
Abstract
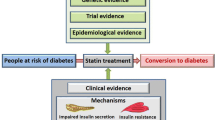
There is growing evidence to suggest that statin therapy is associated with an increased risk of incident diabetes. The risk for statin-related diabetes depends upon many factors including age, pre-existing diabetic risk, type and potency of statin. Several mechanisms have been suggested for the diabetogenic effects of statins involving processes that alter islet ß-cell function, resulting in impaired glucose metabolism. Recent evidence suggests that the association of statin therapy with the development of diabetes may be partly mediated by a statin-induced decrease in circulating adiponectin and coenzyme Q10. The available evidence suggests the benefit of statins in reducing cardiovascular events outweigh the risk of developing diabetes. Moreover, statin therapy does not impair glycemic control in diabetic patients. Expert recommendations for the use of statins in people at risk of developing diabetes have recently been published. However, further research is required to elucidate both the association between statin use and incident diabetes as well as underlying mechanisms.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Type 2 diabetes (T2DM) is a major health epidemic affecting at least 382 million people worldwide. The World Health Organization estimates that the incidence of diabetes will escalate to 471 million by 2035 [1]. People with diabetes are at increased risk for cardiovascular disease (CVD), and low-density lipoprotein cholesterol (LDL-C) is a significant determinant of CVD in this population [2]. Current guidelines for primary and secondary CVD prevention recommend a multifactorial approach, including therapeutic lifestyle changes and cholesterol-lowering therapy, to achieve optimal control of LDL-C [2, 3, 4•]. Statin therapy is the cornerstone of dyslipidemia management in high-risk patients, including T2DM [2, 4•]. Although statins have a good safety record in clinical practice, an association with new-onset diabetes has been suggested [5•, 6, 7, 8••, 9]. We review the current knowledge on the diabetogenic effects of statin, with emphasis on the role of adiponectin and coenzyme Q10 (CoQ10) in developing diabetes.
Statins and Risk of New-Onset Diabetes Mellitus: Clinical Evidence
Statins are the most effective drugs for treating hypercholesterolemia due to increased plasma levels of LDL particles. Large prospective primary and secondary prevention studies have demonstrated the cardiovascular benefit of statins in a wide variety of people, including those with T2DM [3, 10–12]. Although there has never been a prospective, randomized study to support the link between statins and diabetes risk, several retrospective cohort studies have examined this relationship.
Major Statin Trials
The West of Scotland Coronary Prevention Study (WOSCOPs) cohort involved 5974 men aged 45 to 64 (mean age 55.2 years) with follow-up ranging from 3.5 to 6.1 years. Pravastatin therapy resulted in a 30 % risk reduction for incident diabetes [13]. Contrary to this, there was a 32 % higher incidence of diabetes with pravastatin therapy in the Prospective Study of Pravastatin in the Elderly at Risk (PROSPER), a study of elderly patients aged between 70 and 82 years [14]. Pravastatin therapy was not, however, associated with increased risk of T2DM in the Long-term Disease (LIPID) trial (n = 9014, 31 to 75 years of age) [15]. Age-dependent loss of beta cell function has been suggested to explain the observed increase in new-onset diabetes in the PROSPER trial. In the Anglo-Scandinavian Cardiac Outcome Trial-Lipid-Lowering Arm (ASCOT-LLA), low-dose atorvastatin (20 mg) was not associated with the incidence of new-onset diabetes [16]. However, a significant 34 % increase in new-onset T2DM was observed with high-dose atorvastatin (40 mg) in the Stoke Prevention by Aggressive Reduction in Cholesterol Levels (SPARCL) trials [17]. In the Heart Protection Study (HPS), increased new-onset diabetes was not noted with simvastatin (40 mg) [18]. In the JUPITER (Justification for the Use of Statins in Primary Prevention: An Intervention Trial Evaluating Rosuvastatin) trial [19••], rosuvastatin therapy (20 mg) caused a 28 % increased risk of new-onset diabetes in those participants with pre-existing risk factors for diabetes (e.g. metabolic syndrome, impaired fasting glucose, obesity or raised glycated haemoglobin A1c [HbA1c]); no increase in diabetes with rosuvastatin was observed in individuals without major diabetes risk factors. This observation has raised the possibility that people with pre-existing risk factors for T2DM are more likely to develop diabetes with statin therapy. Consistent with this notion, in WOSCOPS, the patients who entered the study did not generally have pre-existing diabetic risks (e.g. obesity and atherogenic dyslipidemia), and those who have developed T2DM were in fact already at risk of diabetes (e.g. a higher body mass index and elevated triglyceride levels) [13].
Meta-analyses
Several meta-analyses have addressed the association between statin therapy and new-onset diabetes. In a meta-analysis of six trials with a total of 57,593 patients [20], Rajpathaket et al. reported that the incidence of diabetes was 13 % higher in patients receiving statin therapy compared with those not receiving a statin. In another meta-analysis, Coleman et al. found that statin therapy resulted in a significant 14 % increase in the relative risk of developing diabetes [21]. However, the relative increase in risk of incident diabetes was not statistically significant in these reports when the WOSCOPS was included. Using a larger database with 91,140 participants in 13 major statin trials including the WOSCOPS trial [6], Sattar et al. demonstrated that the risk of developing diabetes was 9 % higher (95 % confidence interval [CI], 2–17 %) over a 4-year period compared with patients randomized to placebo or standard care.
Low-Dose vs High-Dose Statin Therapy
Compelling evidence indicates that high-potency statin therapy provides a significant benefit in preventing CVD compared with lower potency therapy [12, 22]. However, the adverse effect of intensive statin therapy on new-onset diabetes remains a serious concern. Waters et al. compared the incidence of new-onset diabetes between low-dose (10 mg) and high-dose atorvastatin (80 mg) in the Treating to New Targets (TNT) and Incremental Decrease in Endpoints Through Aggressive Lipid Lowering (IDEAL) trials [17]. Compared with low-dose atorvastatin therapy, high-dose atorvastatin therapy resulted in a 24 % increase in incident diabetes in those participants with 2–4 pre-existing diabetic risk factors (e.g. raised fasting glucose and triglyceride levels, reduced high-density lipoprotein [HDL]-cholesterol, history of hypertension and obesity at baseline). This effect with atorvastatin therapy was not observed in those with 0–2 diabetic risk factor, however. Again, this finding suggests a role of pre-existing diabetic risks in the development of T2DM with statin therapy. In a meta-analysis of five statin trials involving 32,752 participants [23•], intensive-dose statin therapy was associated with an increased risk of new-onset diabetes compared with moderate-dose statin therapy (+12 %). More recently, in a meta-analyses of eight population-based cohort studies involving 136,966 patients aged ≥40 years [24], higher potency statin use (rosuvastatin ≥40 mg, atorvastatin ≥20 mg and simvastatin ≥40 mg) was associated with a 15 % increase in the risk of new-onset diabetes compared with lower potency statins in patients treated for secondary prevention of CVD.
