
Abstract
This review article summarizes recent research into the mechanisms as to how elevated levels of triglyceride (TG) and low levels of high- density- lipoprotein cholesterol (HDL-C) contribute to inflammation and atherosclerosis. Evidence supports the role of TG-rich lipoproteins in signaling mechanisms via apolipoproteins C-III and free fatty acids leading to activation of NFKβ, VCAM-1 and other inflammatory mediators which lead to fatty streak formation and advanced atherosclerosis. Moreover, the cholesterol content in TG-rich lipoproteins has been shown to predict CAD risk better than LDL-C. In addition to reverse cholesterol transport, HDL has many other cardioprotective effects which include regulating immune function. The “functionality” of HDL appears more important than the level of HDL-C. Insulin resistance and central obesity underlie the pathophysiology of elevated TG and low HDL-C in metabolic syndrome and type 2 diabetes. Lifestyle recommendations including exercise and weight loss remain first line therapy in ameliorating insulin resistance and the adverse signaling processes from elevated levels of TG-rich lipoproteins and low HDL-C.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The lowering of low density lipoprotein (LDL) cholesterol (C) with statin drugs has significantly reduced cardiovascular events; however, patients with LDL-C less than 70 mg/dL on statin drugs continue to have cardiovascular events. In the Treating to New Targets (TNT) trial, subjects with coronary heart disease (CHD) were randomized to atorvastatin 10 mg or 80 mg. Those in the lowest quintile of high density lipoprotein (HDL)-C had the highest event rate even with LDL-C < 70 mg/dL [1]. A graded decrease in levels of triglyceride (TG) from the lowest to highest HDL quintile was observed, a finding suggesting that metabolic syndrome and/or insulin resistance may be contributing. Taken together, these findings suggest that high levels of TG and low levels of HDL-C were contributing to residual risk. Therefore, additional treatment options need to be sought to prevent cardiovascular events.
TG levels between 200 and 800 mg/dL are associated with low levels of HDL-C, small, dense LDL particles, atherogenic TG-rich remnants and insulin resistance, all of which are associated with central obesity, metabolic syndrome and type 2 diabetes and increase the risk for CHD [2]. The metabolic syndrome now has a single global definition with at least three of the following: central obesity (waist circumference > 88 cm (35 inches) in women and > 100 cm (40 inches, 90 cm Asian) in men, fasting blood glucose ≥ 5.56 mmol/L (100 mg/dL), TGs ≥ 1.7 mmol/L (150 mg/dL), low HDL-C (< 1.04 mmol/L [40 mg/dL] in men and < 1.7 mmol/L [50 mg/dL] in women), and systolic and/or diastolic blood pressure ≥ 130/ ≥ 85 mm Hg [2]. Elevated serum TGs are most often observed in the metabolic syndrome which has a prevalence of 24 % in US adults and 43 % of adults older than 60 years [3] and increases the risk of cardiovascular outcomes two-fold and all-cause mortality, 1.5-fold. Therefore, reliable assessment of the risk associated with lipid fractions other than LDL is important for the development of accurate screening and treatment strategies.
In this review, we summarize recent research which has provided insight into the mechanisms by which high levels of TG and TG-rich lipoproteins, and low levels of HDL-C contribute to the development of inflammation, atherosclerosis and acute plaque rupture and thrombus formation. On the basis of these mechanisms, we provide suggestions for evaluation, treatment and management.
Epidemiological and Clinical Trial Evidence Linking Triglyceride Levels with Risk for CHD
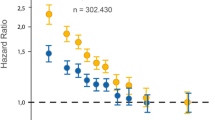
In prospective population-based cohort studies, TG levels were an independent univariate predictor of CHD; however, the risk of TG was generally attenuated or eliminated after adjustment, especially for HDL-C levels [reviewed in 4, 5]. The largest meta-analysis to date, the Emerging Risk Factors Collaboration (ERFC), comprised 302,430 people without initial vascular disease from 68 long-term prospective studies (Europe and North America) for a total of 12,785 cases of CHD (8,857 nonfatal MIs and 3,928 deaths from CHD) during 2.79 million person-years of follow-up (median, 6.1 years to first outcome) [6]. The HR for the primary outcome (nonfatal MI and CHD death) for TG was 1.37 (95 % CI, 1.31-1.42) after adjustment for nonlipid risk factors. However, after further adjustment for HDL-C and non-HDL-C, the HR for TG was reduced to 0.99 (95 % CI, 0.94-1.05). In case–control studies and angiographic studies, TG has remained an independent predictor after adjustment for TC or LDL-C [7–14] and HDL-C [13–18]. In clinical outcome trials, subjects with elevated TG levels showed improvement in CVD risk primarily when high LDL-C and low HDL-C (the atherogenic dyslipidemia triad) accompanied elevated TG at baseline. On the basis of this, a recent AHA statement on TG concluded that TG levels may provide information on risk especially when combined with low HDL-C and elevated LDL-C [19••].
TG-Rich Lipoprotein Metabolism
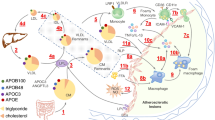
The metabolism of TG-rich containing lipoprotein fractions significantly impacts the levels and composition of other lipoprotein fractions which also contribute to cardiovascular risk [20]. Triacylglycerols are the major source of metabolic energy for utilization in skeletal muscle and storage in adipose tissue and are comprised of a glycerol backbone in which each of the three hydroxyl groups is esterified with a fatty acid [21]. Cholesterol and TGs are almost insoluble in plasma; therefore, they are transported in spherical lipoprotein particles which contain a central core of varying amounts of TG and cholesteryl ester (CE) (both nonpolar lipids) covered on the surface by polar lipids comprised of phospholipids, one or more apolipoproteins (apo) and unesterified cholesterol [21]. They are divided into five major classes based on density, which are inversely related to size and lipid content: chylomicrons (CM), very low-density lipoprotein (VLDL), intermediate density lipoprotein (IDL), LDL and HDL [22]. Containing mainly TG in their core, CMs and VLDL are the major TG carriers in plasma and are the two largest classes of lipoproteins.
ApoB-100 is synthesized by the liver and secreted in the form of VLDL, a TG-rich-lipoprotein which in plasma contains 60 % TG by mass and 20 % CE by mass [22]. The TGs in VLDL come from glycerol with fatty acids taken up from plasma either as albumin-bound fatty acids or as TG-fatty acids in remnant lipoproteins as they return to the liver or are newly synthesized in the liver. Produced in the intestine in response to dietary fat, CMs contain apoB-48, the amino terminal 48 % of apoB-100, which is synthesized by the intestine and produced by a premature stop codon at the apoB-100 codon 2153 by tissue specific mRNA processing (Fig. 1) [23]. Within the intestinal cell, free fatty acids (FFAs) combine with glycerol to form TGs, and cholesterol is esterified by acyl-coenzyme A:cholesterol acyltransferase (ACAT) to form cholesterol esters. Microsomal triglyceride transfer protein (MTP) transfers TGs from the cytosol to the endoplasmic reticulum (ER) containing nascent apoB during the assembly of CM and VLDL in enterocytes and hepatocytes, respectively [24].

