
Abstract
Over the past decade, there have been significant advances in our understanding of the immunopathogenesis and pharmacogenomics of severe immunologically-mediated adverse drug reactions. Such T-cell-mediated adverse drug reactions such as Stevens-Johnson syndrome/toxic epidermal necrolysis (SJS/TEN), drug-induced liver disease (DILI) and other drug hypersensitivity syndromes have more recently been shown to be mediated through interactions with various class I and II HLA alleles. Key examples have included the associations of HLA-B*15:02 and carbamazepine induced SJS/TEN in Southeast Asian populations and HLA-B*57:01 and abacavir hypersensitivity. HLA-B*57:01 screening to prevent abacavir hypersensitivity exemplifies a successful translational roadmap from pharmacogenomic discovery through to widespread clinical implementation. Ultimately, our increased understanding of the interaction between drugs and the MHC could be used to inform drug design and drive pre-clinical toxicity programs to improve drug safety.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Adverse drug reactions (ADRs) are common causes of patient morbidity and mortality and costs to the healthcare system [1–4]. Type A ADRs comprise the majority of reported drug reactions and are predictable based on the dose and pharmacologic activity of the drug. Type B ADRs account for less than 20 % of reported drug reactions and include immunologically-mediated drug reactions. More recently, some of these reactions have been shown to be immunogenetically driven [5]. Delayed hypersensitivity reactions (DHR) are a subgroup of type B ADRs that are also classified according to the Gell-Coombs classification of immunologically-mediated drug reactions as type IVa-d based on the effector cell and the antigen involved. These consist of a variety of phenotypes encompassing milder skin reactions, such as exanthems, to more severe drug hypersensitivity syndromes (DHS). DHS also include severe cutaneous adverse reactions (SCARs) such as Stevens-Johnson syndrome/toxic epidermal necrolysis (SJS/TEN), drug-induced hypersensitivity syndrome (DIHS) also known as drug reaction with eosinophilia and systemic symptoms (DRESS) and acute generalized exanthematous pustulosis (AGEP). DHS may also occur where involvement is limited to one organ such as drug-induced liver disease (DILI). Onset of symptoms typically occurs a few days to 8 weeks after exposure to the antigen or drug.
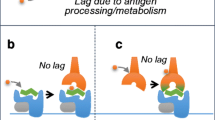
DHS are driven by the inappropriate activation of T-cells. The primary proteins involved in these responses are now known to be human leukocyte antigen (HLA) molecules encoded within the major histocompatibility complex (MHC). MHC class I molecules are encoded by the genes HLA-A, HLA-B and HLA-C; whereas MHC class II molecules are encoded by HLA-DP, HLA-DQ and HLA-DR. Since HLA molecules are highly polymorphic, a systematic nomenclature has been developed. The digits before the first colon describe the allele group and the two digits following the colon signify specific subtypes of the locus. These proteins are responsible for the presentation of peptides to T-cells [5]. More recently, several associations have been characterized between certain HLA molecules and drug hypersensitivity reactions. The classic model proposed to explain these T-cell mediated reactions was the hapten/prohapten hypothesis proposing that drugs or their reactive metabolites covalently bind to host proteins. Alternative models were proposed including the p-i model that proposes drugs bind non-covalently to the T-cell receptor (TCR) or MHC protein, leading to direct activation of T-cells without the presence of a peptide. This theory is supported by evidence that some drugs are able to elicit T-cell activation without proper antigen processing by antigen presenting cells (APCs) [6–8]. Thus, the p-i model provided a possible explanation for how T-cell activation could occur without prior exposure to a given drug [9]. In the altered peptide repertoire model, drugs non-covalently occupy sites within the peptide-binding groove of MHC proteins. This occupation leads to alteration of the specificities of MHC-peptide binding and results in presentation of self-peptide antigens that are distinct from those normally bound by the unaltered MHC protein [5, 10, 11••]. Recent studies have demonstrated that this model may be central to the pathogenesis of phenotypically distinct hypersensitivity reactions to medications such as abacavir hypersensitivity syndrome and carbamazepine SJS/TEN [11••, 12••, 13••, 14••] (Fig. 1).