Types of Statins
The potential diabetogenic effects of statins may differ between statins. In a population-based study [25], Carter et al. examined the risk of incident diabetes among patients treated with different statins. Compared with pravastatin (the reference drug in all analyses), treatments with high-potency statins, including atorvastatin, simvastatin and rosuvastatin, were associated with increased risk of incident diabetes. There was no significant increased risk among patients who received fluvastatin or lovastatin. The study also found that the risk of incident diabetes was similar whether statins were used for primary or secondary prevention of CVD.
Pitavastatin is a newer and more tolerable LDL-C-lowering agent with effect similar to comparable doses of atorvastatin (−40 % at daily standard doses of 4 and 20 mg, respectively) [26, 27]. While most statins show inconsistent effects on HDL-C levels, pitavastatin has been shown to improve HDL function, including elevations in HDL-cholesterol by 15–25 %. More importantly, pitavastatin has demonstrated neutral or favorable effects on glucose control in patients with and without T2DM or metabolic syndrome [27]. In some studies, pitavastatin has been shown to decrease homeostatic model assessment (HOMA) score and HbA1c in T2DM patients [28]. Experimental data has shown that pitavastatin increases glucose uptake and sensitivity to intraperitoneal insulin in KKAy mice [28, 29]. Given the pleiotropic effects of HDL in preventing oxidation, vascular inflammation and pancreatic beta cell dysfunction [30, 31], improvement in HDL function may be associated with enhanced insulin sensitivity in subjects treated with pitavastatin.
Taken together, there is strong evidence indicating that statin therapy is associated with increased risk of new-onset diabetes. However, the risk of statin-related diabetes may depend upon age, pre-existing diabetic risks, type and potency of statins.
Statins and Risk of New-Onset Diabetes Mellitus: Possible Mechanisms
Type 2 diabetes mellitus is a disorder of glucose metabolism and encompasses individuals who have insulin resistance and relative insulin deficiency where insulin secretion is insufficient to compensate for insulin resistance and normal glucose metabolism [2]. Insulin-resistance and insulin-secretory defects caused by beta cell dysfunction are key metabolic characteristics contributing to the pathogenesis of T2DM.
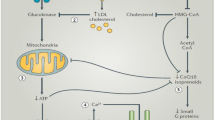
The precise mechanisms of action to explain a cause and effect between statin and T2DM have not yet been identified. Several mechanisms have been suggested for statin-induced impairment of glucose metabolism (Fig. 1) [32••, 33]. (1) Intracellular glucose uptake via glucose transporter 2 (GLUT2) initiates phosphorylation by glucose kinase and subsequently ATP-dependent potassium and voltage-gated calcium channel-mediated signalling cascades for the synthesis and secretion of insulin. Statins can reduce mRNA and protein expression of GLUT2 and voltage-dependent calcium channel, thereby inhibiting insulin synthesis and secretion [34]. (2) Statins can also directly reduce insulin-stimulated glucose uptake by impairing insulin pathway via the inhibition of phosphatidylinositol triphospho-kinase [35]. (3) Statin inhibition of 3-hydroxy-3-methyl-glutaryl-CoA (HMG-CoA) reductase can suppress synthesis of isoprenoids which in turn inhibits the expression of GLUT4 (another rate-limiting protein for glucose transport), leading to impaired glucose uptake [36, 37]. (4) Upregulation of LDL receptor with statins enhances uptake of LDL cholesterol. However, increased abundance of plasma-derived cholesterol can inhibit glucose kinase (the rate-limiting enzyme for intracellular glucose metabolism), thus impairing normal glucose uptake [38]. (5) Oxidation of plasma-derived cholesterol can incite pro-inflammatory and pro-oxidative cascades that compromise the structural integrity and function of the islet beta cells, thereby worsening glucose metabolism [39]. (6) Statin treatment increases the production and bioavailability of nitric oxide (NO) via the upregulation of endothelial nitric oxide synthase. However, overproduction of NO can induce beta cell apoptosis via the activation of calcium-dependent protease (calpain), thereby impairing beta cell function [40, 41]. (7) Statin-induced inflammation and mitochondrial dysfunction in skeletal muscle can impair beta cell function and trigger the development of T2DM [42]. Consistent with these mechanisms, a recent Mendelian randomization study reported that the inhibition of HMG-CoA reductase activity with statin, particularly due to genetic variation in the HMGCR gene (rs17238484 and rs12916 alleles), was associated with an increased risk of T2DM [43, 44]. This implies that the association is an on-target effect of the drug and that the risk of diabetes is causally related to the degree inhibition of HMG-CoA reductase activity and hence to the potency of the statin. However, unlike other statins (e.g. atorvastatin or simvastatin), pravastatin does not suppress glucose-induced elevation of intracellular calcium ion level and glucose-stimulated insulin secretion [34]; it also does not reduce sensitivity to insulin nor attenuate the expression of GLUT4 [45]. As will be discussed later, pravastatin increases the expression of adiponectin mRNA and enhances adiponectin secretion [46, 47]. Whether these effects with pravastatin are related to lower incidence of diabetes remain to be elucidated.

Potential mechanisms of action in the development of diabetes
While the relative roles of contribution of these mechanisms remain to be elucidated, it appears that the potential diabetogenic effects of statins may involve multiple mechanisms that alter islet beta cell function, resulting in impaired glucose metabolism and insulin sensitivity. Recent experimental and clinical evidence also highlights the role of adiponectin and ubiquinone, also known as CoQ10, in the modulation of glucose metabolism. As discussed below, statin may impair glucose metabolism via effects on adiponectin and CoQ10 metabolism.
