
Abstract
Purpose of Review
Recent studies indicate an association between hypertriglyceridemia (HTG) and atherosclerotic cardiovascular disease (ASCVD). The purpose of this review is to discuss the potential mechanism connecting HTG and ASCVD risk and the potential efficacy of HTG-targeting therapies in ASCVD prevention.
Recent Findings
HTG, with elevations in triglyceride-rich lipoproteins (TGRL) and their remnants, are causal ASCVD risk factors. The mechanisms whereby HTG increases ASCVD risk are not well understood but may include multiple factors. Inflammation plays a crucial role in atherosclerosis. TGRL compared to low-density lipoproteins (LDL) correlate better with inflammation. TGRL remnants can penetrate endothelium and interact with macrophages leading to foam cell formation and inflammation in arterial walls, thereby contributing to atherogenesis. In addition, circulating monocytes can take up TGRL and become lipid-laden foamy monocytes, which infiltrate the arterial wall and may also contribute to atherogenesis. Novel therapies targeting HTG or inflammation are in development and have potential of reducing residual ASCVD risk associated with HTG.
Summary
Clinical and preclinical studies show a causal role of HTG in promoting ASCVD, in which inflammation plays a vital role. Novel therapies targeting HTG or inflammation have potential of reducing residual ASCVD risk.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Atherosclerotic cardiovascular disease (ASCVD), including coronary heart disease and stroke, remains the leading cause of morbidity and mortality in the United States and worldwide [1]. Hypercholesterolemia (HCH) has long been recognized as a key risk factor and a therapeutic target for ASCVD [1, 2]. However, there is residual risk of ASCVD even after low-density lipoprotein cholesterol (LDL-C) levels are reduced to normal ranges with a stain therapy or other cholesterol-lowering therapies [3]. From genetic and epidemiologic studies, hypertriglyceridemia (HTG) with elevated triglyceride (TG)-rich lipoproteins (TGRL) is also recognized as a causal ASCVD risk factor [4, 5, 6, 7]. However, the mechanisms by which HTG/TGRL increase the risk of ASCVD remain poorly understood. This review will discuss how inflammation may serve as a mechanistic link between HTG/TGRL and ASCVD and reveal the potential of novel therapies targeting HTG or inflammation for ASCVD prevention.
HTG, TGRL, and RLP
HTG is defined as fasting triglyceride levels ≥ 150 mg/dL and mainly characterized by elevations in TGRL, which include very-low-density lipoproteins (VLDL), chylomicrons (CM), and their remnants. CM are synthesized in intestinal cells and contain apolipoprotein (APO) B48, while VLDL are synthesized in the liver and mainly contain APOB100 (in humans, and also APOB48 in mice) [8•, 9]. After release into the circulation, CM and VLDL are hydrolyzed by lipoprotein lipase (LPL) on the surface of endothelial cells (ECs), resulting in the production of remnant lipoprotein particles (RLP) and free fatty acids (FFAs) [9]. FFAs are transferred into peripheral sites such as adipose tissue for storage as TG or skeletal muscle as an energy source. As hydrolysis proceeds, TG is depleted constantly from RLP. While CM remnants are mainly cleared in the liver, VLDL, and their remnants, intermediate density lipoproteins (IDL), gradually change to cholesteryl esters (CE)-rich lipoproteins, i.e., LDL [8•].
HTG and ASCVD in Population Studies
A large number of clinical studies have shown an association of HTG with ASCVD risk [10, 11, 12, 13, 14]. Because HTG is usually accompanied by reduced high-density lipoprotein cholesterol (HDL-C) and elevated LDL-C, both of which are associated with increased ASCVD risk, it is difficult to ascertain a direct role of HTG in ASCVD risk. For this reason, the causal relationship between HTG and ASCVD risk has been debated for decades. However, recent observational and interventional studies support the hypothesis that HTG is a causal ASCVD risk factor. In statin-treated patients with well-controlled LDL-C levels, those with elevated TG levels (≥ 150 mg/dL) still have higher ASCVD risk than those with well-managed TG (< 150 mg/dL) [15]. Several clinical studies showed that HTG remains associated with ASCVD risk after adjusting for other lipid-associated factors [7, 16, 17]. Genetic studies show that TG levels can contribute to a 50% increase in ASCVD risk [18]. Large meta-analysis or clinical studies show that both fasting and non-fasting serum TG levels can serve as independent predictors of ASCVD risk [14, 19, 20]. However, another earlier meta-analysis including 30,2430 subjects showed no significant association between HTG and ASCVD risk after adjustment for multiple risk factors [21].
While most studies have focused on increased TG in TGRL and their remnants, HTG is also commonly associated with increased TG content in other lipoproteins such as LDL. A prospective study in Atherosclerosis Risk in Communities (ARIC) Study showed a strong association between LDL-TG and ASCVD events after a follow-up of up to 16 years in individuals with no prevalent ASCVD [22••]. An earlier study showed that the association of LDL-TG with ASCVD was stronger than that of LDL-C and that the predictive value of LDL-TG for ASCVD was independent of LDL-C [23]. Therefore, in addition to total TG and TG in TGRL, LDL-TG may also be a risk factor for ASCVD.
Potential Mechanisms for Atherogenesis in HTG
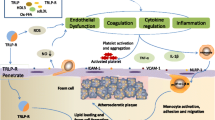
In addition to the clinical evidence for a causal role of HTG in ASCVD, HTG in LDL receptor (Ldlr)-deficient mice due to transgenic expression of human Apoc3 increases atherosclerosis [24, 25]. However, the mechanism by which HTG promotes ASCVD remains poorly understood. In the following, we discuss some potential mechanisms by which HTG accelerates atherosclerosis, with a focus on inflammation (Fig. 1).