The altered peptide repertoire model of abacavir hypersensitivity. Crystal structure of the abacavir-MHC-peptide complex shown to resolution of 0.2 nm (nanometers). a HLA-B*57:01 is shown in gray, peptide V is shown in cyan, and abacavir is shown as a multicolored structure (spheres with orange for carbon, blue for nitrogen, and red for oxygen). b Abacavir binding leads to altered peptide repertoire by changing the conformation of the peptide backbone in the main chain of HLA-B*57:01. Binding of peptide is shown in the absence of abacavir (yellow) and associated with abacavir and HLA-B*57:01 (cyan). c H-bound interactions (black dashes) are shown between abacavir, peptide, and HLA-B*57:01. Specific residues differentiating for abacavir-sensitive HLA-B*57:01 from abacavir-insensitive HLA-B*57:03 are shown in magenta (carbon), blue (nitrogen), and red (oxygen). d Experimental electron density showing abacavir in a Fo-Fc difference map with blue mesh showing the final 2Fo-Fc electron density map of abacavir in the antigen-binding cleft of HLA-B*57:01. H-bond interactions between abacavir and HLA-B*57:01 are shown in yellow. (Adapted from Ostrov et al. [14••])
Over the past decade, there have been numerous publications that have described associations between HLA alleles and DHS offering promise for identification for patients at risk and prevention of DHS that were previously believed to be unpredictable (Table 1; Fig. 2). The best characterized of these HLA-mediated DHS are discussed below.

Key events in the immunogenetics of drug hypersensitivity. Over the past decade, there has been increasing recognition of the key involvement of the major histocompatibility complex in different phenotypes of immunologically-mediated drug reactions. The ability to make these associations in the 1980s was hampered by the limitation of serologically-based HLA typing techniques. With the ability of high resolution 4-digit typing, attempts to standardize clinical phenotyping, and international collaboration, it should be possible to define HLA associations within even rare forms of DHS
Abacavir
The studies of abacavir-induced hypersensitivity (AHS) have created a roadmap for translational pharmacogenomics. Abacavir is a nucleoside reverse-transcriptase inhibitor used in combination antiretroviral treatment for HIV-1 infection. This syndrome was apparent in the pre-marketing phase of drug development of abacavir where an estimated 5–8 % of patients developed AHS characterized by fever, malaise, gastrointestinal symptoms and organ involvement within the first 2–6 weeks of therapy [15]. Unlike DHS related to other drugs, rash was found to be a late feature of AHS and absent in up to 30 % of these patients. Treatment included immediate cessation of abacavir and supportive care. Reports of severe disease including hypotension and even death with repeat ingestion or prolonged dosing led to a stringent pharmacovigilance program surrounding the use of abacavir to encourage early discontinuation in those with potential AHS and to prevent re-exposure in those who had experience a syndrome compatible with AHS [16]. In 2001, during the premarketing phase of abacavir, a study was published that showed a higher frequency of AHS in Caucasians when compared to black and Asian populations [17]. Further studies, published in 2002, demonstrated an association between AHS and the MHC class I allele, HLA-B*57:01 [18, 19]. Due to the higher carriage rate of HLA-B*57:01 in populations of predominantly white race when compared to black individuals, initial studies found a particularly low sensitivity of HLA-B*57:01 for clinically suspected AHS in non-white populations [18, 20]. However, this perceived lack of predictive value across all races was later found to be due to the high rates of false-positive clinical diagnosis of AHS which was particularly problematic in non-white races with a lower prevalence of HLA-B*57:01. Abacavir patch testing was found to be highly specific for the identification of those with true immunologically-mediated AHS and was used in two clinical trials that were performed to establish the clinical utility of pharmacogenetic testing for HLA-B*57:01 [21, 22]. In the PREDICT-1 trial, nearly 2,000 predominantly European Caucasian HIV-positive patients, were randomized to undergo real-time HLA-B*57:01 screening with exclusion of abacavir therapy for HLA-B*57:01-positive patients or abacavir initiation followed by retrospective HLA-B*57:01 genotyping. Clinical and patch test diagnosis of AHS were used as co-primary endpoints. Results revealed a 100 % negative predictive value of HLA-B*57:01 as a screening test for the prevention of abacavir hypersensitivity [22]. The results of the PREDICT-1 trial were further supported by the SHAPE trial; a case-control study of black and white patients that revealed a 100 % negative predictive value of HLA-B*57:01 for AHS across black and white populations and supported the generalizability of HLA-B*57:01 screening across different ethnicities [21]. Additional studies have shown that HLA-B*57:01 is cost-effective and has dramatically reduced the incidence of AHS [23, 24]. HLA-B*57:01 screening prior to abacavir treatment has been widely implemented in routine clinical practice and is part of US FDA and international HIV treatment guidelines [25].