Adiponectin and T2DM
It has been well documented that adipose tissue is not only a store of excess energy, but also a hormonally active metabolic system [48]. Several adipocyte-derived biologically active molecules (adipocytokines) have been identified that can potentially impact on glucose metabolism and contribute to the pathogenesis of insulin resistance and T2DM [49]. These adipocytokines include adiponectin, leptin, resistin, retinol-binding protein-4, interleukin-6 and tumour necrosis factor-α (TNF-α). Of these adipocytokines, adiponectin is a 244-amino acid collagen-like protein that circulates at relatively high concentrations, accounting for 0.01 % of total plasma protein in the circulation [50, 51•]. In a meta-analysis of seven prospective studies involving a total of 1318 CHD cases, higher adiponectin levels were associated with a lower risk of CHD (odds ratio, 0.84 [95 % CI, 0.70 to 1.01]) [52]. However, the association between adiponectin levels and CVD was not found in T2DM patients [53]. Unlike other adipocytokines, plasma adiponectin levels correlate inversely with a wide range of cardiometabolic factors including insulin sensitivity, blood glucose, obesity and dyslipidaemia [50, 54]. Hypoadiponectinaemia has been found in individuals with T2DM and obesity [55]. The precise mechanism for this remains unclear but may be attributable to the inhibition of adiponectin gene transcription by inflammatory and angiogenic factors secreted by hypertrophic adipocytes [56]. Several studies have addressed the association of plasma adiponectin levels and risk of T2DM. In a meta-analysis of 13 prospective studies in 14,598 individuals, higher adiponectin levels were associated with a lower risk of T2DM across diverse populations [57]. Hypoadiponectinaemia is well known to be substantially associated with genetic factors which may contribute to increased diabetic risk. Single nucleotide polymorphisms in the adiponectin gene (e.g. −11377CG and +45T>G) have been identified to be associated with hypoadiponectinaemia and increased T2DM [58]. Moreover, two meta-analyses of genome-wide association studies (GWAS) have demonstrated that several loci associated with adiponectin levels (e.g. variants in ADIPOQ and GPR109A genes) are linked to increased risk of T2DM [59, 60].
The protective role of adiponectin against T2DM remains unclear. Experimental and human evidence suggests that adiponectin may protect against the development of T2DM by improving insulin sensitivity. Animal studies show that adiponectin knockout mice develop insulin resistance and glucose intolerance with a high fat diet whereas mice overexpressing adiponectin are insulin sensitive and are resistant to diet-induced diabetes [61, 62]. Administration of adiponectin to rodents has also been shown to improve insulin sensitivity via stimulation of insulin-induced tyrosine phosphorylation of the insulin receptor in skeletal muscle [63]. Consistent with this, plasma adiponectin concentration has been shown to be associated with skeletal muscle insulin receptor tyrosine phosphorylation [64]. Several molecular mechanisms of action of adiponectin on insulin sensitivity have been proposed, including suppression of hepatic gluconeogenesis, stimulation of fatty acid oxidation in the liver, stimulation of glucose uptake and fatty acid oxidation in skeletal muscle [65, 66]. These effects are at least in part involved in the activation of AMP-kinase and peroxisome proliferator-activated receptor-α in skeletal muscle [67]. Adiponectin also stimulates insulin gene expression and secretion in pancreatic beta cells via the induction of extracellular signal-regulated kinase (ERK) and Akt phosphorylation in insulin signalling pathway [68, 69]. Adiponectin has been shown to increase mitochondrial biogenesis and fatty acid oxidation in skeletal muscle by enhancing mitogen-activated protein kinase (MAPK) and PPAR-ϒ coactivator 1α (PGC-1α) [70]. In addition, the anti-inflammatory effect of adiponectin can protect beta cell function, thereby improving insulin sensitivity. One potential mechanism has also been suggested involving the antagonistic effect of adiponectin to attenuate the adverse effect of TNF-α on beta cell function [71, 72].
Adiponectin and Statins
It has been postulated that the association of statin therapy with the development of T2DM may be partly mediated by a statin-induced decrease in circulating adiponectin levels. This evidence is reviewed below regarding the effect of statins on plasma adiponectin levels.
Lipophilic Statins
There are conflicting results on the effect of atorvastatin on adiponectin levels [73–76]. However, at a basic science level, there is some evidence showing that atorvastatin can attenuate adipocyte maturation, thus decreasing adiponectin production [37]. Another lipophilic statin, simvastatin, appears to not have an affect or reduce adiponectin levels [77–79]. A study by Forst et al. demonstrated a significant 12 % adiponectin reduction on 12 weeks of simvastatin in nondiabetic subjects [78]. However, simvastatin failed to alter adiponectin levels in subjects with T2DM, hypertension or the metabolic syndrome [79]. Fluvastatin showed no effect on adiponectin levels [80].
Hydrophilic Statins
Pravastatin has been shown to improve insulin sensitivity and reduce risk of new-onset T2DM [13]. Several studies have demonstrated that pravastatin therapy increases adiponectin levels [81, 82]. In one study, pravastatin (20 mg/day; 6 months) demonstrated an up to 35 % increase in adiponectin levels in CAD patients with impaired glucose tolerance [81]. Experimental data has suggested that pravastatin increases the expression of adiponectin mRNA and enhances adiponectin secretion in adipocytes [47].
Another hydrophilic statin, rosuvastatin, has been shown to not have an effect or increase plasma adiponectin levels. In one study, rosuvastatin (10 mg/day) resulted in 65 % increase in adiponectin levels in patients with hypercholesterolemia while in other studies, rosuvastatin therapy showed no effect on adiponectin levels [75, 76, 82–84]. Previous studies show that rosuvastatin was not associated with any change in insulin sensitivity [85]. However, in a study employing rosuvastatin, a significant dose-dependent increase (10, 20 and 40 mg) was observed in plasma insulin levels and HOMA score [86]. This result indicates a deterioration of insulin sensitivity, consistent with the outcomes from the JUPITER, showing a 28 % increased risk of new-onset diabetes with rosuvastatin therapy [19••].
Pitavastatin is another relatively potent hydrophilic statin that has consistently been shown to increase adiponectin levels and, in some studies, improve insulin sensitivity [27, 87]. In two studies of patients with hyperlipidemia, pitavastatin therapy (2 mg) showed a significant increase (∼25 %) in adiponectin levels [88, 89]. By contrast to atorvastatin [78], pitavastatin does not impair maturation of pre-adipocytes, thereby preventing adipocyte hypertrophy and adipocytokine dysregulation [29].
Taken together, the lack of consistent effects of statins on adiponectin levels may relate to differences in biophysical properties of statin used. It is unclear whether lipophilic and hydrophilic statins have differential effect on adiponectin metabolism. In contrast to lipophilic statins, hydrophilic statins (pravastatin, rosuvastatin and pitavastatin) are generally more consistent to increase adiponectin levels and insulin resistance. However, the increased incidence of T2DM with rosuvastatin in the JUPITER trial challenges this speculation [19••], but the effect may be dependent on pre-existing risk factors for T2DM. Hence, we maintain that there is no convincing evidence that the effect of statin on adiponectin is associated with changes in insulin sensitivity and, by implication, the development of T2DM. More studies are required to examine the role of adiponectin on insulin sensitivity and risk of diabetes with statins, particularly pitavastatin.
