epicardium-derived cells contribute a novel population to the myocardial wall and the atrioventricular cushions. Circ Res. 1998;82(10):1043–52. https://doi.org/10.1161/01.Res.82.10.1043.
Mikawa T, Gourdie RG. Pericardial mesoderm generates a population of coronary smooth muscle cells migrating into the heart along with ingrowth of the epicardial organ. Dev Biol. 1996;174(2):221–32. https://doi.org/10.1006/dbio.1996.0068.
Dettman RW, Denetclaw W, Ordahl CP, Bristow J. Common epicardial origin of coronary vascular smooth muscle, perivascular fibroblasts, and intermyocardial fibroblasts in the avian heart. Dev Biol. 1998;193(2):169–81. https://doi.org/10.1006/dbio.1997.8801.
Skelly DA, Squiers GT, McLellan MA, Bolisetty MT, P Robson, Rosenthal NA, et al. Single-Cell Transcriptional Profiling Reveals Cellular Diversity and Intercommunication in the Mouse Heart. Cell Rep. 2018;22(3):600–10. https://doi.org/10.1016/j.celrep.2017.12.072.
Ali SR, Ranjbarvaziri S, Talkhabi M, Zhao P, Subat A, Hojjat A, et al. Developmental heterogeneity of cardiac fibroblasts does not predict pathological proliferation and activation. Circ Res. 2014;115(7):625–35. https://doi.org/10.1161/CIRCRESAHA.115.303794.
Huang X, Feng T, Jiang Z, Meng J, Kou S, Lu Z, et al. Dual lineage tracing identifies intermediate mesenchymal stage for endocardial contribution to fibroblasts, coronary mural cells, and adipocytes. J Biol Chem. 2019;294(22):8894–906. https://doi.org/10.1074/jbc.RA118.006994.
Jiang, XB, DH Rowitch, P Soriano, AP McMahon, HM Sucov. Fate of the mammalian cardiac neural crest. Development. 2000;127(8):1607–16.
Zhou B, Honor LB, He H, Ma Q, Oh JH, Butterfield C, et al. Adult mouse epicardium modulates myocardial injury by secreting paracrine factors. J Clin Invest. 2011;121(5):1894–904. https://doi.org/10.1172/JCI45529.
Ruiz-Villalba A, Simon AM, Pogontke C, Castillo MI, Abizanda V, Pelacho B, et al. Interacting Resident Epicardium-Derived Fibroblasts and Recruited Bone Marrow Cells Form Myocardial Infarction Scar. J Am Coll Cardiol. 2015;65(19):2057–66. https://doi.org/10.1016/j.jacc.2015.03.520.
Kanisicak O, Khalil H, Ivey MJ, Karch J, Maliken BD, Correll RN, et al. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat Commun. 2016;7:12260. https://doi.org/10.1038/ncomms12260.
Ali SR, Ranjbarvaziri S, Talkhabi M, Zhao P, Subat A, Hojjat A, et al. Developmental Heterogeneity of Cardiac Fibroblasts Does Not Predict Pathological Proliferation and Activation. Circulation Research. 2014;115(7):625–U81. https://doi.org/10.1161/circresaha.115.303794.
Wu R, Ma F, Tosevska A, Farrell C, Pellegrini M, Deb A. Cardiac fibroblast proliferation rates and collagen expression mature early and are unaltered with advancing age. Jci Insight. 2020;5(24). https://doi.org/10.1172/jci.insight.140628.
•• Fu X, Khalil H, Kanisicak O, Boyer JG, Vagnozzi RJ, Maliken BD, et al. Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart. J Clin Invest. 2018;128(5):2127–43. https://doi.org/10.1172/JCI98215. This study defined the matrifibrocytes for the first time and described the time window of different states of fibroblasts after myocardial infarction induced heart failure.
Hinz B. The myofibroblast: Paradigm for a mechanically active cell. J Biomech. 2010;43(1):146–55. https://doi.org/10.1016/j.jbiomech.2009.09.020.
Ivey MJ, Kuwabara JT, Pai JT, Moore RE, Sun Z, Tallquist MD. Resident fibroblast expansion during cardiac growth and remodeling. J Mol Cell Cardiol. 2018;114:161–74. https://doi.org/10.1016/j.yjmcc.2017.11.012.
Fomovsky GM, Holmes JW. Evolution of scar structure, mechanics, and ventricular function after myocardial infarction in the rat. Am J Physiol Heart Circ Physiol. 2010;298(1):H221–8. https://doi.org/10.1152/ajpheart.00495.2009.