TG-rich lipoprotein and HDL metabolism. In adipose and muscle capillaries, TG in CM (from intestine) and VLDL (from liver) are hydrolyzed into FFA by endothelial-bound LPL and GP1HBP1. ApoC-II activates LPL, thus lowering levels of TG whereas apoC-III inhibits LPL, thus raising TG levels. The apoC-III in TG-rich VLDL can bind to the TLR2 receptor, thus activating NFKβ, a proinflammatory transcription factor, which upregulates VCAM1 on the endothelial cell to which monocytes attach and migrate into the subendothelial space where they are converted to macrophages which take up oxidized lipoproteins and form foam cells and the fatty streak. When TG levels are high, HL converts TG-rich LDL to small, dense LDL which have a longer half-life and are more readily oxidized. HDL particles pick up the CE from foam cells and transfer it via CETP to apoB-containing particles in exchange for TG. These TG-enriched HDL particles are catabolized faster than large, CE-rich HDL, a finding resulting in lower levels of HDL-C in the setting of high TG levels. TF = tissue factor; ox = oxidized lipoprotein; ec = endothelial cell; sd = small dense; CMR = chylomicron remnant
CMs enter lacteals in the intestinal villi and travel via the lymphatics to the thoracic duct and then into the blood stream. In the plasma, both CMs and VLDL particles adhere to glycosaminoglycan molecules on endothelial cells of capillaries, primarily in muscle, lung and adipose tissue [25], where interaction with lipoprotein lipase (LPL) and glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein 1 (GPIHBP1) results in hydrolysis of VLDL and CM TGs to FFAs and glycerol, a process forming smaller particles termed “VLDL remnants” (or IDL) and CM remnants, respectively [26, 27]. The removal of TG from VLDL by LPL exposes the apoE molecules on the lipoprotein surface of VLDL [26]. ApoE functions as a ligand in the receptor-mediated clearance of CM and VLDL remnants in the liver via several receptors- a remnant receptor [28, 29], the LDL receptor (an apoB/apoE receptor), the LDL receptor-related protein (LRP), the VLDL receptor and apoE receptors [22]. Using stable isotopes, Welty et al. [30] showed that 50 % of VLDL are directly removed from plasma; the remaining 50 % are converted to IDL which then interact with hepatic lipase (HL) and are converted to CE-rich LDL, the major cholesterol carrying lipoprotein in normal human plasma. ApoB-100 is the main structural protein of LDL and contains the LDL-receptor-binding domain; therefore, LDL is removed from the circulation by binding mainly to hepatic LDL receptors (Fig. 1) [22].
TG-Rich VLDL ApoC-III and Proinflammatory Signaling
The C apolipoproteins play an important role in VLDL metabolism. ApoC-II is an activator of LPL which hydrolyzes the core TGs, thereby, releasing FFAs and lowering TG levels. ApoC-III, a 79-amino-acid glycoprotein synthesized by the liver and intestines [31], is an inhibitor of LPL and thereby, can raise TG levels [32]. It also stimulates VLDL synthesis and inhibits the binding of remnants to the LDL-R mediated by apoE. Thus, TG-rich apoC-III VLDL particles circulate longer and are converted to TG-rich remnants which are then lipolyzed by HL to small, dense LDL particles [33]. Persons lacking apoC-III have efficient lipolysis of TGs and therefore, low levels of TG. Subjects lacking one allele for apoC-III had TG levels of approximately 50 mg/dL and decreased coronary calcium at CT scanning [34].
ApoC-III plays an important role in inflammation and the development of atherosclerosis by acting as a proinflammatory mediator by activating the Toll-like receptor 2 (TLR2) signaling pathway in mouse atherosclerosis [35]. Activation of TLR2 leads to activation of NFKβ, a master regulator of over 200 genes involved in inflammation. Activation of NFKβ leads to upregulation of vascular cell adhesion molecules (Fig. 1) as VCAM1 which bind peripheral monocytes to endothelial cells [36, 37]. Support for the role of apoC-III comes from human epidemiological studies in which apoC-III containing lipoproteins independently predicted CHD [38]. Genetic variants in apoC-III are also associated with nonalcoholic fatty liver disease (NAFLD) and insulin resistance [39••, 40]. Thus, TG-rich lipoproteins as VLDL apoC-III exhibit proinflammatory properties and may initiate early inflammatory events in atherosclerosis via signaling mechanisms.
Metabolic Syndrome: Role of Insulin in TG-Rich Lipoprotein Metabolism
The pathophysiology of metabolic syndrome appears related to a genetic predisposition created by multiple genes coupled with a sedentary lifestyle and a diet containing excess calories. With development of obesity, the ability of adipocytes to store TGs as fat is exceeded; therefore, fat is stored in liver and skeletal muscle [41]. Increased levels of circulating FFAs released from adipocytes cause insulin resistance in muscle and other tissues [41]. Adipocyte expression of monocyte chemoattractant protein-1 and other cytokines causes infiltration of macrophages and other immune cells into adipocytes [42•]. These macrophages release cytokines which cause insulin resistance in muscle, liver and other tissues by impairing insulin signaling due to increased serine phosphorylation of insulin receptor substrate-1 (IRS-1) [42•].
Insulin resistance leads to increased adipocyte lysis which leads to increased levels of FFAs which flux to the liver where increased rates of hepatic TG and apoC-III synthesis lead to increased secretion of TG-rich VLDL containing apoC-III (Fig. 1) and elevated levels of TG in the plasma [43]. Insulin inhibits apoC-III gene transcription; therefore, insulin resistance leads to overproduction of apoC-III [44]. Down regulation of LPL expression in insulin resistance leads to decreased catabolism of TG-rich VLDL. Insulin causes degradation of apoB in the liver [45]; therefore, in insulin-resistant states, less apoB degradation leads to increased production and secretion of VLDL and increased plasma TG levels [46]. FFAs also block apoB degradation. MTP gene expression is inhibited by insulin; therefore, increased MTP transfer of TG to VLDL apoB is another explanation for why VLDL secretion and TG levels are increased in insulin resistance syndromes [47]. Moreover, insulin resistance is also associated with increases in intrahepatic expression of genes of TG biosynthesis, i.e., sterol regulatory element-binding protein-1C [48••]; increased intrahepatic TGs cause nonalcoholic fatty liver disease.
Lipolysis of TG-rich lipoproteins produces oxidized fatty acids which have a number of adverse effects including eliciting proinflammatory responses in endothelial cells by activating Toll–like receptors [reviewed in 49••], leading to endothelial cell insulin resistance, upregulation of NFKβ and expression of adhesion molecules, macrophage cytotoxicity and promotion of coagulation [reviewed in 50•]. FFAs can also suppress the atheroprotective and anti-inflammatory effects of HDL. In contrast, esterification of FFA to TG reduces inflammation. Therefore, insulin resistance increases production of apoB, FFA, apoC-III and VLDL, all of which lead to increased secretion of TG-rich VLDL which is associated with increased proinflammatory mechanisms which can cause atherosclerosis and thrombosis.