CoQ10 and T2DM
CoQ10 (or ubiquinone) is a lipid-soluble molecule with a side chain of 10 isoprenoid units, endogenously synthesized in the body from phenylalanine and mevalonic acid with some obtained from diet (meat products). CoQ10 is presented in all cellular membranes, blood and lipoproteins, but its concentration is the highest in the heart, kidney, liver and muscles owing to their high energy requirements or metabolic activity. The synthesis of CoQ10 is regulated by the HMG-CoA reductase reaction in the mevalonate pathway [90, 91]. Biologically, CoQ10 serves as an energy transporter in mitochondrial and extra-mitochondrial membranes. It accepts electrons from several donors (such as NADH, succinate and glycerol-3-phosphate) and transfers them to the cytochrome complex which, in turn, drives ATP synthesis [90]. Hence, deficiencies in mitochondrial CoQ10 levels can impair the electron transport rate, thus uncoupling ATP production. CoQ10 is also a potential antioxidant and free-radical scavenger protecting cell membranes and lipoprotein from protein and lipid peroxidation. CoQ10 also acts as a membrane-stabilizing agent that enhances resistance of bacterial cell and liposome membranes to salt stress. CoQ10 deficiency has been implicated in several clinical disorders, including heart failure, hypertension, malignancy and T2DM [92].
CoQ10 is known to be deficient in the diabetic state [93]. The underlying mechanism for CoQ10 deficiency (quantitative or functional) in T2DM may be a consequence of impaired mitochondrial substrate utilization and/or increased oxidative stress. It has been suggested that CoQ10 deficiency can depress islet beta cell function, thus impairing glucose metabolism [94]. Moreover, a reduction in CoQ10 levels in muscle tissue can impair mitochondrial function, thus increasing the risk of statin-induced myopathy [95].
CoQ10 and Statins
HMG-CoA reductase catalyses the conversion of HMG-CoA to mevalonate, a precursor of both cholesterol and CoQ10. Hence, the inhibition of conversion of HMG-CoA to mevalonate with statins reduces not only cholesterol synthesis but also other products downstream of mevalonate such as CoQ10. In the LIPID trial, pravastatin lowered plasma CoQ10 concentrations by 15 % [96]. A more potent statin, atorvastatin, has been shown to reduce plasma CoQ10 concentration by 40 % [97], suggesting a dose-related effect of statins in lowering CoQ10 concentrations. Given the fundamental role of CoQ10 in mitochondrial bioenergetics and its antioxidant properties, the cardiovascular benefits of statin therapy may be attenuated by its inhibition of endogenous CoQ10 production.
Several mechanisms have been proposed to explain the association between statin-induced CoQ10 depletion and diabetes. Statins suppress the synthesis of CoQ10, resulting in reduced insulin secretion due to direct inhibition of ATP production. Sub-optimal beta cell levels of CoQ10 with statin therapy reduce mitochondrial glycerol-3-phosphate-dehydrogenase (G3PD) levels [94], which are critical to the function of mitochondria. As mentioned earlier, statin-induced mitochondrial dysfunction can impair beta cell function and induce insulin resistance in skeletal muscles [98]. Furthermore, the depletion of CoQ10 induces myocyte inflammation and fiber damage. This may be one explanation to underlie the pathophysiology of statin-induced myopathy and potentially contribute to insulin resistance in skeletal muscle [99]. In addition, it has been proposed that the decline in tissue CoQ10 with aging is causally related to oxidative stress and mitochondrial dysfunction in skeletal muscle, thereby accelerating statin-induced peripheral insulin resistance [100]. This speculation is consistent with the increased incidence of diabetes with pravastatin therapy in the PROSPER trial [14].
Supplementation with oral CoQ10 can restore plasma CoQ10 in patients receiving statin therapy. CoQ10 can also improve glycemic control and other metabolic disorders associated with insulin resistance [101–103]. Experimental evidence has demonstrated that CoQ10 administration improves pancreatic beta cell function, increases insulin sensitivity and preserves the mitochondrial function in the islets [104]. The antidiabetic or insulin-sensitizing mechanisms underlying its favourable effects remain unclear but may involve the upregulation of insulin and adiponectin receptor, stimulation of insulin signalling pathways (tyrosine kinase and PI3K) in addition to improvement in the redox system (oxidative stress) and elevation of soluble receptor for advanced glycation end products (sRAGE) and adiponectin levels [105]. In addition, CoQ10 has also been shown to ameliorate the reduction in GLUT4 transporter by simvastatin in adipocytes [106].
Collectively, there is strong and consistent evidence indicating that CoQ10 plays an important role in the regulation of mitochondrial function, which is critical for beta cell function in the islets. However, direct clinical evidence linking between CoQ10 deficiency and the onset of T2DM is not yet available. Statin therapy has been shown to reduce the production of CoQ10, and potentially induce myopathy, but it is unclear whether this contributes to impaired insulin sensitivity and increase risk of T2DM. More research is needed to determine whether supplementation of CoQ10 can prevent the development or progression of T2DM, especially in those with pre-existing diabetic risk and receiving statin therapy.
Cardiovascular Benefits and Diabetes Risks: a Question of Balance
Data from several clinical trials, as well as meta-analyses, consistently indicate that statin therapy reduces the risk of CVD by 25–30 %, with greater effects for those receiving higher doses or potent statins. A meta-analysis by Sattar et al. estimated that the number needed to treat over a 4-year period to cause one excess case of T2DM was 255 [8••]. In contrast, 5.4 fewer deaths from CHD and cases of nonfatal myocardial infarction would be prevented per 255 patients treated over 4 years for each 1-mmol/L reduction in LDL cholesterol compared with controls [8••]. In another meta-analysis, Preiss et al. found that the number needed to treat with high-dose statin per year to produce one excess case of T2DM compared with moderate-dose statin therapy was 498. However, the estimated number of CVD events prevented would be 3.2, in comparison with one excess case of T2DM [23•]. In the JUPITER trial, the primary CVD endpoint was reduced by 36 %, but incident diabetes was increased by 28 % for participants with one or more major diabetes risk factors allocated rosuvastatin. In absolute terms, 134 total vascular events or deaths were avoided for every 54 new case of T2DM [19••]. For participants without major diabetes risk factors allocated rosuvastatin, the primary endpoint was reduced by 53 %, with no increase in incident diabetes. In absolute term, 86 total vascular events or deaths were avoided with no excess incidence of diabetes [19••].
Collectively, the overall CV benefits outweigh the controversy surrounding the potential diabetogenic effects of statin. There is currently no evidence to suggest that glycemic control is impaired in T2DM receiving statin therapy [107]. However, as a matter of best clinical practice, all individuals on a statin who have major risk factors for T2DM need to be informed about the increased diabetic risk, monitored regularly for hyperglycemia, and advised to lose weight and participate in regular physical exercise to mitigate the potential emergence of T2DM.