Hoare M, Narita M. Transmitting senescence to the cell neighbourhood. Nat Cell Biol. 2013;15(8):887–9. https://doi.org/10.1038/ncb2811.
Zhu F, Li Y, Zhang J, Piao C, Liu T, Li HH, et al. Senescent Cardiac Fibroblast Is Critical for Cardiac Fibrosis after Myocardial Infarction. Plos One. 2013;8(9). https://doi.org/10.1371/journal.pone.0074535.
Tallquist MD. Cardiac Fibroblast Diversity. In: Nelson MT, Walsh K, editors. Annu Rev Physiol. 2020;82:63–78.
Yamada S, Nomura S. Review of Single-Cell RNA Sequencing in the Heart. Int J Mol Sci. 2020;21(21). https://doi.org/10.3390/ijms21218345.
McLellan MA, Skelly DA, Dona MS, Squiers GT, Farrugia GE, Gaynor TL, et al. High-Resolution Transcriptomic Profiling of the Heart During Chronic Stress Reveals Cellular Drivers of Cardiac Fibrosis and Hypertrophy. Circulation. 2020;142(15):1448–63. https://doi.org/10.1161/circulationaha.119.045115.
Ceco E, McNally EM. Modifying muscular dystrophy through transforming growth factor-beta. Febs J. 2013;280(17):4198–209. https://doi.org/10.1111/febs.12266.
Smith LR, Barton ER. Regulation of fibrosis in muscular dystrophy. Matrix Biol. 2018;68–69:602–15. https://doi.org/10.1016/j.matbio.2018.01.014.
Frangogiannis NG. The role of transforming growth factor (TGF)-beta in the infarcted myocardium. J Thorac Dis. 2017;9(Suppl 1):S52–63. https://doi.org/10.21037/jtd.2016.11.19.
Biernacka A. M Dobaczewski, NG Frangogiannis TGF-beta signaling in fibrosis. Growth Factors. 2011;29(5):196–202. https://doi.org/10.3109/08977194.2011.595714.
Andreotti F, Pasceri V, Hackett DR, Davies GJ, Haider AW, Maseri A. Preinfarction angina as a predictor of more rapid coronary thrombolysis in patients with acute myocardial infarction. N Engl J Med. 1996;334(1):7–12. https://doi.org/10.1056/nejm199601043340102.
Jenkins DP, Pugsley WB, Alkhulaifi AM, Kemp M, Hooper J, Yellon DM. Ischaemic preconditioning reduces troponin T release in patients undergoing coronary artery bypass surgery. Heart. 1997;77(4):314–8. https://doi.org/10.1136/hrt.77.4.314.
Scheuer J, Greenberg MA, Zohman LR. Exercise training in patients with coronary-artery disease. Mod Concepts Cardiovasc Dis. 1978;47(6):85–90.
Contreras O, Cruz-Soca M, Theret M, Soliman H, Tung LW, Groppa E, et al. Cross-talk between TGF-beta and PDGFRalpha signaling pathways regulates the fate of stromal fibro-adipogenic progenitors. J Cell Sci. 2019;132(19). https://doi.org/10.1242/jcs.232157.
Hinz B. Formation and function of the myofibroblast during tissue repair. J Investig Dermatol. 2007;127(3):526–37. https://doi.org/10.1038/sj.jid.5700613.
Kim KK, Sheppard D, Chapman HA. TGF-beta 1 Signaling and Tissue Fibrosis. Cold Spring Harb Perspect Biol. 2018;10(4). https://doi.org/10.1101/cshperspect.a022293.
David CJ, Massague J. Contextual determinants of TGF beta action in development, immunity and cancer. Nat Rev Mol Cell Biol. 2018;19(7):419–35. https://doi.org/10.1038/s41580-018-0007-0.
Khalil H, Kanisicak O, Prasad V, Correll RN, Fu X, Schips T, et al. Fibroblast-specific TGF-beta-Smad2/3 signaling underlies cardiac fibrosis. J Clin Investig. 2017;127(10):3770–83. https://doi.org/10.1172/jci94753.
Li Q, Pangas SA, Jorgez CJ, Graff JM, Weinstein M, Matzuk MM. Redundant roles of SMAD2 and SMAD3 in ovarian granulosa cells in vivo. Mol Cell Biol. 2008;28(23):7001–11. https://doi.org/10.1128/MCB.00732-08.
Sano M, Fukuda K, Sato T, Kawaguchi H, Suematsu M, Matsuda S, et al. ERK and p38 MAPK, but not NF-kappa B, are critically involved in reactive oxygen species-mediated induction of IL-6 by angiotensin II in cardiac fibroblasts. Circ Res. 2001;89(8):661–9. https://doi.org/10.1161/hh2001.098873.