Role of Lipids in Atherosclerotic Plaque Formation
To transport TG and cholesterol to peripheral tissues, lipoprotein particles cross the endothelial barrier in blood vessels to reach the extracellular space (Fig. 1). The subendothelial retention of apoB-100-containing lipoproteins via a charge-mediated interaction with proteoglycans in the extracellular matrix is thought to be the initiating event in atherogenesis. Smaller, electronegative LDL particles penetrate the endothelial barrier 1.7-fold better than large LDL particles; CM and VLDL remnants also can cross. All of these particles interact with positively charged intimal proteoglycans [51]. Oxidation, by reactive oxygen species, of fatty acids of surface phospholipids of the apoB-containing lipoprotein particles results in modification of lysine residues of apoB (termed Ox-PL). Of note, the presence of Ox-PL precedes the appearance of the monocytes [52]; dietary cholesterol can lead to Ox-PL [53•].
Scavenger receptors on macrophages recognize modified apoB in oxLDL, and unregulated uptake of the modified lipoprotein particle causes macrophage accumulation of lipids, a process leading to a foamy cytoplasm and the term foam cells (Fig. 1) [reviewed in 4]. The most important scavenger receptor is CD36 (also called scavenger receptor B) [54]. As foam cells increase in number, the fatty streak develops. VLDL particles from patients with hypertriglyceridemia are enriched in apoE, which can lead to a conformational change in the VLDL particle that facilitates binding to the macrophage scavenger receptor. CM remnants and IDL are also small enough to enter the subendothelial space where they are taken up in an unregulated fashion by scavenger receptors on macrophages, leading to foam cell formation similar to oxLDL (55 and reviewed in reference 4]. In support of this, remnant lipoproteins have been identified in the arteries of humans [56]. Remnant cholesterol is the cholesterol content of VLDL, IDL and chylomicron remnants (which are all TG-rich lipoproteins) expressed as TC- (HDL-C + LDL-C). Elevated nonfasting TG is a marker of elevated nonfasting remnant cholesterol. In a Mendelian randomization study of 73,513 subjects from Copenhagen, the hazard ratio for CAD was greater for subjects in the highest versus lowest quintile of remnant cholesterol (HR: 2.3;95%CI: 1.7 to 3.1) compared with those in the upper versus lower quintile of LDL-C (HR:1.8;95 % CI: 1.4 to 2.2) [57]. Therefore, remnant cholesterol predicted risk of CAD better than LDL-C.
Endothelium-dependent vasomotor function in human coronary arteries is impaired by both IDL and LDL [58]. Several angiographic trials of cholesterol-lowering therapy have shown that serum IDL concentrations are predictive of an increased incidence of CHD [59] and an increased incidence of coronary events in those with CHD, independently of other factors [60–62] VLDL and IDL have been identified in human atherosclerotic plaques [63], and these particles are associated with progression of mild to moderate coronary lesions. In the Monitored Atherosclerosis Regression Study (MARS) angiographic trial, IDL, but not VLDL or LDL, was associated with progression of carotid artery intima-media thickness [64]. Moreover, the total TG level and markers for TG metabolism predicted risk of progression of low-grade but not high-grade coronary artery lesions [65, 66].
Effect of TG-Rich Lipoproteins on Thrombosis, Necrotic Core Formation and Plaque Rupture
In vitro studies have implicated TG-rich lipoproteins (and in some experiments, TG lipolysis products) in promotion of coagulation [67]. VLDL increases transcription of the plasminogen activator-1 gene, leading to decreased lysis of thrombi [68]. Insulin resistance in macrophages promotes formation of a necrotic core in atherosclerotic plaques by enhancing macrophage apoptosis [69]. This leads to rupture of the shoulder of the plaque, causing plaque rupture and exposure of the necrotic core to circulating blood and tissue factor which precipitates thrombosis, leading to unstable angina pectoris and myocardial infarction [70••]. Macrophages and smooth muscle cells in plaque overexpress the potent procoagulant tissue factor. Nishi et al. [71] found that the vulnerability of plaques was related to the amount of LDL containing oxidized phosphatidylcholine in the lesion, a finding highlighting the importance of ox-PL in plaque rupture.
Effect of TG-Rich Lipoproteins on Level of HDL-C in Humans
Levels of TG-rich lipoproteins also impact HDL level and particle size. The cardioprotective effect of HDL has been largely attributed to its role in reverse cholesterol transport (RCT) which involves removing cholesterol from peripheral cells including macrophages in the arterial wall and directing to the liver for recycling or excretion [72]. Cholesterol efflux is the first step in the formation of HDL particles which occurs when lipid-poor apoA-I (which has been secreted from the liver) interacts with the ATP binding cassette transporter (ABC) A1 receptor on the surface of peripheral cells as macrophages to produce nascent HDL (nHDL) (Fig. 1) [21, 73•]. This results in transfer of free cholesterol and phospholipid to the apoA-I, forming a pre-beta particle. The free cholesterol is then esterified under the action of lecithin-cholesterol-acyl transferase (LCAT) and ABCG1 to form a mature HDL particle.
There are two pathways by which RCT can occur. In the first, the scavenger receptor class B type 1 (SRB-1) mediates hepatic uptake of CE from HDL particles without uptake of apoA-I or the whole HDL particle [74]. In the second pathway, cholesteryl ester transfer protein (CETP) catalyzes the transfer of CE from HDL to apoB-containing lipoproteins (VLDL and LDL) in exchange for TG from the apoB-containing lipoproteins (Fig. 1) [21, 75]. This exchange results in apoB-containing lipoproteins which are enriched with CEs and depleted of TGs, and HDL particles which are depleted of CEs and enriched with TGs. The TG-rich and CE-poor HDL particles are catabolized faster than large, CE-rich HDL (apoA-I FCR is increased as noted in Fig. 1), a finding resulting in lower levels of HDL-C in the setting of high TG levels [76]. The apoB-containing lipoproteins, now enriched in CE, can also be taken up by the liver receptors as previously described [75]. When TG levels are high, the apoB particles are TG-enriched and hepatic lipase then hydrolyzes the TGs within the TG-rich LDL to release FFAs, a process which remodels the LDL particles into smaller and denser LDL particles which can enter the arterial intima more easily than larger LDL particles, thus making them more atherogenic (Fig. 1). Small, dense LDL particles also bind less avidly to the LDL receptor, thus prolonging their half-life in the circulation and making these particles more susceptible to oxidative modification and to subsequent uptake by the macrophage scavenger receptors [77].