Guidance for Clinical Practice
Expert recommendations for the use of statins in people with or at risk of developing T2DM have recently been published [8••]. Briefly, overall CV risk using a validated score (e.g. Framingham Risk Calculator [FRC] or Systematic COronary Risk Evaluation [SCORE]) should be assessed in all patients prior to starting statin therapy [108, 109•]. All with high CV risk considered for statins should have their assessment of the risk of developing T2DM (e.g. Finnish Diabetes Risk Score [FINDRISC]) [109•]. The possible consequences of statin treatment should be discussed with the patient who should be strongly encouraged to reduce both their CV and T2DM risk through lifestyle changes. For patients with low to moderate T2DM risk (e.g. FINDRISC score <15 or equivalent using other T2DM risk scores), measurement of HbA1c and/or fasting glucose levels are not required. In those with CVD and/or high CV risk, and high to very high T2DM risk score, HbA1c or fasting glucose should be measured pre-statin and re-assessed 3 months after statin initiation. Patients with a high T2DM risk score and/or fasting glucose >5.6 mmol/L (>100 mg/dL), or HbA1c 6.0–6.4 %, should be given intensive lifestyle advice to reduce the risk of conversion from pre-diabetes to T2DM. Patients who develop T2DM during statin therapy should be treated according to national guidelines on diabetes management [2].
Conclusions
Statins have been demonstrated to be beneficial for primary and secondary prevention of atherosclerotic cardiovascular events in adults. However, post hoc analyses of major statin trials indicate a definite but small increase in the development of diabetes with statin therapy, especially in those with pre-existing diabetic risk factors, such as older age, obesity, impaired glycemic control and insulin resistance. Results from recent risk/benefit analyses support the continual use of statin indicating that the absolute benefit of treatment far outweighs the diabetes risk with statin therapy. Several hypotheses have been proposed to underlie the association between statin use and increased risk of diabetes, but more research is required to substantiate the mechanisms. Although adiponectin and CoQ10 appear to be key players in the regulation of glucose metabolism, more solid evidence or research data is required to support their roles in linking the association between statin and incidence of diabetes. Further studies are required to investigate whether the diabetogenic effects differ between statins and whether some patient groups (e.g. elderly and obese subjects) are at a higher risk of developing T2DM with statin than others. Whether combination therapy of statin and CoQ10 will prevent increased risk of diabetes, particularly in the elderly and those with pre-existing diabetic risks, merits further investigation. All patients should be assessed for CV and diabetic risks prior to starting statin therapy. They should be educated about the risk of statin therapy and encouraged to reduce diabetic risk through lifestyle changes. Patients who develop T2DM during statin therapy should be managed according to national guidelines.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major important
International Diabetes Federation (IDF). IDF diabetes atlas. Epidemiology and morbidity. [http://www.idf.org/].
American Diabetes Association. Standards of medical care in diabetes—2014. Diabetes Care. 2014;37(Supplement 1):S14–80.
Collins R, Armitage J, Parish S, Sleigh P, Peto R. MRC/BHF Heart Protection Study of cholesterol-lowering with simvastatin in 5963 people with diabetes: a randomised placebo-controlled trial. Lancet. 2003;361:2005–16.
Stone NJ, Robinson JG, Lichtenstein AH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129(25 suppl 2):S1–S45. Very recent guideline on the treatment of lipid disorders.
Ridker PM, Danielson E, Fonseca FA, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. 2008;359:2195. This study supports the notion that statin therapy increases the risk of incident diabetes.
Sattar N, Preiss D, Murray HM, et al. Statins and risk of incident diabetes: a collaborative meta-analysis of randomised statin trials. Lancet. 2010;375:735–42.
Shah RV, Goldfine AB. Statins and risk of new-onset diabetes mellitus. Circulation. 2012;126:e282–e4.
Sattar NA, Ginsberg H, Ray K, et al. The use of statins in people at risk of developing diabetes mellitus: evidence and guidance for clinical practice. Atheroscler Suppl. 2014;15:1–15. Very recent and comprehensive review on the diabetogenic effect of statin and guidance for clinical practice.
Bell DS, Dinicolantonio J, O’Keefe J. Is statin-induced diabetes clinically relevant? A comprehensive review of the literature. Diabetes Obes Metab. 2014;16:689–94.
Cholesterol Treatment Trialists’ (CTT) Collaborators. Efficacy of cholesterol-lowering therapy in 18 686 people with diabetes in 14 randomised trials of statins: a meta-analysis. Lancet. 2008;371:117–25.
Ray KK, Seshasai SRK, Erqou S, et al. Statins and all-cause mortality in high-risk primary prevention: a meta-analysis of 11 randomized controlled trials involving 65 229 participants. Arch Intern Med. 2010;170:1024–31.
Baigent C, Blackwell L, Emberson J, et al. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet. 2010;376:1670–81.
Freeman DJ, Norrie J, Sattar N, et al. Pravastatin and the development of diabetes mellitus evidence for a protective treatment effect in the West of Scotland Coronary Prevention Study. Circulation. 2001;103:357–62.
Shepherd J, Blauw GJ, Murphy MB, et al. Pravastatin in elderly individuals at risk of vascular disease (PROSPER): a randomised controlled trial. Lancet. 2002;360:1623–30.
The Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group. Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. N Engl J Med. 1998;339:1349–57.
Sever PS, Dahlöf B, Poulter NR, et al. Prevention of coronary and stroke events with atorvastatin in hypertensive patients who have average or lower-than-average cholesterol concentrations, in the Anglo-Scandinavian Cardiac Outcomes Trial-Lipid Lowering Arm (ASCOT-LLA): a multicentre randomised controlled tria. Lancet. 2003;361:1149–58.
Waters DD, Ho JE, DeMicco DA, et al. Predictors of new-onset diabetes in patients treated with atorvastatin: results from 3 large randomized clinical trials. J Am Coll Cardiol. 2011;57:1535–45.
Collins R, Armitage J, Parish S, Sleight P, Peto R. MRC/BHF heart protection study of cholesterol lowering with simvastatin in 20 536 high-risk individuals: a randomized placebo-controlled trial. Lancet. 2002;360:7–22.
Ridker PM, Pradhan A, MacFadyen JG, Libby P, Glynn RJ. Cardiovascular benefits and diabetes risks of statin therapy in primary prevention: an analysis from the JUPITER trial. Lancet. 2012;380:565–71. This study supports the notion that the absolute benefit of treatment outweighs the diabetic risk with statin therapy statin therapy increase the risk of incident diabetes.
Rajpathak SN, Kumbhani DJ, Crandall J, Barzilai N, Alderman M, Ridker PM. Statin therapy and risk of developing type 2 diabetes: a meta-analysis. Diabetes Care. 2009;32:1924–9.
Coleman CI, Reinhart K, Kluger J, White MC. The effect of statins on the development of new-onset type 2 diabetes: a meta-analysis of randomized controlled trials. Curr Med Res Opin. 2008;24:1359–62.