Recent studies have provided new insight into the molecular pathogenesis underlying AHS. Activated cytotoxic CD8+ T-cells have been shown to be the main driving factor in AHS. Initially, activation of these T-cells were thought to be dependent upon processing and presentation on MHC Class-I molecules via the drug/hapten theory [26]. More recently published studies provide evidence for an “altered peptide repertoire” model in which abacavir undergoes a metabolism-independent, direct and non-covalent interaction with the HLA-B*57:01 binding cleft to form a new antigenic structure capable of eliciting a T-cell response [11••, 13••, 14••, 27••]. In 2012, Adam and colleagues produced abacavir-specific T-cell clones from HLA-B*57:01-positive individuals and showed that proteasome inhibition did not prevent T-cell reactivity to abacavir, providing evidence that T-cell stimulation does not require covalent bonds to produce new antigenic determinants [27••]. Moreover, activation of T-cells have been shown to be dependent on the TCR avidity, drug concentration, and level of HLA-B*57:01 molecules expressed on APCs [14••, 27••]. Additional studies have provided evidence that specific residues in the peptide binding cleft are required to elicit T-cell responses [26]. It has been shown that peptides eluted from abacavir-treated APCs expressing HLA-B*57:01 in cell culture models include many neo-epitopes [11••, 13••, 14••]. For example, Illing et al. demonstrated that 20–25 % of the recovered peptides from abacavir-treated HLA-B*57:01 APCs were distinct from those isolated from untreated cells [13••]. These novel peptides were shown to lack typical carboxyl terminal aromatic hydrophobic amino acids and instead contained predominantly hydrophobic aliphatic amino acids such as isoleucine, leucine or valine residues. These differences can be explained by a change in the shape of the antigen-binding cleft from abacavir binding within the floor of the HLA-B*57:01 F pocket peptide-binding groove [11••, 13••, 14••]. Further support for the altered peptide repertoire model in AHS has been recently provided by two structural analyses of the abacavir-peptide-HLA-B*57:01 complex [13••, 14••]. In each of these studies, abacavir association with the HLA-B*57:01 molecule was shown to alter the chemistry and topography of the peptide binding groove, resulting in a neo-antigen capable of eliciting an allo-reactive T-cell response [13••, 14••].