The exchange via CETP action is thought responsible for the inverse relationship between levels of TG and HDL-C. Specifically, the larger the VLDL pool (higher TG level), the greater the CETP-mediated transfer of CE from HDL to VLDL in exchange for TG, resulting in TG-rich small, dense HDL which are catabolized more rapidly, leading to low levels of HDL-C [78, 79]. These small, dense HDL also have reduced antioxidant and anti-inflammatory properties. Thus, the greater the increase in hepatic VLDL-TG synthesis and secretion that characterizes insulin-resistant/hyperinsulinemic individuals, the lower will be the HDL-C concentration.
HDL and Cholesterol Efflux
Although the risk of developing CHD is inversely correlated with plasma levels of HDL-C [8, 80], recent studies suggest that the amount of cholesterol efflux from cells is a better predictor of CHD than HDL-C concentration [81••]. In statin-treated men with LDL-C <70 mg/dL who had high TG (mean 215 mg/dL) and low levels of HDL-C (mean 31 mg/dL), cholesterol efflux was reduced compared to normolipidemic subjects [82••]. This finding suggests that decreased cholesterol efflux may contribute to the residual risk observed in statin-treated subjects with low HDL-C even when LDL-C < 70 mg/dL. Decreased cholesterol efflux has also been found to be inversely related to carotid IMT and angiographic coronary artery disease independent of HDL-C level [8]. Taken together, the evidence suggests that the functional status of HDL is more important than its level.
Three human infusion studies have been done with apoA-I Milano or reconstituted HDL with the goal of increasing the number of functional HDL particles. Reduction in atheroma volume was 4.2 %, and 3.4 %, respectively, and decreased expression of VCAM-1 (a measure of macrophage activation) occurred in the third [reviewed in [83••]. However, there is no clinical assay yet for the measurement of HDL functionality.
Anti-Inflammatory Effects of HDL on T Cell Function
In addition to participating in RCT, HDL and apoA-I can exert anti-inflammatory effects by modulating T cell activation. “Lipid rafts” are regions of the plasma membrane which contain free cholesterol (FC), sphingomyelin (SM), and gangliosides [84•]. Cholesterol efflux by HDL can reduce the raft-like regions in the membrane, a process which may affect immune cell responses. T cells can participate in the atherosclerotic process [85–88]; the T regulatory cell (Treg-CD4 + CD25 + FoxP3+) is reported to suppress atherosclerosis by inhibiting proinflammatory T cells [89–91]. Similar to macrophages, T and B cells can also take up CE, leading to their dysfunction and the development of an autoimmune phenotype [92, 93]. Higher levels of plasma membrane cholesterol enhance the inflammatory T helper response, thus increasing inflammation mediated via T cells. In contrast, reduction of plasma membrane FC or SM via HDL and RCT has been correlated with the attenuation of inflammatory responses [94, 95, 96••].
HDL carries apoB-bound sphingosine-1-phosphate (S1P), a lipid mediator with anti-inflammatory properties when present at low concentration, thus providing another cardioprotective effect of HDL [reviewed in [97••]. S1P promotes the development of inflammatory T helper 1 cells while suppressing differentiation of Treg cells. Therefore, HDL and apoA-I can suppress inflammation by modifying the lipid-raft environment by promoting FC efflux from tissues and inflammatory cells, such as T cells and macrophages, resulting in an increased fraction of Treg cells that attenuate inflammation.
Additional antiatherogenic properties of HDL include antioxidative effects (protecting apoB lipoproteins from oxidation), antithrombotic and anti-inflammatory properties and maintenance of endothelial function. Due to space limitations, we refer the reader to two excellent very recent reviews summarizing these areas [83••, 98••].
Recommendations for Evaluation and Management
Table 1 outlines a management approach to patients with hypertriglyceridemia based on the TG cut points defined by ATPIII and the AHA statement on TG [2, 99]. Elevated levels of non-HDL-C are predictive of cardiovascular disease and cardiovascular disease mortality, similar to the predictive value of apoB and as good as, or better than, that of LDL-C [6] and reviewed in reference 4]. Non–HDL-C is total cholesterol minus HDL-C, which is the sum of VLDL-C, IDL-C and LDL-C. Therefore, non-HDL-C includes the cholesterol in all of the atherogenic apoB-containing lipoproteins: TG-enriched lipoproteins, CMs, CM remnants, VLDL and VLDL remnants, IDL, LDL and lipoprotein (a). Non-HDL-C is accurate and reliable in a nonfasting state; therefore, measurement of non-HDL is practical and easy [99]. When lipid levels are normal, non-HDL-C is highly correlated with apoB levels [100]. Because high LDL-C and TG confer greater risk for CHD than high LDL-C alone, NCEP ATP III guidelines and the European Society of Cardiology (ESC) and European Atherosclerosis Society (EAS) guidelines recommend the use of non–HDL-C as a secondary target of therapy when the serum TG level is ≥ 2.26 mmol/L (200 mg/dl) after achieving LDL-C target with non-HDL-C goal 0.8 mmol/L (30 mg) higher than LDL-C goal [99, 101••].
The prevalence of small, dense LDL particles (pattern B) increases substantially as fasting plasma TG levels rise above 1.7 mmol/L (150 mg/dL) [102]; therefore, there is no need to measure LDL particle size. Measurement of waist circumference and elevation of TGs will identify patients at high risk [101••].
Lifestyle recommendations remain first line therapy (Table 1). By improving insulin sensitivity, weight loss decreases TG levels. TG levels can be lowered 20 to 30 % with weight reduction [103]. Exercise is also very important to lower TG levels. As noted earlier, LPL hydrolyzes VLDL, thus lowering TG levels. Physical activity increases LPL activity whereas inactivity leads to loss of skeletal muscle LPL, thus, shifting from fatty acid to glucose oxidation and leading to a redistribution of TG to heart and liver, thereby, increasing TG in these tissues and leading to insulin resistance. Factors that reduce LPL activity produce dramatic hypertriglyceridemia. Therefore, exercise is extremely important in upregulating LPL and lowering TG levels.
Medications
Table 2 summarizes the major medications which lower elevated TG concentrations: statins, fibrates, niacin, omega 3-fatty acids and PCSK9 inhibitors. Several studies have shown that monotherapy with various fibrates reduces risk of CHD events in patients with high TGs and low HDL-C, especially in patients with diabetes mellitus or characteristics of the metabolic syndrome (Table 3) [104–107]. In a post-hoc subgroup analysis in the Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) trial, those with metabolic syndrome and marked dyslipidemia (TG ≥ 2.3 mmol/l [204 mg/dL] and low HDL-C) had the highest risk (17.8 %) of developing CVD over 5 years (106). Fenofibrate had the greatest benefit in this group in whom a 27 % relative risk reduction in total CVD events occurred (HR 0.73; 95 % CI: 0.58-0.91, p = 0.005, number needed to treat = 23) compared to 6 % in all others (HR 0.94; 95 % CI: 0.83-1.06, p = 0.321, number needed to treat =143) [105]. However, clinical trials have shown no additional benefit when either fenofibrate or niacin were added to statin drugs in lowering risk for CHD (Table 3) [108, 109]. In a subset analysis of the Action to Control Cardiovascular Risk in Diabetes (ACCORD), those with TG ≥ 2.3 mmol/L (204 mg/dL) (TG was lowered an average of 35 %) and HDL ≤ 0.88 mmol/L (34 mg/dL) (HDL-C increased an average of 12.9 %) randomized to fenofibrate had a 28 % reduction in relative risk (12.4 % event rate compared to 17.3 % in placebo [p = 0.057]) compared to rates of 10.1 % in both study groups for all other subjects (Table 3) [108]. Therefore, the addition of fenofibrate to a statin may provide additional benefit in type 2 diabetics only in those with high TG and low HDL-C.