Lu X, Law BK, Chytil AM, Brown KA, Aakre ME, Moses HL. Activation of the Erk pathway is required for TGF-beta 1-induced EMT in vitro. Neoplasia. 2004;6(5):603–10. https://doi.org/10.1593/neo.04241.
Nagoshi T, Matsui T, Aoyama T, Leri A, Anversa P, Li L, et al. PI3K rescues the detrimental effects of chronic Akt activation in the heart during ischemia/reperfusion injury. J Clin Investig. 2005;115(8):2128–38. https://doi.org/10.1172/jci23073.
Lin Z, Zhou P, von Gise A, Gu F, Ma Q, Chen J, et al. Pi3kcb Links Hippo-YAP and PI3K-AKT Signaling Pathways to Promote Cardiomyocyte Proliferation and Survival. Circ Res. 2015;116(1):35–U98. https://doi.org/10.1161/circresaha.115.304457.
Codelia VA, Sun G, Irvine KD. Regulation of YAP by Mechanical Strain through Jnk and Hippo Signaling. Curr Biol. 2014;24(17):2012–7. https://doi.org/10.1016/j.cub.2014.07.034.
Ikeuchi M, Tsutsui H, Shiomi T, Matsusaka H, Matsushima S, Wen J, et al. Inhibition of TGF-beta signaling exacerbates early cardiac dysfunction but prevents late remodeling after infarction. Cardiovasc Res. 2004;64(3):526–35. https://doi.org/10.1016/j.cardiores.2004.07.017.
Leask A. TGFbeta, cardiac fibroblasts, and the fibrotic response. Cardiovasc Res. 2007;74(2):207–12. https://doi.org/10.1016/j.cardiores.2006.07.012.
Rosenkranz S. TGF-beta1 and angiotensin networking in cardiac remodeling. Cardiovasc Res. 2004;63(3):423–32. https://doi.org/10.1016/j.cardiores.2004.04.030.
Rosenkranz S, Flesch M, Amann K, Haeuseler C, Kilter H, Seeland U, et al. Alterations of beta-adrenergic signaling and cardiac hypertrophy in transgenic mice overexpressing TGF-beta(1). Am J Physiol Heart Circ Physiol. 2002;283(3):H1253–H62. https://doi.org/10.1152/ajpheart.00578.2001.
Morine KJ, Qiao XY, York S, Natov PS, Paruchuri V, Zhang YL, et al. Bone Morphogenetic Protein 9 Reduces Cardiac Fibrosis and Improves Cardiac Function in Heart Failure. Circulation. 2018;138(5):513–26. https://doi.org/10.1161/circulationaha.117.031635.
Zeisberg M, Kalluri R. Reversal of experimental renal fibrosis by BMP7 provides insights into novel therapeutic strategies for chronic kidney disease. Pediatr Nephrol. 2008;23(9):1395–8. https://doi.org/10.1007/s00467-008-0818-x.
Duan J, Gherghe C, Liu D, Hamlett E, Srikantha L, Rodgers L, et al. Wnt1/beta catenin injury response activates the epicardium and cardiac fibroblasts to promote cardiac repair. Embo J. 2012;31(2):429–42. https://doi.org/10.1038/emboj.2011.418.
Blyszczuk P, Mueller-Edenborn B, Valenta T, Osto E, Stellato M, Behnke S, et al. Transforming growth factor-beta-dependent Wnt secretion controls myofibroblast formation and myocardial fibrosis progression in experimental autoimmune myocarditis. Eur Heart J. 2017;38(18):1413–25. https://doi.org/10.1093/eurheartj/ehw116.
Gordon MD, Nusse R. Wnt signaling: Multiple pathways, multiple receptors, and multiple transcription factors. J Biol Chem. 2006;281(32):22429–33. https://doi.org/10.1074/jbc.R600015200.
Eisenberg LM, Eisenberg CA. Wnt signal transduction and the formation of the myocardium. Dev Biol. 2006;293(2):305–15. https://doi.org/10.1016/j.ydbio.2006.02.014.
Xiang FL, Fang M, Yutzey KE. Loss of beta-catenin in resident cardiac fibroblasts attenuates fibrosis induced by pressure overload in mice. Nat Commun. 2017;8(1):712. https://doi.org/10.1038/s41467-017-00840-w.
Francisco J, Zhang Y, Jeong JI, Mizushima W, Ikeda S, Ivessa A, et al. Blockade of Fibroblast YAP Attenuates Cardiac Fibrosis and Dysfunction Through MRTF-A Inhibition. Jacc-Basic to Translational Science. 2020;5(9):931–45. https://doi.org/10.1016/j.jacbts.2020.07.009.