Carbamazepine
Carbamazepine is an aromatic amine anticonvulsant used to treat bipolar disorder, epilepsy, and trigeminal neuralgia. Carbamazepine has been associated with maculopapular eruption, DHIS/DRESS, and less commonly SJS/TEN that most typically occurs 2–8 weeks following drug initiation [28]. SJS/TEN is estimated to occur in 1–6 out of every 10,000 individuals receiving carbamazepine but has been previously noted to occur tenfold more commonly in those of Southeast Asian descent [5, 29•, 30]. In 2004, Chung et al. showed that 100 % of Han Chinese patients with carbamazepine-associated SJS/TEN carried the HLA-B*15:02 allele as compared to only 3 % in carbamazepine-tolerant controls and 8.6 % of the general population [30]. Subsequent studies have supported the association between the HLA-B*15:02 and carbamazepine-induced SJS/TEN in Chinese, Thai, Malaysian and Indian populations [31–36]. An observational study examined the utility of HLA-B*15:02 screening in 4,855 Han Chinese patients and subsequent avoidance of carbamazepine for those who were positive. Although rashes and other hypersensitivity syndromes occurred, there were no cases of carbamazepine-associated SJS/TEN in this group compared to an incidence of 0.23 % (i.e. 10 cases) expected based on historical data [37]. Approximately 3 % of patients carrying the HLA-B*15:02 allele will develop carbamazepine associated SJS/TEN. Ko et al. characterized the TCR repertoire of patients with carbamazepine-associated SJS/TEN and showed that VB-11-ISGSY, a dominant TCR clonotype, was present in 84 % of HLA-B*15:02 positive patients with carbamazepine-associated SJS/TEN and only 14 % of carbamazepine-naïve controls. Additionally, this clonotype was absent in all carbamazepine-tolerant controls, including two individuals who were HLA-B*15:02 carriers. Cell culture experiments on T-cells from carbamazepine-naïve subjects revealed a cytotoxic phenotype following stimulation with carbamazepine in those who were both HLA-B*15:02- and VB-11-ISGSY-positive. Moreover, addition of a VB-11-ISGSY-specific antibody blocked stimulation following carbamazepine exposure [38•]. These studies imply that the presence of both HLA-B*15:02 and a specific TCR clonotype are required for the development of carbamazepine-induced SJS/TEN. HLA-A*31:01 which is present in approximately 7 % of Han Chinese, 7–12 % of Japanese, 5 % of Koreans, and 2–5 % in northern European populations [allelefrequencies.net] has more recently been associated with DRESS/DIHS in Taiwanese, Japanese and European populations [29•, 39, 40, 41•, 42]. Some but not all studies have also suggested an association between HLA-A*31:01 and CBZ SJS/TEN in predominantly European populations [29•, 43].
Multiple in vitro studies have shed light onto the molecular mechanisms underlying carbamazepine-associated ADRs. In 2012, Wei and colleagues published data that carbamazepine presentation to T-cells in the context of the HLA-B*15:02 protein is independent of processing but does require peptide binding to stabilize the class I MHC complex on the cell surface [12••]. Additionally, APCs expressing other members of the HLA-B75 family, which includes HLA-B*15:02, showed T-cell activation in the context of carbamazepine presentation [12••]. Modifications of the carbamazepine ring structure lead to alteration in HLA-B*15:02 binding and T-cell response. Furthermore, mutagenesis was used to show that the region of carbamazepine association with HLA-B*15:02 maps to the B pocket of the peptide binding groove with a contact at the Arg62 residue, a conserved amino acid among B75 serotypes [12••]. An additional study by Illing et al. demonstrated that 15 % of isolated peptides eluted from carbamazepine-treated APCs expressing HLA-B*15:02 were distinct from those obtained in untreated cells [13••]. These findings support the altered peptide repertoire model as a feasible explanation for carbamazepine-induced SJS/TEN.
Allopurinol
Allopurinol is a xanthine oxidase inhibitor that has been used primarily to treat hyperuricemia and its complications, including chronic gout, since the early 1960s [44, 45]. Allopurinol hypersensitivity is characterized by a variety of cutaneous manifestations from benign maculopapular rashes to more severe forms, including SJS/TEN, and DHIS/DRESS [46]. The incidence of true allopurinol hypersensitivity is rare, with estimates to be approximately 0.1 % [47]. In 1989, a serological study revealed associations of the HLA haplotype AW33-BW58 with cutaneous allopurinol hypersensitivity in Southern Chinese [48]. In 2005, a case-controlled study revealed HLA-B*58:01 carriage in 100 % of allopurinol-induced SJS/TEN/DIHS/DRESS compared to 15 % of allopurinol-tolerant patients and 20 % of healthy controls [49]. Similar findings have been replicated in other ethnic groups including Japanese, Thai, and Korean populations [50–54]. In the first study of European patients, only 61 % of patients with allopurinol-induced SJS/TEN carried the HLA-B*58:01 genotype, likely reflecting the lower carriage rate of HLA-B*58:01 in European populations (1-6 %) versus 10–15 % in Southeast Asian populations and further work has also suggested that HLA alleles in addition to HLA-B*58:01 may be implicated [55–58].
The exact mechanism by which allopurinol causes different DHS phenotypes remains elusive, although it has been proposed that allopurinol or its metabolite oxypurinol may be small enough molecules to bind multiple sites on the HLA molecule, possibly leading to direct TCR binding and activation [59].