The potential inflammatory mechanisms for atherogenesis in hypertriglyceridemia (HTG). Chylomicrons (CM) and very-low-density lipoproteins (VLDL) are secreted into the circulation from intestinal cells (1) and liver (2), respectively, and then hydrolyzed by lipoprotein lipase (LPL) on the surface of endothelial cells (ECs), resulting in the production of free fatty acids (FFAs) and remnant lipoprotein particles (RLP), which include CM remnants derived from CM (3), VLDL remnants derived from VLDL (4a), and intermediate-density lipoproteins (IDL) derived further from VLDL remnants (4b). IDL subsequently change to low-density lipoproteins (LDL) (4c), which can be cleared in the liver (4d). Circulating monocytes can take up TGRL/RLP (5) and possibly FFAs and become foamy monocytes (6), which express high levels of TNFα, IL-1β (7a), and CD11c; adhere firmly to VCAM-1/ICAM-1 expressed on activated ECs; and infiltrate into plaques (7b), where foamy monocytes differentiate into foam macrophages (7c), thereby contributing to atherosclerosis. RLP (8a) and FFAs (8b) can also enter arterial walls directly, be engulfed by lesional macrophages, and increase macrophage lipid accumulation, promoting foam macrophage formation (9) and contributing to atherogenesis. Macrophages (10a) including foam macrophages (10b) secrete cytokines, which, along with those derived from foamy monocytes (10c), can, in turn, further increase inflammation in macrophages (10a, 10b) and other cells in plaques and promote atherosclerosis progression. TGRL, RLP (11a), and FFAs (11b) can interact with ECs and induce EC inflammation and activation, with upregulation of cytokines and adhesion molecules such as ICAM-1 and VCAM-1, which mediate monocyte adhesion and recruitment (7b) into atherosclerotic plaques. RLP and FFAs also impair the balance of nitric oxide (NO) and reactive oxygen species (ROS) in ECs (12), leading to endothelial dysfunction
Role of TGRL and RLP
In recent years, it has been frequently suggested that TGRL and TGRL-derived RLP rather than TG per se are the main mediators for HTG-linked risk for ASCVD [22••, 26]. A cross-sectional study showed a positive association between fasting plasma APOB48 (unique to intestinal CM-derived RLP) and other cardiometabolic risk factors [27]. Elevated RLP-C and type III hyperlipoproteinemia, which has elevated IDL (VLDL remnant), are associated with increased ASCVD risk or incidences of peripheral arterial disease [8•, 22••, 26, 28, 29, 30, 31]. Enhanced ASCVD risk of elevated postprandial HTG is possibly caused by increased accumulation of RLP in the circulation [32]. Depending on the extent of lipolysis, the cholesterol content in RLP (cholesterol molecule numbers per lipoprotein particle) is 2–40 times higher than that in LDL [32], highly supporting a proatherogenic role of RLP. Because of its smaller size vs. TGRL per se, RLP can infiltrate into sub-endothelial space by transcytosis [8•]. In support of this, APOB48- and APOB100-containing lipoprotein particles occur in human aortic intimal lesion [33]. APOE and APOC3, which are enriched in TGRL and RLP, have been implicated in binding of RLP and TGRL to the arterial wall and promote RLP deposition and retention in atherosclerotic plaques [8•, 34]. The entry rates of RLP into the vessel wall are 2.8 times faster than their efflux rate, whereas the ratio of LDL entry to efflux rate is 1.4 [35], also supporting a pivotal role of RLP in atherogenesis. Increasing evidence shows that APOB48- and APOB100-containing RLP play interactive and complementary roles in atherogenesis [8•].
Foam Cell Formation and Inflammation in Arterial Walls
Macrophages, which are mainly derived from local proliferation of resident macrophages and infiltration of circulating monocytes, play key roles in atherogenesis. Once having entered arterial walls, RLP can be engulfed by lesional macrophages thereby increasing macrophage lipid accumulation and foam cell formation, a hallmark of atherosclerosis [3, 36, 37]. TGRL/RLP–macrophage interactions may require APOE on TGRL/RLP and VLDL receptor (VLDLR) on macrophages and can induce macrophages to a classically activated M1-like proinflammatory phenotype [8•, 38]. Indeed, macrophage-associated VLDLR has been implicated in foam cell formation and atherogenesis in mice [39, 40]. Compared to LDL, TGRL and RLP are more proinflammatory. Clinical studies show that HTG and elevated RLP-C are more causally associated than LDL-C with low-grade inflammation [8•]. TGRL and their LPL-mediated lipolytic products, i.e., RLP and FFAs, increase adhesion-molecule and cytokine expression, which contribute to the atherogenesis [3, 36, 41, 42]. Indeed, macrophage LPL is involved in foam cell formation and atherogenesis [43]. Furthermore, in vitro treatment with FFA induces TG accumulation and foam cell formation in macrophages, which are associated with upregulation of CD36 and a proinflammatory phenotype [44]. The scavenger receptor, CD36, binds modified LDL, another type of atherogenic lipoproteins, and many other ligands. Consistently, upregulation of CD36 on FFA-treated macrophages correlates with increased uptake of modified LDL [44]. In addition, TG synthesis within macrophages can drive macrophage inflammation by regulation of prostaglandin E2 (PGE2) [45]. TG accumulation in macrophages also upregulates paraoxonase 2 expression via JNK/c-Jun signaling pathway, resulting in the mitochondrial generation of reactive oxygen species (ROS) and inflammation [46]. Moreover, cholesterol and FFAs activate the NLRP3 inflammasome in macrophages [47, 48]. The NLRP3 inflammasome is crucial for maturation and secretion of IL-1β and IL-18 from inflammatory cells including macrophages and plays an important role in atherogenesis [47].
Foamy Monocyte Formation and Inflammation in the Circulation
In addition to the lesion-macrophages that have been the traditional focus of foam-cell studies, infiltration of circulating monocytes into arterial walls, where monocytes differentiate into macrophages, is a key step in atherogenesis [49, 50, 51, 52•, 53]. Monocytes are heterogeneous. Based on CD14 and CD16, human monocytes are classified into CD14+CD16– classical, CD14+CD16+ intermediate, and CD14dimCD16+ nonclassical monocytes [54, 55]. Based on Ly-6c, mouse monocytes include Ly-6chigh classical and Ly-6clow nonclassical monocytes [56]. Classical monocytes are CCR2high and CD11c–/low, while nonclassical and intermediate monocytes are CX3CR1high and CD11c+/high [49, 50, 55, 56, 57]. In mice, the initial study and many other studies focused on classical monocytes in inflammation, whereas nonclassical monocytes also play a pivotal role in inflammation, particularly initiation of inflammation by rapidly moving to inflamed sites and mediating other cell infiltration [49, 56, 58, 59, 60, 61, 62]. In humans, CD16+ intermediate and nonclassical monocytes, analogous to mouse nonclassical monocytes [54, 63], appear to be more proinflammatory [64, 65, 66].