Two clinical trials testing whether niacin in combination with a statin provides additional benefit to a statin alone have been stopped early. The Atherothrombosis Intervention in Metabolic Syndrome with Low HDL/High Triglycerides: Impact on Global Health Outcomes (AIM-HIGH) trial was stopped after 3 years due to no significant difference in the primary outcome (Table 3) but a significant increase in stroke in those on niacin [109]. In a post-hoc subset analysis of AIM HIGH, patients with a baseline HDL < 0.88 mmol/L (34 mg/dL) and TG > 2.3 mmol/L (204 mg/dL) had a significant reduction in cardiovascular events on niacin (unpublished results). The Heart Protection Study 2-Treatment of HDL to Reduce the Incidence of Vascular Events (HPS-2 THRIVE) study was a secondary-prevention trial testing whether the addition of extended-release niacin to a statin provided additional benefit to a statin alone. It was stopped early due to a significant increased risk of nonfatal but serious side effects thought related to the extended-release niacin preparation which included laropiprant, a DP1 antagonist which blocks PGD2 mediated vasodilation to prevent flushing (marketed as Tredaptive) [110]. Therefore, Tredaptive has been withdrawn from the market, and the role of niacin in lipid-lowering therapy is unclear.
Conclusion
Hypertriglyceridemia is associated with other lipid abnormalities that predispose to atherosclerosis including low levels of HDL-C, the presence of small, dense LDL particles and atherogenic TG-rich lipoprotein remnants and insulin resistance. High levels of TG lead to TG-rich lipoproteins enriched with apoC-III which affect signaling pathways which activate NFKβ and increase inflammatory molecules leading to development of the fatty streak and advanced atherosclerosis. Moreover, the cholesterol content in TG-rich lipoproteins has been shown to predict CAD risk better than LDL-C. The “functionality” of HDL appears more important than level in affecting atherosclerosis and immunity. Insulin resistance related to central obesity appears to underlie the pathophysiology of elevated TGs and low HDL-C. Lifestyle recommendations including exercise and weight loss remain first line therapy. Clearly, preventing obesity and insulin resistance is most important in terms of preventing the adverse sequelae from high TG and low HDL-C.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Barter P, Gotto AM, LaRosa JC, et al. Treating to New Targets Investigators. HDL cholesterol, very low levels of LDL cholesterol and cardiovascular events. N Engl J Med. 2007;357:1301–10.
Alberti KG, Eckel RH, Grundy SM, et al. Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation. 2009;120:1640–5.
Ford ES, Giles WH, Dietz WH. Prevalence of the metabolic syndrome among US adults: findings from the Third National Health and Nutrition Examination Survey. JAMA. 2002;287:356–9.
Welty FK. The Contribution of Triglycerides and Triglyceride-Rich Lipoproteins to Atherosclerotic Cardiovascular Disease. In: Blumenthal RG, editor. Preventive Cardiology, a Companion to Braunwald’s Heart Disease. Philadelphia: Elsevier Saunders; 2011. p. 230–51.
Sarwar N, Danesh J, Eiriksdottir G, et al. Triglycerides and the risk of coronary heart disease: 10,158 incident cases among 262,525 participants in 29 Western prospective studies. Circulation. 2007;115:450–8.
Emerging Risk Factors Collaboration. Major lipids, apolipoproteins, and risk of vascular disease. JAMA. 2009;302:1993–2000.
Brunner D, Altman S, Loebl K, Schwartz S, Levin S. Serum cholesterol and triglycerides in patients suffering from ischemic heart disease and in healthy subjects. Atherosclerosis. 1977;28:197–204.
Wilhelmsen L, Bengtsson C, Elmfeldt D, et al. Multiple risk prediction of myocardial infarction in women as compared with men. Br Heart J. 1977;39:1179–85.
Scott DW, Gotto AM, Cole JS, Gorry GA. Plasma lipids as collateral risk factors in coronary artery disease: a study of 371 males with chest pain. J Chronic Dis. 1978;31:337–45.
Gotto AM, Gorry GA, Thompson JR, et al. Relationship between plasma lipid concentrations and coronary artery disease in 496 patients. Circulation. 1977;56:875–83.
Anderson AJ, Barboriak JJ, Rimm AA. Risk factors and angiographically determined coronary occlusion. Am J Epidemiol. 1978;107:8–14.
Cabin HS, Roberts WC. Relation of serum total cholesterol and triglyceride levels to the amount and extent of coronary arterial narrowing by atherosclerotic plaque in coronary heart disease: quantitative analysis of 2,037 five mm segments of 160 major epicardial coronary arteries in 40 necropsy patients. Am J Med. 1982;73:227–34.
Reardon MF, Nestel PJ, Craig IH, Harper RW. Lipoprotein predictors of the severity of coronary artery disease in men and women. Circulation. 1985;71:881–8.
Freedman DS, Gruchow HW, Anderson AJ, Rimm AA, Barboriak JJ. Relation of triglyceride levels to coronary artery disease: the Milwaukee Cardiovascular Data Registry. Am J Epidemiol. 1988;127:1118–30.
Castelli WP, Doyle JT, Gordon T, et al. HDL cholesterol and other lipids in coronary heart disease: the Cooperative Lipoprotein Phenotyping Study. Circulation. 1977;55:767–72.
Fager G, Wiklund O, Olofsson SO, Wilhelmsen L, Bondjers G. Multivariate analyses of serum apolipoproteins and risk factors in relation to acute myocardial infarction. Arteriosclerosis. 1981;1:273–9.
Kukita H, Imamura Y, Hamada M, Joh T, Kokubu T. Plasma lipids and lipoproteins in Japanese male patients with coronary artery disease and in their relatives. Atherosclerosis. 1982;42:21–9.
Hamsten A, Walldius G, Dahlén G, Johansson B, De Faire U. Serum lipoproteins and apolipoproteins in young male survivors of myocardial infarction. Atherosclerosis. 1986;59:223–35.