Cannon CP, Steinberg BA, Murphy SA, Mega JL, Braunwald E. Meta-analysis of cardiovascular outcomes trials comparing intensive versus moderate statin therapy. J Am Coll Cardiol. 2006;48:438–45.
Preiss D, Seshasai SRK, Welsh P, et al. Risk of incident diabetes with intensive-dose compared with moderate-dose statin therapy. JAMA. 2011;305:2556–64. A high impact meta-analysis showing that intense-dose statin therapy was associated with an increased risk of incident diabetes.
Dormuth CR, Filion KB, Paterson JM, et al. Higher potency statins and the risk of new diabetes: multicentre, observational study of administrative databases. BMJ. 2014;348:g3244.
Carter AA, Gomes T, Camacho X, Juurlink DN, Shah BR, Mamdani MM. Risk of incident diabetes among patients treated with statins: population based study. BMJ. 2013;346:f2610. A population based cohort study with time to event analyses to estimate the relation between use of different statins and incident diabetes.
Masana L. Pitavastatin in cardiometabolic disease: therapeutic profile. Cardiovasc Diabetol. 2013;12 Suppl 1:S2.
Teramoto T. Pitavastatin: clinical effects from the LIVES study. Atheroscler Suppl. 2011;12:285–8.
Kawai Y, Sato-Ishida R, Motoyama A, Kajinami K. Place of pitavastatin in the statin armamentarium: promising evidence for a role in diabetes mellitus. Drug Des Dev Ther. 2011;5:283–97.
Ishihara Y, Ohmori K, Mizukawa M, Ul Hasan A, Noma T, Kohno M. Beneficial direct adipotropic actions of pitavastatin in vitro and their manifestations in obese mice. Atherosclerosis. 2010;212:131–8.
Navab M, Reddy ST, Van Lenten BJ, Fogelman AM. HDL and cardiovascular disease: atherogenic and atheroprotective mechanisms. Nat Rev Cardiol. 2011;8:222–32.
Fryirs M, Barter PJ, Rye K-A. Cholesterol metabolism and pancreatic β-cell function. Curr Opin Lipidol. 2009;20:159–64.
Sampson UK, Linton MF, Fazio S. Are statins diabetogenic? Curr Opin Cardiol. 2011;26:342–7. A comprehensive review on the mechanisms of action by which statins increase the risk of developing diabetes.
Sattar N, Taskinen M-R. Statins are diabetogenic—myth or reality? Atheroscler Suppl. 2012;13:1–10.
Yada T, Nakata M, Shiraishi T, Kakei M. Inhibition by simvastatin, but not pravastatin, of glucose‐induced cytosolic Ca2+ signalling and insulin secretion due to blockade of L‐type Ca2+ channels in rat islet β‐cells. Br J Pharmacol. 1999;126:1205–13.
Mcguire TF, Xu X-Q, Corey SJ, Romero GG, Sebti SM. Lovastatin disrupts early events in insulin signaling: a potential mechanism of lovastatins anti-mitogenic activity. Biochem Biophys Res Commun. 1994;204:399–406.
Chamberlain LH. Inhibition of isoprenoid biosynthesis causes insulin resistance in 3T3-L1 adipocytes. FEBS Lett. 2001;507:357–61.
Nakata M, Nagasaka S, Kusaka I, Matsuoka H, Ishibashi S, Yada T. Effects of statins on the adipocyte maturation and expression of glucose transporter 4 (SLC2A4): implications in glycaemic control. Diabetologia. 2006;49:1881–92.
Hao M, Head WS, Gunawardana SC, Hasty AH, Piston DW. Direct effect of cholesterol on insulin secretion. A novel mechanism for pancreatic β-cell dysfunction. Diabetes. 2007;56(9):2328–38.
Donath MY, Böni-Schnetzler M, Ellingsgaard H, Ehses JA. Islet inflammation impairs the pancreatic β-cell in type 2 diabetes. Physiology. 2009;24:325–31.
Nakata M, Uto N, Maruyama I, Yada T. Nitric oxide induces apoptosis via Ca2+−dependent processes in the pancreatic β-cell line MIN6. Cell Struct Funct. 1999;24:451–5.
Laufs U, La Fata V, Plutzky J, Liao JK. Upregulation of endothelial nitric oxide synthase by HMG CoA reductase inhibitors. Circulation. 1998;97:1129–35.
Sirvent P, Fabre O, Bordenave S, et al. Muscle mitochondrial metabolism and calcium signaling impairment in patients treated with statins. Toxicol Appl Pharmacol. 2012;259:263–8.
Swerdlow DI, Preiss D, Kuchenbaecker KB, et al. HMG-coenzyme a reductase inhibition, type 2 diabetes, and bodyweight: evidence from genetic analysis and randomized trials. Lancet. 2014. doi:10.1016/S0140-6736(14)61183-1.
Frayling TM. Statins and type 2 diabetes: genetic studies on target. Lancet. 2014. doi:10.1016/S0140-6736(14)61639-1.
Takaguri A, Satoh K, Itagaki M, Tokumitsu Y, Ichihara K. Effects of atorvastatin and pravastatin on signal transduction related to glucose uptake in 3T3L1 adipocytes. J Pharmacol Sci. 2008;107:80–9.
Koh KK, Quon MJ, Han SH, et al. Differential metabolic effects of pravastatin and simvastatin in hypercholesterolemic patients. Atherosclerosis. 2009;204:483–90.
Takagi T, Matsuda M, Abe M, et al. Effect of pravastatin on the development of diabetes and adiponectin production. Atherosclerosis. 2008;196:114–21.
Frayn K, Karpe F, Fielding B, Macdonald I, Coppack S. Integrative physiology of human adipose tissue. Int J Obes. 2003;27:875–88.
Pittas AG, Joseph NA, Greenberg AS. Adipocytokines and insulin resistance. J Clin Endocrinol Metab. 2004;89:447–52.
Matsuzawa Y, Funahashi T, Kihara S, Shimomura I. Adiponectin and metabolic syndrome. Arterioscler Thromb Vasc Biol. 2004;24:29–33.
Kishida K, Funahashi T, Shimomura I. Molecular mechanisms of diabetes and atherosclerosis: role of adiponectin. Endocr Metab Immune Disord Drug Targets. 2012;12:118–31. A comprehensive review on the role of adiponectin in diabetes and atherosclerosis.
Sattar N, Wannamethee G, Sarwar N, et al. Adiponectin and coronary heart disease. A prospective study and meta-analysis. Circulation. 2006;114:623–9.
Wu Z, Cheng Y, Aung LHH, Li B. Association between adiponectin concentrations and cardiovascular disease in diabetic patients: a systematic review and meta-analysis. PLoS ONE. 2013;8:e78485.