Besides lesion-macrophages, some circulating monocytes take up lipoproteins and take on a foamy aspect. Foamy monocytes were first reported in the 1960s as “lipophages” in rats fed an atherogenic diet [67, 68]. According to electron microscopy, circulating lipophages pass through the endothelium and were therefore postulated to play a critical role in the formation of atherosclerotic lesions in the rats fed atherogenic diet [67, 68]. However, after the initial observations, a direct role of the circulating lipophages in atherosclerosis was not widely studied. In 2009, we reported that these lipophages, which we termed foamy monocytes, also occurred in the circulation of mice with HCH [49]. Foamy monocytes, which contain numerous lipid droplets, are readily detected by flow cytometry in which their high degree of “granularity” is indicated by elevations in side-scatter pattern [49, 50]. Importantly, foamy monocytes emerge early and consistently in the circulation of mice with HCH including Apoe–/– and Ldlr–/– mice fed a western high-fat diet [49, 50]. Of note, not all circulating monocytes have the same degree of lipid accumulation, with foamy monocytes (containing abundant lipid droplets) reaching up to ~ 50% of total monocytes in mice with HCH [49, 50]. Compared to “nonfoamy” monocytes, which contain less lipid and are mostly CD11c–CD36– and Ly-6chigh, i.e., classical monocytes, in the same mice, foamy monocytes are mostly CD11c+CD36+ and Ly-6clow, indicating nonclassical monocytes [49, 50], which are elevated or tend to be higher than Ly-6chigh classical monocytes in mice with HCH [49, 50, 69, 70, 71, 72] despite the initial reports showing reductions or no changes in nonclassical monocytes in mice with HCH [51, 73]. CD36 plays an important role in foamy monocyte formation in mice with HCH [50, 74]. Foamy monocytes express high levels of TNFα and IL-1β [50], adhere with high affinity to VCAM-1, an adhesion molecule expressed on activated ECs [75, 76, 77], and also preferentially form aggregates with platelets [78]. All these phenotypes support a proatherogenic role for foamy monocytes. Indeed, foamy monocytes infiltrate into atherosclerotic lesions, become CD11c+ foam cells, and contribute to atherogenesis in mice with HCH [50]. In addition, CD11c, which is a β2 integrin highly expressed on foamy monocytes, contributes to foamy monocyte adhesion on VCAM-1 via regulation of VLA-4 affinity for VCAM-1 [75, 76, 77]. Consistent with this, CD11c ablation reduces monocyte adhesion and suppresses atherosclerosis in mice [49, 50]. It is highly relevant that foamy monocytes (or leukocytes) also occur in the circulation of humans with familial HCH [79, 80], exhibit an inflammatory phenotype [80], and likely contribute to the development of premature ASCVD in familial HCH.
In addition to HCH, HTG is also associated with increased lipid accumulation in circulating monocytes. Postprandial HTG in humans following a single high-saturated fat meal induces lipid accumulation within monocytes and foamy monocyte formation in the circulation [76, 81, 82]. HTG in humans with metabolic syndrome (MetS) or type III hyperlipoproteinemia correlates with increased lipid accumulation in circulating monocytes and inflammation in arterial walls [57, 76, 81, 82, 83, 84, 85]. Furthermore, increased lipid accumulation within monocytes in humans with HTG is associated with monocyte phenotypic drift, with elevations in adhesion molecules including CD11c and CD11b and cytokines such as TNFα and IL-1β, particularly in intermediate and/or nonclassical monocytes [57, 76, 83, 86]. Importantly, intermediate and nonclassical monocytes are increased in humans with HTG or MetS and correlate positively with ASCVD or progression of carotid intimal-medial thickness [55, 64, 87, 8889, 90]. Therefore, lipid accumulation and phenotypic changes of circulating monocytes, intermediate and nonclassical monocytes, in particular, play a crucial role in development and progression of ASCVD associated with HTG. Data on the effects of HTG on monocytes and their roles in atherosclerosis in HTG in mouse models are limited. One study showed that mice with HTG induced by an LPL inhibitor, Ly-6clow nonclassical, but not Ly-6chigh classical, monocytes extravasate into various tissues including the heart, liver, and kidney and differentiate into foam macrophages [91]. However, the relevance of this observation to atherogenesis in the context of HTG remains to be determined.
The mechanisms by which HTG and TGRL/RLP interact with circulating monocytes leading to foamy monocyte formation are poorly defined. While LPL-mediated lipolysis has been implicated in HTG/TGRL-induced lipid accumulation and foam cell formation in tissue macrophages, HTG/TGRL sometimes induces lipid accumulation in circulating monocytes independent of LPL [92]. Monocytes express LDL receptor-related protein-1 (LRP-1), which mediates monocyte internalization of TGRL in vitro [76]. Monocytes also express VLDLR [40, 93] so that it is conceivable that VLDLR may also mediate monocyte-TGRL/RLP interactions leading to lipid accumulation in monocytes. A major ligand for LRP-1 and VLDLR is APOE, which is abundant in TGRL and RLP. Of note, HTG increases atherogenesis in Ldlr-deficient, but not in Apoe-deficient, mice, suggesting a possible role of APOE in HTG-induced acceleration of atherogenesis [24, 94] and also likely foamy monocyte formation. Consistently, plasma APOE levels correlate positively with ASCVD mortality [95]. In addition to RLP-C and FFAs, which are proinflammatory and may thus induce inflammation in monocytes, APOC3, abundant in TGRL and RLP, can induce inflammation in monocytes and other cells independent of lipids or lipid metabolism [96, 97].
Therefore, in HTG, monocytes can interact with TGRL/RLP and become foamy in the circulation. Foamy monocytes have an inflammatory phenotype and can infiltrate the arterial wall and also possibly other tissues, where foamy monocytes differentiate into foam macrophages and may therefore play a pivotal role in the development of atherosclerosis and also inflammation in other tissues such as eruptive xanthomas in the skin, which are commonly observed in severe HTG including familial chylomicronemia syndrome.
Endothelial Inflammation and Dysfunction
During the whole pathological course of atherogenesis, endothelial inflammation and dysfunction may begin early and persistently. TGRL and their lipolytic products, RLP and FFAs, can interact with ECs and induce EC inflammation and activation, resulting in upregulation of cytokines and adhesion molecules such as ICAM-1 and VCAM-1 [98], which promote the recruitment of monocytes to inflammatory sites including atherosclerotic lesions, thereby contributing to atherogenesis [3, 36, 41, 42]. Bleda S et al. reported that plasma isolated from patients with elevated TG levels or elevated VLDL-cholesterol promotes activation of the NLRP1 inflammasome in ECs, which may also contribute to arterial inflammation [99]. In addition, increased TGRL or RLP including postprandial elevations in TGRL and RLP compared to LDL-C are more closely associated with oxidative stress and endothelial dysfunctions [3, 100, 101], which can impair vasorelaxation and vasoconstriction, decrease flow-mediated dilatation of brachial artery, increase intima-media thickness of artery, and trigger superficial erosion [3, 36, 100, 101] that can cause arterial thrombosis blocking blood flow and resulting in myocardial infarction and stroke [102]. In support of this model, one clinical study revealed a stronger correlation of the oxidative stress markers with TG levels than with total cholesterol levels [103]. In vitro studies showed that RLP and FFAs impair the balance of nitric oxide and ROS in ECs, leading to endothelial dysfunction and increased vascular endothelial permeability [100, 104]. RLP also promote NAD(P)H oxidase-dependent superoxide formation and production of TNF-α and IL-1β in ECs via activation of lectin-type oxidized LDL receptor 1 (LOX-1), resulting in EC death and dysfunction [105]. These findings may partially explain how TGRL, especially RLP, cause endothelial inflammation and dysfunction, thereby contributing to the development of atherosclerosis [9].
In addition, HTG can increase TG contents in HDL and promote HDL remodeling, which may dampen the anti-inflammatory and atheroprotective properties of HDL [9]. Studies in mouse models demonstrated that TGRL can also promote smooth muscle cell proliferation, which may also accelerate atherogenesis [25].