•• Miller M, Stone NJ, Ballantyne C, on behalf of the American Heart Association Clinical Lipidology, Thrombosis, and Prevention Committee of the Council on Nutrition, Physical Activity and Metabolism, Council on Arteriosclerosis, Thrombosis and Vascular Biology, Council on Cardiovascular Nursing, and Council on the Kidney in Cardiovascular Disease, et al. Triglycerides and cardiovascular disease: a scientific statement from the American Heart Association. Circulation. 2011;123:2292–333. This AHA statement summarizes evidence supporting the role of triglycerides in cardiovascular risk and provides recommendations for management.
Ginsberg HN. New perspectives on atherogenesis: role of abnormal triglyceride-rich lipoprotein metabolism. Circulation. 2002;106:2137–42.
Havel RJ, Kane JP. Introduction: structure and metabolism of plasma lipoproteins. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. 7th ed. New York: McGraw-Hill; 1995. p. 1841–51.
Young SG. Recent progress in understanding apolipoprotein B. Circulation. 1990;82(5):1574–94.
Chen SH, Habib G, Yang CY, et al. Apolipoprotein B-48 is the product of a messenger RNA with an organ-specific in-frame stop codon. Science. 1987;238(4825):363–6.
Berriot-Varoqueaux N, Aggerbeck LP, Samson-Bouma M, et al. The role of the microsomal triglyceride transfer protein in abetalipoproteinemia. Annu Rev Nutr. 2000;20:663–97.
Weisgraber KH, Rall Jr SC. Human apolipoprotein B-100 heparin-binding sites. J Biol Chem. 1987;262(23):11097–103.
Sehayek E, Lewin-Velvert U, Chajek-Shaul T, Eisenberg S. Lipolysis exposes unreactive endogenous apolipoprotein E-3 in human and rat plasma very low density lipoprotein. J Clin Invest. 1991;88(2):553–60.
Beigneux AP, Davies BS, Gin P, Weinstein MM, et al. Glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein 1 plays a critical role in the lipolytic processing of chylomicrons. Cell Metab. 2007;5(4):279–91.
Kowal RC, Herz J, Goldstein L, et al. Low density lipoprotein receptor-related protein mediates uptake of cholesteryl esters derived from apoprotein E-enriched lipoproteins. Proc Natl Acad Sci USA. 1989;86(15):5810–4.
Mahley RW, Hussain M. Chylomicron and chylomicron remnant catabolism. Curr Opin Lipidol. 1991;2:170–6.
Welty FK, Lichtenstein AH, Barrett PHR. Human apolipoprotein (Apo) B-48 and ApoB-100 kinetics with stable isotopes. Arterioscler Thromb Vasc Biol. 1999;19(12):2966–74.
Ooi EM, Barrett PH, Chan DC, Watts GF. Apolipoprotein C-III: understanding an emerging cardiovascular risk factor. Clin Sci (Lond). 2008;114:611–24.
Zheng C, Khoo C, Furtado J, Sacks FM. Apolipoprotein C-III and the metabolic basis for hypertriglyceridemia and the dense low-density lipoprotein phenotype. Circulation. 2010;121:1722–34.
Rapp RJ. Hypertriglyceridemia: a review beyond low-density lipoprotein. Cardiol Rev. 2002;10:163–72.
Pollin TI, Damcott CM, Shen H, et al. A null mutation in human APOC3 confers a favorable plasma lipid profile and apparent cardioprotection. Science. 2008;322:1702–5.
Kawakami A, Osaka M, Aikawa M, et al. Toll-like receptor 2 mediates apolipoprotein CIII-induced monocyte activation. Circ Res. 2008;103:1402–9.
Kawakami A, Aikawa M, Libby P, et al. Apolipoprotein CIII in apolipoprotein B lipoproteins enhances the adhesion of human monocytic cells to endothelial cells. Circulation. 2006;113:691–700.
Kawakami A, Aikawa M, Alcaide P, et al. Apolipoprotein CIII induces expression of vascular cell adhesion molecule-1 in vascular endothelial cells and increases adhesion of monocytic cells. Circulation. 2006;114:681–7.
Lee SJ, Campos H, Moye LA, Sacks FM. LDL containing apolipoprotein CIII is an independent risk factor for coronary events in diabetic patients. Arterioscler Thromb Vasc Biol. 2003;23:853–8.
•• Petersen KF, Dufour S, Hariri A, et al. Apolipoprotein C3 gene variants in nonalcoholic fatty liver disease. N Engl J Med. 2010;362:1082–9. Important research providing insight into role of ApoCIII gene variants in pathophysiology of nonalcoholic fatty liver disease.
Tsai MY, Ordovas JM. APOC3 mutation, serum triglyceride concentrations, and coronary heart disease. Clin Chem. 2009;55:1274–6.
Guilherme A, Virbasius JV, Puri V, Czech MP. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat Rev Mol Cell Biol. 2008;9:367–77.
• Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol. 2011;11:98–107. Excellent review of inflammatory mechanisms and signaling underlying Type 2 diabetes.
Pavlic M, Valero R, Duez H, et al. Triglyceride-rich lipoprotein-associated apolipoprotein C-III production is stimulated by plasma free fatty acids in humans. Arterioscler Thromb Vasc Biol. 2008;28:1660–5.
Fabbrini E, Mohammed BS, Magkos F, et al. Alterations in adipose tissue and hepatic lipid kinetics in obese men and women with nonalcoholic fatty liver disease. Gastroenterology. 2008;134:424–31.
Sparks JD, Sparks CE. Insulin modulation of hepatic synthesis and secretion of apolipoprotein B by rat hepatocytes. J Biol Chem. 1990;265:8854–62.
Ginsberg HN, Fisher EA. The ever-expanding role of degradation in the regulation of apolipoprotein B metabolism. J Lipid Res. 2009;50(suppl):S162–6.
Sato R, Miyamoto W, Inoue J, et al. Sterol regulatory element-binding protein negatively regulates microsomal triglyceride transfer protein gene transcription. J Biol Chem. 1999;274:24714–20.
•• Eckel RH. The Complex Metabolic Mechanisms Relating Obesity to Hypertriglyceridemia. Arterioscler Thromb Vasc Biol. 2011;31:1946–8. Comprehensive review summarizing relation between obesity, insulin resistance and hypertriglyceridemia.
•• Goldberg IJ, Eckel RH, McPherson R. Triglycerides and heart disease: Still a hypothesis? Arterioscler Thromb Vasc Biol. 2011;31:1716–25. Outstanding review of triglyceride metabolism and relation to cardiovascular disease with update on genetics and clinical trials of triglyceride-lowering therapies.
• Rask-Madsen C, Kahn CR. Tissue-specific insulin signaling, metabolic syndrome and cardiovascular disease. Arterioscler Thromb Vasc Biol. 2012;32:2052–9. Review summarizing known mechanisms of insulin signaling on various organ systems.
Hurt-Camejo E, Olsson U, Wiklund O, et al. Cellular consequences of the association of apoB lipoproteins with proteoglycans. Potential contribution to atherogenesis. Arterioscler Thromb Vasc Biol. 1997;17:1011.