Ikeda S, Mizushima W, Sciarretta S, Abdellatif M, Zhai P, Mukai R, et al. Hippo Deficiency Leads to Cardiac Dysfunction Accompanied by Cardiomyocyte Dedifferentiation During Pressure Overload. Circ Res. 2019;124(2):292–305. https://doi.org/10.1161/circresaha.118.314048.
Zhou Q, Li L, Zhao B, Guan KL. The Hippo Pathway in Heart Development, Regeneration, and Diseases. Circ Res. 2015;116(8):1431–47. https://doi.org/10.1161/circresaha.116.303311.
Xiao Y, Hill MC, Zhang M, Martin TJ, Morikawa Y, Wang S, et al. Hippo Signaling Plays an Essential Role in Cell State Transitions during Cardiac Fibroblast Development. Dev Cell. 2018;45(2):153–69 e6. https://doi.org/10.1016/j.devcel.2018.03.019.
Xiao Y, Hill MC, Li L, Deshmukh V, Martin TJ, Wang J, et al. Hippo pathway deletion in adult resting cardiac fibroblasts initiates a cell state transition with spontaneous and self-sustaining fibrosis. Genes Dev. 2019;33(21-22):1491–505. https://doi.org/10.1101/gad.329763.119.
Heallen T, Zhang M, Wang J, Bonilla-Claudio M, Klysik E, Johnson RL, et al. Hippo Pathway Inhibits Wnt Signaling to Restrain Cardiomyocyte Proliferation and Heart Size. Science. 2011;332(6028):458–61. https://doi.org/10.1126/science.1199010.
Gulati A, Jabbour A, Ismail TF, Guha K, Khwaja J, Raza S, et al. Association of Fibrosis With Mortality and Sudden Cardiac Death in Patients With Nonischemic Dilated Cardiomyopathy. Jama-J Am Med Assoc. 2013;309(9):896–908. https://doi.org/10.1001/jama.2013.1363.
Meng Q, Bhandary B, Bhuiyan MS, James J, Osinska H, Valiente-Alandi I, et al. Myofibroblast-Specific TGF beta Receptor II Signaling in the Fibrotic Response to Cardiac Myosin Binding Protein C-Induced Cardiomyopathy. Circ Res. 2018;123(12):1285–97. https://doi.org/10.1161/circresaha.118.313089.
Zmajkovicova K, Bauer Y, Menyhart K, Schnoebelen M, Freti D, Boucher M, et al. GPCR-induced YAP activation sensitizes fibroblasts to profibrotic activity of TGF beta 1. Plos One. 2020;15(2). https://doi.org/10.1371/journal.pone.0228195.
Molkentin JD, Bugg D, Ghearing N, Dorn LE, Kim P, Sargent MA, et al. Fibroblast-Specific Genetic Manipulation of p38 Mitogen-Activated Protein Kinase In Vivo Reveals Its Central Regulatory Role in Fibrosis. Circulation. 2017;136(6):549-+. https://doi.org/10.1161/circulationaha.116.026238.
Parichatikanond W, Luangmonkong T, Mangmool S, Kurose H. Therapeutic Targets for the Treatment of Cardiac Fibrosis and Cancer: Focusing on TGF-beta Signaling. Front Cardiovasc Med. 2020;7. https://doi.org/10.3389/fcvm.2020.00034.
Tan SM, Zhang Y, Connelly KA, Gilbert RE, Kelly DJ. Targeted inhibition of activin receptor-like kinase 5 signaling attenuates cardiac dysfunction following myocardial infarction. Am J Physiol Heart Circ Physiol. 2010;298(5):H1415–H25. https://doi.org/10.1152/ajpheart.01048.2009.
Engebretsen KVT, Skardal K, Bjornstad S, Marstein HS, Skrbic B, Sjaastad I, et al. Attenuated development of cardiac fibrosis in left ventricular pressure overload by SM16, an orally active inhibitor of ALK5. J Mol Cell Cardiol. 2014;76:148–57. https://doi.org/10.1016/j.yjmcc.2014.08.008.
Wu QQ, Xiao Y, Jiang XH, Yuan Y, Yang Z, Chang, W, et al. Evodiamine attenuates TGF-beta1-induced fibroblast activation and endothelial to mesenchymal transition. Mol Cell Biochem. 2017;430(1-2):81–90. https://doi.org/10.1007/s11010-017-2956-6.
Tamai H, Katoh O, Suzuki S, Fujii K, Aizawa T, Takase S, et al. Impact of tranilast on restenosis after coronary angioplasty: Tranilost restenosis following angioplasty trial (TREAT). Am Heart J. 1999;138(5):968–75. https://doi.org/10.1016/s0002-8703(99)70025-6.