Farvid M, Ng T, Chan D, Barrett P, Watts G. Association of adiponectin and resistin with adipose tissue compartments, insulin resistance and dyslipidaemia. Diabetes Obes Metab. 2005;7:406–13.
Weyer C, Funahashi T, Tanaka S, et al. Hypoadiponectinemia in obesity and type 2 diabetes: close association with insulin resistance and hyperinsulinemia. J Clin Endocrinol Metab. 2001;86:930–5.
Bruun JM, Lihn AS, Verdich C, et al. Regulation of adiponectin by adipose tissue-derived cytokines: in vivo and in vitro investigations in humans. Am J Physiol Gastr L Physiol. 2003;285:E527–33.
Li S, Shin HJ, Ding EL, van Dam RM. Adiponectin levels and risk of type 2 diabetes: a systematic review and meta-analysis. JAMA. 2009;302:179–88.
Han L, Wu Q, Jiao M, et al. Associations between single-nucleotide polymorphisms (+45T>G, +276G>T, −11377C>G, −11391G>A) of adiponectin gene and type 2 diabetes mellitus: a systematic review and meta-analysis. Diabetologia. 2011;54:2303–14.
Dastani Z, Hivert M-F, Timpson N, et al. Novel loci for adiponectin levels and their influence on type 2 diabetes and metabolic traits: a multi-ethnic meta-analysis of 45,891 individuals. PLoS Genet. 2012;8:e1002607.
Mori Y, Otabe S, Dina C, et al. Genome-wide search for type 2 diabetes in Japanese affected sib-pairs confirms susceptibility genes on 3q, 15q, and 20q and identifies two new candidate loci on 7p and 11p. Diabetes. 2002;51:1247–55.
Maeda N, Shimomura I, Kishida K, et al. Diet-induced insulin resistance in mice lacking adiponectin/ACRP30. Nat Med. 2002;8:731–7.
Kim J-Y, van de Wall E, Laplante M, et al. Obesity-associated improvements in metabolic profile through expansion of adipose tissue. J Clin Invest. 2007;117:2621–37.
Yamauchi T, Kamon J, Waki H, et al. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat Med. 2001;7:941–6.
Stefan N, Vozarova B, Funahashi T, et al. Plasma adiponectin concentration is associated with skeletal muscle insulin receptor tyrosine phosphorylation, and low plasma concentration precedes a decrease in whole-body insulin sensitivity in humans. Diabetes. 2002;51:1884–8.
Kadowaki T, Yamauchi T, Kubota N, Hara K, Ueki K, Tobe K. Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J Clin Invest. 2006;116:1784–92.
Rabe K, Lehrke M, Parhofer KG, Broedl UC. Adipokines and insulin resistance. Mol Med. 2008;14:741–51.
Yoon MJ, Lee GY, Chung J-J, Ahn YH, Hong SH, Kim JB. Adiponectin increases fatty acid oxidation in skeletal muscle cells by sequential activation of AMP-activated protein kinase, p38 mitogen-activated protein kinase, and peroxisome proliferator–activated receptor α. Diabetes. 2006;55:2562–70.
Okamoto M, Ohara-Imaizumi M, Kubota N, et al. Adiponectin induces insulin secretion in vitro and in vivo at a low glucose concentration. Diabetologia. 2008;51:827–35.
Wijesekara N, Krishnamurthy M, Bhattacharjee A, Suhail A, Sweeney G, Wheeler MB. Adiponectin-induced ERK and Akt phosphorylation protects against pancreatic beta cell apoptosis and increases insulin gene expression and secretion. J Biol Chem. 2010;285:33623–31.
Qiao L, Kinney B, Yoo HS, Lee B, Schaack J, Shao J. Adiponectin increases skeletal muscle mitochondrial biogenesis by suppressing mitogen-activated protein kinase phosphatase-1. Diabetes. 2012;61:1463–70.
Tsatsanis C, Zacharioudaki V, Androulidaki A, et al. Adiponectin induces TNF-α and IL-6 in macrophages and promotes tolerance to itself and other pro-inflammatory stimuli. Biochem Biophys Res Commun. 2005;335:1254–63.
Argiles J, Lopez-Soriano J, Lopez-Soriano F. Cytokines and diabetes: the final step? Involvement of TNF-alpha in both type I and II diabetes mellitus. Horm Metab Res. 1994;26:447–9.
Miyagishima K, Hiramitsu S, Kato S, et al. Efficacy of atorvastatin therapy in ischaemic heart disease—effects on oxidized low-density lipoprotein and adiponectin. J Int Med Res. 2007;35:534–9.
Koh KK, Quon MJ, Han SH, et al. Additive beneficial effects of fenofibrate combined with atorvastatin in the treatment of combined hyperlipidemia. J Am Coll Cardiol. 2005;45:1649–53.
Anagnostis P, Selalmatzidou D, Polyzos SA, et al. Comparative effects of rosuvastatin and atorvastatin on glucose metabolism and adipokine levels in non-diabetic patients with dyslipidaemia: a prospective randomised open-label study. Int J Clin Pract. 2011;65:679–83.
Thongtang N, Ai M, Otokozawa S, et al. Effects of maximal atorvastatin and rosuvastatin treatment on markers of glucose homeostasis and inflammation. Am J Cardiol. 2011;107:387–92.
Koh KK, Quon MJ, Han SH, et al. Simvastatin improves flow-mediated dilation but reduces adiponectin levels and insulin sensitivity in hypercholesterolemic patients. Diabetes Care. 2008;31:776–82.
Forst T, Pfützner A, Lübben G, et al. Effect of simvastatin and/or pioglitazone on insulin resistance, insulin secretion, adiponectin, and proinsulin levels in nondiabetic patients at cardiovascular risk—the PIOSTAT study. Metabolism. 2007;56:491–6.
Devaraj S, Siegel D, Jialal I. Simvastatin (40 mg/day), adiponectin levels, and insulin sensitivity in subjects with the metabolic syndrome. Am J Cardiol. 2007;100:1397–9.
Sonmez A, Dogru T, Tasci I, et al. The effect of fluvastatin on plasma adiponectin levels in dyslipidaemia. Clin Endocrinol (Oxf). 2006;64:567–72.
Sugiyama S, Fukushima H, Kugiyama K, et al. Pravastatin improved glucose metabolism associated with increasing plasma adiponectin in patients with impaired glucose tolerance and coronary artery disease. Atherosclerosis. 2007;194:e43–51.
Yokoyama H, Saito S, Daitoku K, et al. Effects of pravastatin and rosuvastatin on the generation of adiponectin in the visceral adipose tissue in patients with coronary artery disease. Fundam Clin Pharmacol. 2011;25:378–87.