In summary, HTG with elevations in TGRL and their lipolytic products, i.e., RLP and FFAs, may promote ASCVD development and progression through multiple mechanisms. Inflammation in foamy monocytes and macrophages and inflammation and activation of ECs with acceleration of monocyte adhesion and transendothelial migration into arterial walls may play important roles in ASCVD associated with HTG [9, 36].
HTG-Targeting Therapies for ASCVD Prevention
Given the putative role of HTG in ASCVD, therapies targeting HTG may represent a new strategy for ASCVD prevention in HTG. First-line treatment of HTG is lifestyle intervention such as controlling body mass index in a proper range (20–25 kg/m2), eating healthy diet, reducing alcohol intake, and taking more regular physical activity [1, 106]. Regarding TG-targeting medications, please refer to another review article [19] for detailed discussion. In brief, fibrates, niacin, and high-dose omega-3 polyunsaturated FAs (PUFAs) have been used to lower TG levels [19, 106]. However, the efficacy of these medications in ASCVD prevention has been inconsistent in different trials. The discrepancy in outcomes may derive from differences in patient populations studied, formulations and doses of TG-lowering medications, placebo controls selected, use of other medications, duration of the studies, and also other factors that have not been identified. For example, fibrates effectively reduce TG levels and lower ASCVD risk in individuals with dyslipidemia and MetS, but not in patients with type 2 diabetes mellitus [107]. Omega-3 PUFAs have also been proved to lower TG levels, but their effects on ASCVD prevention have been debated [108]. While the REDUCE-IT trial and the Japan EPA Lipid Intervention Study (JELIS) showed that, compared to placebo or no omega-3 PUFA treatment control, treatment with EPA at 4 g/day or 1.8 g/day reduced ASCVD risk in statin-treated patients with HTG [109, 110], the results of other clinical trials such as ORIGIN, VITAL, ASCEND, OMEMI, and STRENGTH trials using omega-3 PUFAs at the doses of 1 g/day to 4 g/day did not show benefits of a mixture of EPA and DHA in prevention of ASCVD in patients with prediabetes or diabetes or elderly patients with a history of myocardial infarction [111, 112, 113, 114, 115].
In addition to the traditional HTG-targeting medications, several novel therapeutics such as anti-sense oligonucleotides (ASOs), small interfering RNAs (siRNA), or antibodies that target APOC3 or angiopoietin-like protein 3 (ANGPTL3) have been developed [116, 117, 118, 119, 120, 121, 122]. These new therapeutics profoundly lower plasma TG levels and also LDL-C in humans and rodents [116, 117, 118, 119, 120, 121, 122]. Treatment with ASO targeting APOC3 or ANGPTL3 reduces atherosclerosis progression in mice [118, 123]. Therefore, these new agents may be promising therapies for HTG and HTG-related diseases including ASCVD. However, their efficacy in ASCVD prevention in humans remains unknown and warrant additional clinical investigation.
Recently, several clinical trials including CANTOS, COLCOT, and LoDoCo/LoDoCo2 trials have shown benefits of anti-inflammatory therapies in ASCVD prevention [124, 125, 126]. Moreover, in addition to TG-lowering effects, several TG-lowering drugs have anti-inflammatory effects. Gemfibrozil reduces the incidence of ASCVD via decreasing TG levels, raising HDL-C levels, and also diminishing inflammation in both primary and secondary intervention studies [127]. Pemafibrate, firstly launched in 2018, decreases plasma levels of TG, RLP-C, non-HDL-C, APOB, and APOC3 in clinical trials and has anti-inflammatory and anti-atherogenesis effects in animal studies [128]. EPA may also benefit ASCVD prevention in humans with HTG by reducing inflammation [110]. Notably, short-term treatment with EPA in humans with HTG improves monocyte phenotypes [129], which may also contribute to the beneficial effects of EPA on ASCVD prevention. Because HTG is more strongly associated with inflammation than HCH, future studies should assess whether anti-inflammatory therapies have greater benefits in prevention of ASCVD associated with HTG.
Conclusions
HCH with elevations in LDL-C has long been recognized as a key risk factor and a therapeutic target for ASCVD. Recent genetic and epidemiological studies have revealed that HTG with elevated TGRL and RLP is also an important causal risk factor for ASCVD. Elevations in TGRL and RLP compared to LDL are more strongly associated with inflammation, which may be the mechanism that links HTG and with ASCVD. In vivo and ex vivo/in vitro studies show that TGRL and RLP induce lipid accumulation and inflammation in both tissue macrophages and circulating monocytes, which may play pivotal roles in atherogenesis. In addition, TGRL and RLP induce endothelial dysfunction and inflammation, which promote monocyte recruitment into atherosclerotic lesions, thereby accelerating development and progression of atherosclerosis. Moreover, TGRL and RLP can induce HDL remodeling and dysfunctions, which further promote vascular inflammation and atherosclerosis. Therapies targeting TG are being actively studied for their potential beneficial effects on ASCVD prevention. While clinical studies with the traditional TG-lowering medications have generated inconsistent data in ASCVD prevention, the newly developed therapeutics including those targeting APOC3 or ANGPTL3 have been highly effective in reducing plasma levels of TG and also LDL-C and bring promising potential for treatment of HTG-related diseases including ASCVD. The benefits of anti-inflammatory therapies in ASCVD prevention in several clinical trials have also opened up a new avenue for development of new anti-inflammatory therapies for prevention of ASCVD associated with HTG. In addition, with the continuation of the genetic and mechanistic studies, identification of new targets associated with HTG will provide more opportunities of developing additional novel therapies specific for ASCVD in HTG.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Arnett DK, Blumenthal RS, Albert MA, et al. 2019 ACC/AHA Guideline on the primary prevention of cardiovascular disease: A report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation. 2019;140(11):e596–646.
Domanski MJ, Tian X, Wu CO, et al. Time course of LDL cholesterol exposure and cardiovascular disease event risk. J Am Coll Cardiol. 2020;76(13):1507–16.
Sandesara PB, Virani SS, Fazio S, Shapiro MD. The forgotten lipids: Triglycerides, remnant cholesterol, and atherosclerotic cardiovascular disease risk. Endocr Rev. 2019;40(2):537–57.
Reiner Z. Hypertriglyceridaemia and risk of coronary artery disease. Nat Rev Cardiol. 2017;14(7):401–11.
Gill PK, Dron JS, Hegele RA. Genetics of hypertriglyceridemia and atherosclerosis. Curr Opin Cardiol. 2021;36(3):264–71.
Laufs U, Parhofer KG, Ginsberg HN, Hegele RA. Clinical review on triglycerides. Eur Heart J. 2020;41(1):99–109c.
Lawler PR, Kotrri G, Koh M, et al. Real-world risk of cardiovascular outcomes associated with hypertriglyceridaemia among individuals with atherosclerotic cardiovascular disease and potential eligibility for emerging therapies. Eur Heart J. 2020;41(1):86–94.