Czepiel M, Diviani D, Jazwa-Kusior A, Tkacz K, Rolski F, Smolenski RT, et al. Angiotensin II receptor 1 controls profibrotic Wnt/beta-catenin signalling in experimentalautoimmune myocarditis. Cardiovasc Res. 2021. https://doi.org/10.1093/cvr/cvab039.
Bujak M, Frangogiannis NG. The role of TGF-beta signaling in myocardial infarction and cardiac remodeling. Cardiovasc Res. 2007;74(2):184–95. https://doi.org/10.1016/j.cardiores.2006.10.002.
Xavier S, Piek E, Fujii M, Javelaud D, Mauviel A, Flanders KC, et al. Amelioration of radiation-induced fibrosis - Inhibition of transforming growth factor-beta signaling by halofuginone. J Biol Chem. 2004;279(15):15167–76. https://doi.org/10.1074/jbc.M309798200.
Wang SN, Wilkes MC, Leof EB, Hirschberg R. Imatinib mesylate blocks a non-smad TGF-beta pathway and reduces renal fibrogenesis in vivo. Faseb J. 2005;19(1):1–11. https://doi.org/10.1096/fj.04-2370com.
Daniels CE, Wilkes MC, Edens M, Kottom TJ, Murphy SJ, Limper AH, et al. Imatinib mesylate inhibits the profibrogenic activity of TGF-beta and prevents bleomycin-mediated lung fibrosis. J Clin Investig. 2004;114(9):1308–16. https://doi.org/10.1175/jci200419603.
Liu JC, Wang F, Xie ML, Cheng ZQ, Qin Q, Chen L, et al. Osthole inhibits the expressions of collagen I and III through Smad signaling pathway after treatment with TGF-beta1 in mouse cardiac fibroblasts. Int J Cardiol. 2017;228:388–93. https://doi.org/10.1016/j.ijcard.2016.11.202.
Haak AJ, Tsou PS, Amin MA, Ruth JH, Campbell P, Fox DA, et al. Targeting the Myofibroblast Genetic Switch: Inhibitors of Myocardin-Related Transcription Factor/Serum Response Factor-Regulated Gene Transcription Prevent Fibrosis in a Murine Model of Skin Injury. J Pharmacol Exp Ther. 2014;349(3):480–6. https://doi.org/10.1124/jpet.114.213520.
Yokota T, McCourt J, Ma F, Ren S, Li S, Kim TH, et al. Type V Collagen in Scar Tissue Regulates the Size of Scar after Heart Injury. Cell. 2020;182(3):545-+. https://doi.org/10.1016/j.cell.2020.06.030.
Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, et al. Selective inhibition of BET bromodomains. Nature. 2010;468(7327):1067–73. https://doi.org/10.1038/nature09504
Ruiz-Villalba A, Romero JP, Hernandez SC, Vilas-Zornoza A, Fortelny N, Castro-Labrador L, et al. Single-Cell RNA Sequencing Analysis Reveals a Crucial Role for CTHRC1 (Collagen Triple Helix Repeat Containing 1) Cardiac Fibroblasts After Myocardial Infarction. Circulation. 2020;142(19):1831–47. https://doi.org/10.1161/circulationaha.119.044557.
• Alexanian M, Przytycki PF, Micheletti R, Padmanabhan A, Ye L, Travers JG, et al. A transcriptional switch governs fibroblast activation in heart disease. Nature. 2021;595(7867):438–43. https://doi.org/10.1038/s41586-021-03674-1. This study proved that targeting the mediator of fibroblast activation regulated the cardiac dysfunction by using scRNA-seq and scATAC-seq.
Vagnozzi RJ, Johansen AKZ, Molkentin JD. CARdiac Immunotherapy: T Cells Engineered to Treat the Fibrotic Heart. Mol Ther. 2019;27(11):1869–71. https://doi.org/10.1016/j.ymthe.2019.09.021.
Funding
This work was supported by the National key Research & Development Program of China (2019YFA0110403, 2019YFA0802000, 2021YFA080021).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors confirm that there is no conflict of interest between them.
Human and Animal Rights and Informed Consent
The present study did not involve human or animal subjects.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Regenerative Medicine
Rights and permissions
About this article

Cite this article
Han, M., Zhou, B. Role of Cardiac Fibroblasts in Cardiac Injury and Repair. Curr Cardiol Rep 24, 295–304 (2022). https://doi.org/10.1007/s11886-022-01647-y
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11886-022-01647-y