Qu HY, Xiao YW, Jiang GH, Wang ZY, Zhang Y, Zhang M. Effect of atorvastatin versus rosuvastatin on levels of serum lipids, inflammatory markers and adiponectin in patients with hypercholesterolemia. Pharm Res. 2009;26:958–64.
Kim W, Hong MJ, Woo JS, Kang WY, Hwang SH, Kim W. Rosuvastatin does not affect fasting glucose, insulin resistance, or adiponectin in patients with mild to moderate hypertension. Chonnam Med J. 2013;49:31–7.
Baker WL, Talati R, White CM, Coleman CI. Differing effect of statins on insulin sensitivity in non-diabetics: a systematic review and meta-analysis. Diabetes Res Clin Pract. 2010;87:98–107.
Kostapanos M, Milionis H, Agouridis AD, Rizos C, Elisaf M. Rosuvastatin treatment is associated with an increase in insulin resistance in hyperlipidaemic patients with impaired fasting glucose. Int J Clin Pract. 2009;63:1308–13.
Mita T, Nakayama S, Abe H, et al. Comparison of effects of pitavastatin and atorvastatin on glucose metabolism in type 2 diabetic patients with hypercholesterolemia. J Diabetes Investig. 2013;4:297–303.
Nomura S, Taniura T, Shouzu A, et al. Effects of pitavastatin on plasminogen activator inhibitor-1 in hyperlipidemic patients. Int J Gen Med. 2012;5:535–40.
Inami N, Nomura S, Shouzu A, et al. Effects of pitavastatin on adiponectin in patients with hyperlipidemia. Pathophysiol Haemost Thromb. 2007;36:1–8.
Littarru GP, Tiano L. Bioenergetic and antioxidant properties of coenzyme Q10: recent developments. Mol Biotechnol. 2007;37:31–7.
Chew G, Watts G. Coenzyme Q10 and diabetic endotheliopathy: oxidative stress and the ‘recoupling hypothesis’. QJM. 2004;97:537–48.
Molyneux SL, Young JM, Florkowski CM, Lever M, George PM. Coenzyme Q10: is there a clinical role and a case for measurement? Clin Biochem Rev. 2008;29:71–82.
Ates O, Bilen H, Keles S, et al. Plasma coenzyme Q10 levels in type 2 diabetic patients with retinopathy. Int J Ophthalmol. 2013;6:675–9.
McCarty M. Can correction of sub-optimal coenzyme Q status improve b-cell function in type II diabetics? Med Hypotheses. 1999;52:397–400.
Littarru GP, Langsjoen P. Coenzyme Q10 and statins: biochemical and clinical implications. Mitochondrion. 2007;7:S168–74.
Stocker R, Pollicino C, Gay CA, et al. Neither plasma coenzyme Q10 concentration, nor its decline during pravastatin therapy, is linked to recurrent cardiovascular disease events: a prospective case–control study from the LIPID study. Atherosclerosis. 2006;187:198–204.
Mabuchi H, Higashikata T, Kawashiri M, et al. Reduction of serum ubiquinol-10 and ubiquinone-10 levels by atorvastatin in hypercholesterolemic patients. J Atheroscler Thromb. 2005;12:111–9.
Maechler P, Li N, Casimir M, Vetterli L, Frigerio F, Brun T. Role of mitochondria in β-cell function and dysfunction. Adv Exp Med Biol. 2010;654:193–216.
Draeger A, Monastyrskaya K, Mohaupt M, et al. Statin therapy induces ultrastructural damage in skeletal muscle in patients without myalgia. J Physiol. 2006;210:94–102.
Khamseh ME, Malek M, Aghili R, Emami Z. Sarcopenia and diabetes: pathogenesis and consequences. Br J Diabetes Vasc Dis. 2011;11:230–4.
Hodgson J, Watts G, Playford D, Burke V, Croft K. Coenzyme Q10 improves blood pressure and glycaemic control: a controlled trial in subjects with type 2 diabetes. Eur J Clin Nutr. 2002;56:1137–42.
Shimura Y, Hogimoto S. Significance of coenzyme Q10 on the treatment of diabetes mellitus. Jpn J Clin Exp Med. 1981;58:1349–52.
Singh R, Niaz M, Rastogi S, Shukla P, Thakur A. Effect of hydrosoluble coenzyme Q10 on blood pressures and insulin resistance in hypertensive patients with coronary artery disease. J Hum Hypertens. 1999;13:203–8.
Schroeder MM, Belloto Jr RJ, Hudson RA, McInerney MF. Effects of antioxidants coenzyme Q10 and lipoic acid on interleukin-1β-mediated inhibition of glucose-stimulated insulin release from cultured mouse pancreatic islets. Immunopharmacol Immunotoxicol. 2005;27:109–22.
Amin MM, Asaad GF, Salam RMA, El-Abhar HS, Arbid MS. Novel CoQ10 antidiabetic mechanisms underlie its positive effect: modulation of insulin and adiponectine receptors, tyrosine kinase, PI3K, glucose transporters, sRAGE and visfatin in insulin resistant/diabetic rats. PLoS ONE. 2014;9:e89169.
Ganesan S, Ito MK. Coenzyme Q10 ameliorates the reduction in GLUT4 transporter expression induced by simvastatin in 3T3-L1 adipocytes. Metab Syndr Relat Disord. 2013;11:251–5.
Zhou Y, Yuan Y, Cai R-R, et al. Statin therapy on glycaemic control in type 2 diabetes: a meta-analysis. Expert Opin Pharmacother. 2013;14:1575–84.
Reiner Z, Catapano AL, De Backer G, et al. ESC/EAS guidelines for the management of dyslipidaemias: the Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and the European Atherosclerosis Society (EAS). Eur Heart J. 2011;32:1769–818.
Rydén L, Grant PJ, Anker SD, et al. ESC guidelines on diabetes, pre-diabetes, and cardiovascular diseases developed in collaboration with the EASD: the Task Force on diabetes, pre-diabetes, and cardiovascular diseases of the European Society of Cardiology (ESC) and developed in collaboration with the European Association for the Study of Diabetes (EASD). Eur Heart J. 2013;34:3035–87. A recent guideline on the management of diabetes, pre-diabetes and cardiovascular disease.
Compliance with Ethics Guidelines
Conflict of Interest
Dick C. Chan, Jing Pang and Gerald F. Watts declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Statin Drugs
Rights and permissions
About this article

Cite this article
Chan, D.C., Pang, J. & Watts, G.F. Pathogenesis and Management of the Diabetogenic Effect of Statins: a Role for Adiponectin and Coenzyme Q10?. Curr Atheroscler Rep 17, 472 (2015). https://doi.org/10.1007/s11883-014-0472-7
Published:
DOI: https://doi.org/10.1007/s11883-014-0472-7