Napoli C, D’Armiento FP, Mancini FP. Fatty streak formation occurs in human fetal aortas and is greatly enhanced by maternal hypercholesterolemia. Intimal accumulation of low density lipoprotein and its oxidation precede monocyte recruitment into early atherosclerotic lesions. J Clin Invest. 1997;100:2680–90.
• Fang L, Green SR, Baek JS, et al. In vivo visualization and attenuation of oxidized lipid accumulation in hypercholesterolemic zebrafish. J Clin Invest. 2011;121:4861–9. Very interesting research showing that cholesterol feeding of zebrafish larvae leads to oxidized lipid accumulation, a finding suggesting that diet-induced inflammation is wide-spread in nature.
Podrez EA, Febbraio M, Sheibani N, et al. Macrophage scavenger receptor CD36 is the major receptor for LDL modified by monocyte-generated reactive nitrogen species. J Clin Invest. 2000;105:1095.
Zilversmith DB. A proposal linking atherogenesis to the interaction of endothelial lipoprotein lipase with triglyceride-rich lipoproteins. Circ Res. 1973;33:633–8.
Mamo JC, Proctor SD, Smith D. Retention of chylomicron remnants by arterial tissue; importance of an efficient clearance mechanism from plasma. Atherosclerosis. 1998;141 suppl 1:S63–9.
Varbo A, Benn M, Tybjaerg-Hansen A, et al. Remnant cholesterol as a causal risk factor for ischemic heart disease. J Am Coll Cardiol. 2013;61:427–36.
Kugiyama K, Doi H, Motoyama T, et al. Association of remnant lipoprotein levels with impairment of endothelium-dependent vasomotor function in human coronary arteries. Circulation. 1998;97:2519.
Krauss RM, Lindgren FT, Williams PT, et al. Intermediate-density lipoproteins and progression of coronary artery disease in hypercholesterolaemic men. Lancet. 1987;2:62.
Kugiyama K, Dori H, Takazoe K, et al. Remnant lipoprotein levels in fasting serum predict coronary events in patients with coronary artery disease. Circulation. 1999;99:2858.
Fukushima H, Kugiyama K, Sugiyama S, et al. Comparison of remnant-like lipoprotein particles in postmenopausal women with and without coronary artery disease and in men with coronary artery disease. Am J Cardiol. 2001;88:1370.
Masuoka H, Kamei S, Wagayama H, et al. Association of remnant-like particle cholesterol with coronary artery disease in patients with normal total cholesterol levels. Am Heart J. 2000;139:305.
Williams KJ, Petrie KA, Broacia RW, Swenson TL. Lipoprotein lipase modulates net secretory output of apolipoprotein B in vitro. J Clin Invest. 1991;88:1300.
Hodis HN, Mack WJ, Dunn M, et al. Intermediate-density lipoproteins and progression of carotid arterial wall intima-media thickness. Circulation. 2022;1997:95.
Hodis HN, Mack WJ. Triglyceride-rich lipoproteins and the progression of coronary artery disease. Curr Opin Lipidol. 1995;6:209.
Mack WJ, Krauss RM, Hodis HN. Lipoprotein subclasses in the Monitored Atherosclerosis Regression Study (MARS). Treatment effects and relation to coronary angiographic progression. Arterioscler Thromb Vasc Biol. 1996;16:697.
Tsuda T, Yoshimura H, Hamasaki N. Effect of phosphatidylcholine, phosphatidylethanolamine and lysophosphatidylcholine on the activated factor X-prothrombin system. Blood Coagul Fibrinolysis. 2006;17:465–9.
Eriksson P, Nilsson L, Karpe F, Hamsten A. Very-low-density lipoprotein response element in the promoter region of the human plasminogen activator inhibitor-1 gene implicated in the impaired fibrinolysis of hypertriglyceridemia. Arterioscler Thromb Vasc Biol. 1998;18:20–6.
Han S, Liang CP, DeVries-Seimon T, et al. Macrophage insulin receptor defi-ciency increases ER stress-induced apoptosis and necrotic core formation in advanced atherosclerotic lesions. Cell Metab. 2006;3:257–66.
•• Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011;145:341–55. This is an excellent review of mechanisms by which macrophages lead to acute coronary sydromes.
Nishi K, Itabe H, Uno M, et al. Oxidized LDL in carotid plaques and plasma associates with plaque instability. Arterioscler Thromb Vasc Biol. 2002;22:1649–54.
Yancey PG, Bortnick AE, Kellner-Weibel G, et al. Importance of different pathways of cellular cholesterol efflux. Arterioscler Thromb Vasc Biol. 2003;23:712–9.
• Francis GA. The complexity of HDL. Biochim Biophys Acta. 2010;1801:1286–93. Comprehensive overview of HDL composition and function.
Acton S, Rigotti A, Landschulz KT, et al. Identification of scavenger receptor SR-BI as a high-density lipoprotein receptor. Science. 1996;271:518–20.
Steinberg D. A docking receptor for HDL cholesterol esters. Science. 1996;271:460–1.
Lamarche B, Uffelman KD, Carpentier A, et al. Triglyceride enrichment of HDL enhances in vivo metabolic clearance of HDL apo A-I in healthy men. J Clin Invest. 1999;103(8):1191–9.
Nigon F, Lesnik P, Rouis M, et al. Discrete subspecies of human low density lipoprotein are heterogeneous in their interaction with cellular LDL receptor. J Lipid Res. 1991;32:1741.
Swenson TL. The role of the cholesteryl ester transfer protein in lipoprotein metabolism. Diabetes Metab Rev. 1991;7:139–53.
Brinton EA, Eisenberg S, Breslow JL. Human HDL cholesterol levels are determined by apoA-I fractional catabolic rate, which correlates inversely with estimates of HDL particle size. Effects of gender, hepatic and lipoprotein lipases, triglyceride and insulin. Arterioscler Thromb. 1994;14(5):707–20.
Duffy D, Rader DJ. Update on strategies to increase HDL quantity and function. Nat Rev Cardiol. 2009;6:455–63.
•• Khera AV, Cuchel M, de la Llera-Moya M, et al. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N Engl J Med. 2011;364:127–35. First report showing that role of HDL in reverse cholesterol transport is more important predictor of CAD than level of HDL-C; thus, suggesting that functionality of HDL may be more important than HDL-C level.
•• Posadas-Sanchez R, Posadas-Romero C, Mendoza-Perez E, et al. Cholestrol efflux and metabolic abnormalities associated with low high-density-lipoprotein-cholesterol and high triglycerides in statin-treated coronary men with low-density lipoprotein-cholestrol < 70 mg/dL. Am J Cardiol. 2012;109:636–41. First report to show that decreased cholesterol efflux may account for residual risk observed in statin-treated patients with high triglycerde level and low HDL-C level.