• Ginsberg HN, Packard CJ, Chapman MJ, et al: Triglyceride-rich lipoproteins and their remnants: metabolic insights, role in atherosclerotic cardiovascular disease, and emerging therapeutic strategies-a consensus statement from the European Atherosclerosis Society. Eur Heart J 2021. A nice review paper discusses the role and possible mechanisms of triglyceride-rich lipoproteins and their remnants in ASCVD.
Peng J, Luo F, Ruan G, Peng R, Li X. Hypertriglyceridemia and atherosclerosis. Lipids Health Dis. 2017;16(1):233.
CARDIoGRAMplusC4D Consortium, Deloukas P, Kanoni S, et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet. 2013;45(1):25–33.
Angelakopoulou A, Shah T, Sofat R, et al. Comparative analysis of genome-wide association studies signals for lipids, diabetes, and coronary heart disease: Cardiovascular biomarker genetics collaboration. Eur Heart J. 2012;33(3):393–407.
Triglyceride Coronary Disease Genetics Consortium. Collaboration ERF: Triglyceride-mediated pathways and coronary disease: collaborative analysis of 101 studies. Lancet. 2010;375(9726):1634–9.
Miller M, Stone NJ, Ballantyne CM, et al. Triglycerides and cardiovascular disease: A scientific statement from the American Heart Association. Circulation. 2011;123(20):2292–333.
Raposeiras-Roubin S, Rosselló X, Oliva B, et al. Triglycerides and residual atherosclerotic risk. J Am Coll Cardiol. 2021;77(24):3031–41.
Toth PP, Philip S, Hull M, Granowitz C. Association of elevated triglycerides with increased cardiovascular risk and direct costs in statin-treated patients. Mayo Clin Proc. 2019;94(9):1670–80.
Aberra T, Peterson ED, Pagidipati NJ, et al. The association between triglycerides and incident cardiovascular disease: What is “optimal”? J Clin Lipidol. 2020;14(4):438-447.e433.
Arca M, Veronesi C, D'Erasmo L, et al: Association of hypertriglyceridemia with all-cause mortality and atherosclerotic cardiovascular events in a low-risk Italian population: The TG-REAL retrospective cohort Analysis. J Am Heart Assoc 2020, 9(19):e015801.
Do R, Willer CJ, Schmidt EM, et al. Common variants associated with plasma triglycerides and risk for coronary artery disease. Nat Genet. 2013;45(11):1345–52.
Hussain A, Ballantyne CM, Saeed A, Virani SS. Triglycerides and ASCVD risk reduction: Recent insights and future directions. Curr Atheroscler Rep. 2020;22(7):25.
Nordestgaard BG, Benn M, Schnohr P, Tybjaerg-Hansen A. Nonfasting triglycerides and risk of myocardial infarction, ischemic heart disease, and death in men and women. JAMA. 2007;298(3):299–308.
Di Angelantonio E, Sarwar N, Perry P, et al. Major lipids, apolipoproteins, and risk of vascular disease. JAMA. 2009;302(18):1993–2000.
•• Saeed A, Feofanova EV, Yu B, et al. Remnant-like particle cholesterol, low-density lipoprotein triglycerides, and incident cardiovascular disease. J Am Coll Cardiol. 2018;72(2):156–69. This paper reports novel data from ARIC (atherosclerosis risk in communities) study suggesting that LDL-TG and RLP-C levels were associated with plasma levels of inflammatory markers and predictive of incident ASCVD.
Marz W, Scharnagl H, Winkler K, et al. Low-density lipoprotein triglycerides associated with low-grade systemic inflammation, adhesion molecules, and angiographic coronary artery disease: the Ludwigshafen Risk and Cardiovascular Health study. Circulation. 2004;110(19):3068–74.
Masucci-Magoulas L, Goldberg IJ, Bisgaier CL, et al. A mouse model with features of familial combined hyperlipidemia. Science. 1997;275(5298):391–4.
Li H, Han Y, Qi R, et al. Aggravated restenosis and atherogenesis in ApoCIII transgenic mice but lack of protection in ApoCIII knockouts: The effect of authentic triglyceride-rich lipoproteins with and without ApoCIII. Cardiovasc Res. 2015;107(4):579–89.
Castaner O, Pinto X, Subirana I, et al. Remnant Cholesterol, Not LDL cholesterol, is associated with incident cardiovascular disease. J Am Coll Cardiol. 2020;76(23):2712–24.
Krysa JA, Vine DF, Beilin LJ, et al. ApoB48-remnant lipoproteins are associated with increased cardiometabolic risk in adolescents. Atherosclerosis. 2020;302:20–6.
Koopal C, Retterstøl K, Sjouke B, et al. Vascular risk factors, vascular disease, lipids and lipid targets in patients with familial dysbetalipoproteinemia: A European cross-sectional study. Atherosclerosis. 2015;240(1):90–7.
Wadström BN, Wulff AB, Pedersen KM, Jensen GB, Nordestgaard BG: Elevated remnant cholesterol increases the risk of peripheral artery disease, myocardial infarction, and ischaemic stroke: A cohort-based study. Eur Heart J 2021.
Varbo A, Benn M, Tybjaerg-Hansen A, Jorgensen AB, Frikke-Schmidt R, Nordestgaard BG. Remnant cholesterol as a causal risk factor for ischemic heart disease. J Am Coll Cardiol. 2013;61(4):427–36.
Jorgensen AB, Frikke-Schmidt R, West AS, Grande P, Nordestgaard BG, Tybjaerg-Hansen A. Genetically elevated non-fasting triglycerides and calculated remnant cholesterol as causal risk factors for myocardial infarction. Eur Heart J. 2013;34(24):1826–33.
Borén J, Matikainen N, Adiels M, Taskinen MR. Postprandial hypertriglyceridemia as a coronary risk factor. Clinica Chimica Acta International Journal of Clinical Chemistry. 2014;431:131–42.
Nakano T, Nakajima K, Niimi M, et al. Detection of apolipoproteins B-48 and B-100 carrying particles in lipoprotein fractions extracted from human aortic atherosclerotic plaques in sudden cardiac death cases. Clinica chimica acta international journal of clinical chemistry. 2008;390(1–2):38–43.
Mullick AE, Deckelbaum RJ, Goldberg IJ, Al-Haideri M, Rutledge JC. Apolipoprotein E and lipoprotein lipase increase triglyceride-rich particle binding but decrease particle penetration in arterial wall. Arterioscler Thromb Vasc Biol. 2002;22(12):2080–5.
Proctor SD, Vine DF, Mamo JC. Arterial permeability and efflux of apolipoprotein B-containing lipoproteins assessed by in situ perfusion and three-dimensional quantitative confocal microscopy. Arterioscler Thromb Vasc Biol. 2004;24(11):2162–7.
Reiner Ž. Hypertriglyceridaemia and risk of coronary artery disease. Nat Rev Cardiol. 2017;14(7):401–11.
Chait A, Ginsberg HN, Vaisar T, Heinecke JW, Goldberg IJ, Bornfeldt KE. Remnants of the triglyceride-rich lipoproteins, diabetes, and cardiovascular disease. Diabetes. 2020;69(4):508–16.
Nordestgaard BG. Triglyceride-rich lipoproteins and atherosclerotic cardiovascular disease: New insights from epidemiology, genetics, and biology. Circ Res. 2016;118(4):547–63.
Eck MV, Oost J, Goudriaan JR, et al. Role of the macrophage very-low-density lipoprotein receptor in atherosclerotic lesion development. Atherosclerosis. 2005;183(2):230–7.
Takahashi S. Role of VLDL receptor in atherogenesis. Curr Opin Lipidol. 2021;32(4):219–25.
Wang YI, Bettaieb A, Sun C, et al. Triglyceride-rich lipoprotein modulates endothelial vascular cell adhesion molecule (VCAM)-1 expression via differential regulation of endoplasmic reticulum stress. PloS one. 2013;8(10):e78322.
Doi H, Kugiyama K, Oka H, et al. Remnant lipoproteins induce proatherothrombogenic molecules in endothelial cells through a redox-sensitive mechanism. Circulation. 2000;102(6):670–6.
Takahashi M, Yagyu H, Tazoe F, et al. Macrophage lipoprotein lipase modulates the development of atherosclerosis but not adiposity. J Lipid Res. 2013;54(4):1124–34.
Lloyd EE, Gaubatz JW, Burns AR, Pownall HJ. Sustained elevations in NEFA induce cyclooxygenase-2 activity and potentiate THP-1 macrophage foam cell formation. Atherosclerosis. 2007;192(1):49–55.
Castoldi A, Monteiro LB, van Teijlingen BN, et al. Triacylglycerol synthesis enhances macrophage inflammatory function. Nat Commun. 2020;11(1):4107.
Rosenblat M, Volkova N, Paland N, Aviram M. Triglyceride accumulation in macrophages upregulates paraoxonase 2 (PON2) expression via ROS-mediated JNK/c-Jun signaling pathway activation. BioFactors (Oxford, England). 2012;38(6):458–69.
Duewell P, Kono H, Rayner KJ, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464(7293):1357–61.
Wen H, Gris D, Lei Y, et al. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol. 2011;12(5):408–15.
Wu H, Gower RM, Wang H, et al. Functional role of CD11c+ monocytes in atherogenesis associated with hypercholesterolemia. Circulation. 2009;119(20):2708–17.
Xu L, Dai Perrard X, Perrard JL, et al. Foamy monocytes form early and contribute to nascent atherosclerosis in mice with hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2015;35(8):1787–97.
Tacke F, Alvarez D, Kaplan TJ, et al. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J Clin Invest. 2007;117(1):185–94.
• Clemente C, Rius C, Alonso-Herranz L, et al. MT4-MMP deficiency increases patrolling monocyte recruitment to early lesions and accelerates atherosclerosis. Nat Commun. 2018;9(1):910. Findings from this paper show that MT4-MMP deficiency accelerates atherogenesis in Ldlr–/––/– mice by enhancing recruitment of patrolling monocytes (which include lipid droplets and are foamy in Ldlr–/––/– mice as demonstrated in our lab and others) into atherosclerotic lesions.
Joffre J, Potteaux S, Zeboudj L, et al. Genetic and pharmacological inhibition of TREM-1 limits the development of experimental atherosclerosis. J Am Coll Cardiol. 2016;68(25):2776–93.
Saha P, Geissmann F. Toward a functional characterization of blood monocytes. Immunol Cell Biol. 2011;89(1):2–4.
Poitou C, Dalmas E, Renovato M, et al. CD14dimCD16+ and CD14+CD16+ Monocytes in obesity and during weight loss: Relationships with fat mass and subclinical atherosclerosis. Arterioscler Thromb Vasc Biol. 2011;31(10):2322–30.
Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19(1):71–82.
Khan IM, Pokharel Y, Dadu RT, et al. Postprandial monocyte activation in individuals with metabolic syndrome. J Clin Endocrinol Metab. 2016;101(11):4195–204.
Auffray C, Fogg D, Garfa M, et al. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science. 2007;317(5838):666–70.
Misharin AV, Cuda CM, Saber R, et al. Nonclassical Ly6C(-) monocytes drive the development of inflammatory arthritis in mice. Cell Rep. 2014;9(2):591–604.
Imhof BA, Jemelin S, Ballet R, et al. CCN1/CYR61-mediated meticulous patrolling by Ly6Clow monocytes fuels vascular inflammation. Proc Natl Acad Sci U S A. 2016;113(33):E4847-4856.
Zheng Z, Chiu S, Akbarpour M, et al. Donor pulmonary intravascular nonclassical monocytes recruit recipient neutrophils and mediate primary lung allograft dysfunction. Sci Transl Med 2017, 9(394).
Finsterbusch M, Hall P, Li A, et al. Patrolling monocytes promote intravascular neutrophil activation and glomerular injury in the acutely inflamed glomerulus. Proc Natl Acad Sci U S A. 2016;113(35):E5172-5181.
Wong KL, Tai JJ, Wong WC, et al. Gene expression profiling reveals the defining features of the classical, intermediate, and nonclassical human monocyte subsets. Blood. 2011;118(5):e16-31.
Schlitt A, Heine GH, Blankenberg S, et al. CD14+CD16+ monocytes in coronary artery disease and their relationship to serum TNF-alpha levels. Thromb Haemost. 2004;92(2):419–24.
Chimen M, Yates CM, McGettrick HM, et al. Monocyte subsets coregulate inflammatory responses by integrated signaling through TNF and IL-6 at the endothelial cell interface. J Immunol. 2017;198(7):2834–43.
Cros J, Cagnard N, Woollard K, et al. Human CD14dim monocytes patrol and sense nucleic acids and viruses via TLR7 and TLR8 receptors. Immunity. 2010;33(3):375–86.
Simon RC, Still WJ, O’Neal RM. The circulating lipophage and experimental atherosclerosis. J Atheroscler Res. 1961;1:395–400.
Suzuki M, O’Neal RM. Circulating lipophages, serum lipids, and atherosclerosis in rats. Arch Pathol. 1967;83(2):169–74.
Chipont A, Esposito B, Challier I, et al. MicroRNA-21 Deficiency alters the survival of Ly-6C(lo) monocytes in ApoE(-/-) mice and reduces early-stage atherosclerosis-brief report. Arterioscler Thromb Vasc Biol. 2019;39(2):170–7.
Combadière C, Potteaux S, Rodero M, et al. Combined inhibition of CCL2, CX3CR1, and CCR5 abrogates Ly6Chi and Ly6Clo monocytosis and almost abolishes atherosclerosis in hypercholesterolemic mice. Circulation. 2008;117(13):1649–57.
Potteaux S, Gautier EL, Hutchison SB, et al. Suppressed monocyte recruitment drives macrophage removal from atherosclerotic plaques of Apoe-/- mice during disease regression. J Clin Invest. 2011;121(5):2025–36.