•• Fisher EA, Feig JE, Hewing B, et al. High-density lipoprotein function, dysfunction, and reverse cholesterol transport. Arterioscler Thromb Vasc Biol. 2012;32:2813–20. Outstanding review of beneficial, cardioprotective effects of HDL including animal models, in vitro and clinical trials.
• Owen DM, Magenau A, Williamson D, Gaus K. The lipid raft hypothesis revisited - New insights on raft composition and function from super-resolution fluorescence microscopy. Bioessays. 2012;34:739–47. Provides summary of lipid raft composition and function.
Haraba R, Antohe F. T cells are active participants in the progression of atherosclerotic plaques. Dig J Nanomater Bios. 2011;6:1529–34.
Taleb S, Tedgui A, Mallat Z. Adaptive T cell immune responses and atherogenesis. Curr Opin Pharmacol. 2010;10:197–202.
Lichtman AH. T cell costimulatory and coinhibitory pathways in vascular inflammatory diseases. Front Physiol. 2012;3:18.
Lahoute C, Herbin O, Mallat Z, Tedgui A. Adaptive immunity in atherosclerosis: mechanisms and future therapeutic targets. Nat Rev Cardiol. 2011;8:348–58.
Foks AC, Frodermann V, ter Borg M, et al. Differential effects of regulatory T cells on the initiation and regression of atherosclerosis. Atherosclerosis. 2011;218:53–60.
Ait-Oufella H, Salomon BL, Potteaux S, et al. Natural regulatory T cells control the development of atherosclerosis in mice. Nat Med. 2006;12:178–80.
Maganto-García E, Tarrio ML, Grabie N, Bu DX, Lichtman AH. Dynamic changes in regulatory T cells are linked to levels of diet-induced hypercholesterolemia. Circulation. 2011;124:185–95.
Wilhelm AJ, Zabalawi M, Owen JS, et al. Apolipoprotein A-I modulates regulatory T cells in autoimmune LDLr−/−, ApoA-I−/− mice. J Biol Chem. 2010;285:36158–69.
Wilhelm AJ, Zabalawi M, Grayson JM, et al. Apolipoprotein A-I and its role in lymphocyte cholesterol homeostasis and autoimmunity. Arterioscler Thromb Vasc Biol. 2009;29:843–9.
Dong L, Watanabe K, Itoh M, et al. CD4+ T-cell dysfunctions through the impaired lipid rafts ameliorate concanavalin A-induced hepatitis in sphingomyelin synthase 1-knockout mice. Int Immunol. 2012;24:327–37.
Yin K, Chen WJ, Zhou ZG, et al. Apolipoprotein A-I inhibits CD40 proinflammatory signaling via ATP-binding cassette transporter A1-mediated modulation of lipid raft in macrophages. J Atheroscler Thromb. 2012;19:823–36.
•• Cheng AM, Handa P, Tateya S, et al. Apolipoprotein A-I attenuates palmitate-mediated NF-KB activation by reducing Toll-like receptor-4 recruitment into lipid rafts. PLoS One. 2012;7:e33917. Excellent research showing how ApoA-I reduces inflammation via TLR4 receptor induced by the fatty acid palmitate.
•• Sorci-Thomas MG, Thomas MJ. High density lipoprotein biogenesis, cholesterol efflux and immune cell function. Arterioscler Thromb Vasc Biol. 2012;32:2561–5. Excellent review of role of HDL in cholesterol efflux and regulation of T cell function to reduce atherosclerosis.
•• Navab M, Reddy ST, Van Lenten BJ, et al. High-density lipoprotein and 4F peptide reduce systemic inflammation by modulating intestinal oxidized lipid metabolism: Novel hypotheses and review of literature. Arterioscler Thromb Vasc Biol. 2012;32:2553–60. Informative review of cardioprotective and anti-inflammatory effects of HDL with emphasis on antioxidative properties.
Expert Panel on Detection, Evaluation and Treatment of High Blood Cholesterol in Adults. Third report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). Circulation. 2002;106:3143–421.
Abate N, Vega GL, Grundy SM. Variability in cholesterol content and physical properties of lipoproteins containing apolipoprotein B-100. Atherosclerosis. 1993;104:159–71.
•• Reiner Z, Catapan AL, De Backer G, et al. ESC/EAS Guidelines for the management of dyslipidaemias. Eur Heart J. 2011;32:1769–818. Outstanding summary of the clinical trial evidence supporting scoring for risk factor assessment and recommendations for evaluation and management of dyslipidaemias.
Austin MA, King MC, Vranizan KM, Krauss RM. Atherogenic lipoprotein phenotype. A proposed genetic marker for coronary heart disease risk. Circulation. 1990;82:495–506.
Shaw K, Gennat H, O’Rourke P, Del Mar C. Exercise for overweight or obesity. Cochrane Database Syst Rev. 2006, 4CD003817.
Frick MH, Elo O, Haapa K, et al. Helsinki Heart Study: primary-prevention trial with gemfibrozil in middle-aged men with dyslipidemia: safety of treatment, changes in risk factors, and incidence of coronary heart disease. N Engl J Med. 1987;317(20):1237–45.
Scott R, O’Brien R, Fulcher G, on behalf of the Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) Study Investigators, et al. Effects of fenofibrate treatment on cardiovascular disease risk in 9,795 individuals with type 2 diabetes and various components of the metabolic syndrome. Diabetes Care. 2009;32:493–8.
Rubins HB, Robins SJ, Collins D, Veterans Affairs High-Density Lipoprotein Cholesterol Intervention Trial Study Group, et al. Gemfibrozil for the secondary prevention of coronary heart disease in men with low levels of high-density lipoprotein cholesterol. N Engl J Med. 1999;341(6):410–8.
The BIP Study Group. Secondary prevention by raising HDL cholesterol and reducing triglycerides in patients with coronary heart disease: the Bezafibrate Infarction Prevention (BIP) study. Circulation 2000, 102: 21– 27.
ACCORD Study Group, Ginsberg HN, Elam MB, Lovato LC, et al. Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med. 2010;362:1563–74.
AIM-HIGH Investigators, Boden WE, Probstfield JL, Anderson T, et al. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365:2255–67.
http://www.theheart.org/article accessed 2/3/2013.
Koren MJ, Scott R, Kim JB, et al. Efficacy, safety and tolerability of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 as monotherapy in patients with hypercholesterolaemia (MENDEL): a randomized, double-blind, placebo-controlled, phase 2 study. Lancet. 2012;380:1995–2006.
Compliance with Ethics Guideline
Conflict of Interest
Francine K. Welty declares that she has no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Lipid Abnormalities and Cardiovascular Prevention
Rights and permissions
About this article
Cite this article
Welty, F.K. How Do Elevated Triglycerides and Low HDL-Cholesterol Affect Inflammation and Atherothrombosis?. Curr Cardiol Rep 15, 400 (2013). https://doi.org/10.1007/s11886-013-0400-4
Published:
DOI: https://doi.org/10.1007/s11886-013-0400-4