He S, Kahles F, Rattik S, et al. Gut intraepithelial T cells calibrate metabolism and accelerate cardiovascular disease. Nature. 2019;566(7742):115–9.
Swirski FK, Libby P, Aikawa E, et al. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. 2007;117(1):195–205.
Marcovecchio PM, Thomas GD, Mikulski Z, et al. Scavenger receptor CD36 directs nonclassical monocyte patrolling along the endothelium during early atherogenesis. Arterioscler Thromb Vasc Biol. 2017;37(11):2043–52.
Foster GA, Xu L, Chidambaram AA, et al. CD11c/CD18 signals very late antigen-4 activation to initiate foamy monocyte recruitment during the onset of hypercholesterolemia. J Immunol. 2015;195(11):5380–92.
Gower RM, Wu H, Foster GA, et al. CD11c/CD18 expression is upregulated on blood monocytes during hypertriglyceridemia and enhances adhesion to vascular cell adhesion molecule-1. Arterioscler Thromb Vasc Biol. 2011;31(1):160–6.
Hernandez AA, Foster GA, Soderberg SR, et al. An allosteric shift in CD11c affinity activates a proatherogenic state in arrested intermediate monocytes. J Immunol 2020.
Zhou H, Ran Y, Da Q, et al. Defective association of the platelet glycoprotein Ib-IX complex with the glycosphingolipid-enriched membrane domain inhibits murine thrombus and atheroma formation. J Immunol. 2016;197(1):288–95.
Dresel HA, Via DP, Stohr M, et al. Observations on leukocytes from patients with severe familial hypercholesterolemia. Arteriosclerosis. 1986;6(3):259–64.
Bernelot Moens SJ, Neele AE, Kroon J, et al. PCSK9 monoclonal antibodies reverse the pro-inflammatory profile of monocytes in familial hypercholesterolaemia. Eur Heart J. 2017;38(20):1584–93.
Varela LM, Ortega A, Bermudez B, et al. A high-fat meal promotes lipid-load and apolipoprotein B-48 receptor transcriptional activity in circulating monocytes. Am J Clin Nutr. 2011;93(5):918–25.
den Hartigh LJ, Connolly-Rohrbach JE, Fore S, Huser TR, Rutledge JC. Fatty acids from very low-density lipoprotein lipolysis products induce lipid droplet accumulation in human monocytes. J Immunol. 2010;184(7):3927–36.
Bernelot Moens SJ, Verweij SL, Schnitzler JG, et al. Remnant cholesterol elicits arterial wall inflammation and a multilevel cellular immune response in humans. Arterioscler Thromb Vasc Biol. 2017;37(5):969–75.
Guijas C, Meana C, Astudillo AM, Balboa MA, Balsinde J. Foamy monocytes are enriched in cis-7-hexadecenoic fatty acid (16:1n–9), a possible biomarker for early detection of cardiovascular disease. Cell Chem Biol. 2016;23(6):689–99.
Varela LM, Ortega-Gomez A, Lopez S, Abia R, Muriana FJ, Bermudez B. The effects of dietary fatty acids on the postprandial triglyceride-rich lipoprotein/apoB48 receptor axis in human monocyte/macrophage cells. J Nutr Biochem. 2013;24(12):2031–9.
Wu H, Perrard XD, Wang Q, et al. CD11c expression in adipose tissue and blood and its role in diet-induced obesity. Arterioscler Thromb Vasc Biol. 2010;30(2):186–92.
Rogacev KS, Cremers B, Zawada AM, et al. CD14++CD16+ monocytes independently predict cardiovascular events: a cohort study of 951 patients referred for elective coronary angiography. J Am Coll Cardiol. 2012;60(16):1512–20.
Ozaki Y, Imanishi T, Taruya A, et al. Circulating CD14+CD16+ monocyte subsets as biomarkers of the severity of coronary artery disease in patients with stable angina pectoris. Circ J. 2012;76(10):2412–8.
Rogacev KS, Ulrich C, Blomer L, et al. Monocyte heterogeneity in obesity and subclinical atherosclerosis. Eur Heart J. 2010;31(3):369–76.
Feinstein MJ, Doyle MF, Stein JH, et al: Nonclassical monocytes (CD14dimCD16+) are associated with carotid intima-media thickness progression for men but not women: The multi-ethnic study of atherosclerosis. Arterioscler Thromb Vasc Biol 2021:ATVBAHA120315886.
Saja MF, Baudino L, Jackson WD, et al. Triglyceride-rich lipoproteins modulate the distribution and extravasation of Ly6C/Gr1(low) monocytes. Cell Rep. 2015;12(11):1802–15.
Chang HR, Josefs T, Scerbo D, et al. Role of LpL (lipoprotein lipase) in macrophage polarization in vitro and in vivo. Arterioscler Thromb Vasc Biol. 2019;39(10):1967–85.
Nakazato K, Ishibashi T, Nagata K, et al. Expression of very low density lipoprotein receptor mRNA in circulating human monocytes: its up-regulation by hypoxia. Atherosclerosis. 2001;155(2):439–44.
Ebara T, Ramakrishnan R, Steiner G, Shachter NS. Chylomicronemia due to apolipoprotein CIII overexpression in apolipoprotein E-null mice. Apolipoprotein CIII-induced hypertriglyceridemia is not mediated by effects on apolipoprotein E. J Clin Invest. 1997;99(11):2672–81.
Rasmussen KL, Tybjaerg-Hansen A, Nordestgaard BG, Frikke-Schmidt R. Plasma levels of apolipoprotein E, APOE genotype, and all-cause and cause-specific mortality in 105 949 individuals from a white general population cohort. Eur Heart J. 2019;40(33):2813–24.
Kawakami A, Aikawa M, Libby P, Alcaide P, Luscinskas FW, Sacks FM. Apolipoprotein CIII in apolipoprotein B lipoproteins enhances the adhesion of human monocytic cells to endothelial cells. Circulation. 2006;113(5):691–700.
Zewinger S, Reiser J, Jankowski V, et al. Apolipoprotein C3 induces inflammation and organ damage by alternative inflammasome activation. Nat Immunol. 2020;21(1):30–41.
Ting HJ, Stice JP, Schaff UY, et al. Triglyceride-rich lipoproteins prime aortic endothelium for an enhanced inflammatory response to tumor necrosis factor-a. Circ Res. 2007;100(3):381–90.
Bleda S, de Haro J, Varela C, Ferruelo A, Acin F. Elevated levels of triglycerides and vldl-cholesterol provoke activation of nlrp1 inflammasome in endothelial cells. Int J Cardiol. 2016;220:52–5.
Anderson RA, Evans ML, Ellis GR, et al. The relationships between post-prandial lipaemia, endothelial function and oxidative stress in healthy individuals and patients with type 2 diabetes. Atherosclerosis. 2001;154(2):475–83.
Matsumoto S, Gotoh N, Hishinuma S, et al. The role of hypertriglyceridemia in the development of atherosclerosis and endothelial dysfunction. Nutrients. 2014;6(3):1236–50.
Libby P. The changing landscape of atherosclerosis. Nature. 2021;592(7855):524–33.
Klafke JZ, Porto FG, Batista R, et al. Association between hypertriglyceridemia and protein oxidation and proinflammatory markers in normocholesterolemic and hypercholesterolemic individuals. Clinica chimica acta international journal of clinical chemistry. 2015;448:50–7.
Wang L, Gill R, Pedersen TL, Higgins LJ, Newman JW, Rutledge JC. Triglyceride-rich lipoprotein lipolysis releases neutral and oxidized FFAs that induce endothelial cell inflammation. J Lipid Res. 2009;50(2):204–13.
Shin HK, Kim YK, Kim KY, Lee JH, Hong KW. Remnant lipoprotein particles induce apoptosis in endothelial cells by NAD(P)H oxidase-mediated production of superoxide and cytokines via lectin-like oxidized low-density lipoprotein receptor-1 activation: Prevention by cilostazol. Circulation. 2004;109(8):1022–8.
Mach F, Baigent C, Catapano AL, et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur Heart J. 2020;41(1):111–88.
Tarantino N, Santoro F, Correale M, et al. Fenofibrate and dyslipidemia: Still a place in therapy? Drugs. 2018;78(13):1289–96.
Wu H, Xu L, Ballantyne CM: Dietary and pharmacological fatty acids and cardiovascular health. J Clin Endocrinol Metab 2020, 105(4).
Yokoyama M, Origasa H, Matsuzaki M, et al. Effects of eicosapentaenoic acid on major coronary events in hypercholesterolaemic patients (JELIS): A randomised open-label, blinded endpoint analysis. Lancet. 2007;369(9567):1090–8.
Bhatt DL, Steg PG, Miller M, et al. Cardiovascular risk reduction with icosapent ethyl for hypertriglyceridemia. N Engl J Med. 2019;380(1):11–22.
Group ASC, Bowman L, Mafham M, et al. Effects of n-3 fatty acid supplements in diabetes mellitus. N Engl J Med. 2018;379(16):1540–50.
Manson JE, Cook NR, Lee IM, et al. Marine n-3 fatty acids and prevention of cardiovascular disease and cancer. N Engl J Med. 2019;380(1):23–32.
Investigators OT, Bosch J, Gerstein HC, et al. n-3 fatty acids and cardiovascular outcomes in patients with dysglycemia. N Engl J Med. 2012;367(4):309–18.
Kalstad AA, Myhre PL, Laake K, et al. Effects of n-3 fatty acid supplements in elderly patients after myocardial infarction: A randomized, controlled trial. Circulation. 2021;143(6):528–39.
Nicholls SJ, Lincoff AM, Garcia M, et al. Effect of high-dose omega-3 fatty acids vs corn oil on major adverse cardiovascular events in patients at high cardiovascular risk: The STRENGTH Randomized Clinical Trial. JAMA. 2020;324(22):2268–80.
Alexander VJ, Xia S, Hurh E, et al. N-acetyl galactosamine-conjugated antisense drug to APOC3 mRNA, triglycerides and atherogenic lipoprotein levels. Eur Heart J 2019.
Rosenson RS, Burgess LJ, Ebenbichler CF, et al. Evinacumab in patients with refractory hypercholesterolemia. N Engl J Med. 2020;383(24):2307–19.
Graham MJ, Lee RG, Brandt TA, et al. Cardiovascular and metabolic effects of ANGPTL3 antisense oligonucleotides. N Engl J Med. 2017;377(3):222–32.
Pechlaner R, Tsimikas S, Yin X, et al. Very-low-density lipoprotein-associated apolipoproteins predict cardiovascular events and are lowered by Inhibition of APOC-III. J Am Coll Cardiol. 2017;69(7):789–800.
Graham MJ, Lee RG, Bell TA 3rd, et al. Antisense oligonucleotide inhibition of apolipoprotein C-III reduces plasma triglycerides in rodents, nonhuman primates, and humans. Circ Res. 2013;112(11):1479–90.
Clifton P, Sullivan D, Baker J, et al. Abstract 12594: Pharmacodynamic effect of ARO-APOC3, an investigational hepatocyte-targeted RNA interference therapeutic targeting apolipoprotein C3, in patients with hypertriglyceridemia and multifactorial chylomicronemia. Circulation. 2020;142 SUPPL_3:A12594.
Watts GF, Schwabe C, Scott R, et al: Abstract 15751: Pharmacodynamic effect of ARO-ANG3, an investigational RNA interference targeting hepatic angiopoietin-like protein 3, in patients with hypercholesterolemia. Circulation 2020, 142.suppl_3:A15751.
Kanter JE, Shao B, Kramer F, et al. Increased apolipoprotein C3 drives cardiovascular risk in type 1 diabetes. J Clin Invest. 2019;129(10):4165–79.
Ridker PM, Everett BM, Thuren T, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377(12):1119–31.
Tardif JC, Kouz S, Waters DD, et al. Efficacy and safety of low-dose colchicine after myocardial infarction. N Engl J Med. 2019;381(26):2497–505.
Nidorf SM, Fiolet ATL, Mosterd A, et al: Colchicine in patients with chronic coronary disease. N Engl J Med 2020.
Chapman MJ. Fibrates in 2003: Therapeutic action in atherogenic dyslipidaemia and future perspectives. Atherosclerosis. 2003;171(1):1–13.
Yamashita S, Masuda D, Matsuzawa Y. Pemafibrate, A new selective PPARα modulator: Drug concept and its clinical applications for dyslipidemia and metabolic diseases. Curr Atheroscler Rep. 2020;22(1):5.
Dai Perrard XY, Lian Z, Bobotas G, Dicklin MR, Maki KC, Wu H. Effects of n-3 fatty acid treatment on monocyte phenotypes in humans with hypertriglyceridemia. J Clin Lipidol. 2017;11(6):1361–71.
Funding
This work was supported by The Medical and Health Science and Technology Plan Project of Zhejiang Province (2021436111) and The Hangzhou Health Science and Technology Planning Project (A20210149) to X.P. and grants from the National Institute of Diabetes and Digestive and Kidney Diseases (R01-DK-121348), National Heart, Lung, and Blood Institute (R01-HL-098839), National Institute on Aging (R01-AG-065197), American Heart Association (16GRNT30410012), and American Diabetes Association (1–17-IBS-082) to H.W.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article is a review article in the field of hypertriglyceridemia and atherosclerotic cardiovascular disease. It contains previous publications based on human and animal studies.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Vascular Biology
Rights and permissions
About this article

Cite this article
Peng, X., Wu, H. Inflammatory Links Between Hypertriglyceridemia and Atherogenesis. Curr Atheroscler Rep 24, 297–306 (2022). https://doi.org/10.1007/s11883-022-01006-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11883-022-01006-w