Nevirapine
Nevirapine is a non-nucleoside reverse transcriptase inhibitor used in combination with antiretroviral treatment for HIV-1 infection. DHS with varying clinical phenotypes occurs within the first 6 weeks in approximately 5 % of patients who initiate nevirapine. These reactions may be characterized by a combination of rash, fever and/or hepatitis, and nevirapine has also been associated with SJS/TEN [60, 61]. Uncovering the mechanisms underlying nevirapine-related hypersensitivity has been difficult due to the varying phenotypes and multiple genetic associations across differing ethnicities. Both class I and class II MHC molecules have been implicated in varying clinical syndromes of nevirapine HSRs. A Western Australian prospective cohort study associated carriage of the Class II allele HLA-DRB1*0101 with nevirapine-induced rash associated hepatitis and this effect was abrogated by a CD4 count of <25 % [62]. Later studies have also supported the association between DRB1 supertype alleles: HLA-DRB1*01:01 and HLA-DRB1*01:02 and nevirapine hepatitis DHS phenotypes [63••, 64, 65••]. MHC class I molecules have been primarily associated with nevirapine-associated cutaneous phenotype DHS [66–71]. HLA-B*35:05 and HLA-Cw*04 variants were shown to be involved in cutaneous-only nevirapine reactions in several populations including African-Americans, Thai, and Han Chinese [68–71]. More recently, HLA-C*04:01 has also been associated with nevirapine SJS/TEN in a case-control study of 117 HIV-positive patients in Malawi and 151 controls [72••].
Polymorphisms in the CYP2B6 gene, the main cytochrome p450 isoform involved in nevirapine metabolism, were shown to be associated with nevirapine-associated cutaneous drug reactions in blacks and whites supporting that the parent drug and concentration effects may be important in the immunopathogenesis of nevirapine DHS [63••]. Although much progress has been achieved in uncovering the immunologic basis of nevirapine hypersensitivity, many questions still remain regarding the complex interactions between immunologic, genetic, and metabolic factors.
Amoxicillin-Clavulanate
Amoxicillin-clavulanate (AC) is one of the most frequently prescribed treatments for bacterial infections. Amoxicillin-clavulanate DILI may present as cholestatic, hepatocellular, or a mixed pattern with an onset of symptoms ranging from a few days to several weeks after initial drug exposure, and rare cases require transplantation or are fatal [73].
Initial studies revealed an association between the HLA class II haplotype HLA-DRB1*15:01 and patients with AC-associated DILI [74–76]. Later, a genome wide association study of 201 patients demonstrated that AC-induced DILI is strongly associated with the SNP rs9274407, a SNP that is highly correlated with DRB1*15:01-DQB1*06:02 [77]. Additionally, there was an association between HLA-A*02:01 and AC-associated DILI in Northwestern European populations, but not in those of Spanish descent [77]. The clinical phenotypes of AC-associated DILI may be dependent on the ethnicity of the affected individual. For example, cases of AC-associated DILI in Spanish populations were characterized by hepatocellular injury, cholestatic damage, and a mixed pattern at almost equal frequencies [78]. In contrast, a cholestatic pattern of liver injury was observed in over 65 % of cases reported in French and Belgian populations [78]. A study performed by Stephens et al. evaluated 75 Spanish individuals with AC-associated DILI, of which 37 % had a hepatocellular toxicity pattern. Findings revealed an association between HLA-A*30:02 and hepatocellular liver injury, whereas the class II haplotype DRB1*15:01-DQB1*06:02 was significantly associated with a cholestatic or mixed pattern of DILI [79]. Research to date has been unable to establish a high positive or negative predictive value to make screening for AC-associated DILI useful. Due to the overall rare cases of AC-associated DILI and the large number of different HLA associations, HLA typing would not currently serve a purposeful role in predicting those at risk for DILI prior to initiation of AC at this time.
Flucloxacillin
Flucloxacillin is a narrow-spectrum anti-Staphylococcal semi-synthetic penicillin in wide use in Europe and Australia. DILI, typically manifests as cholestatic hepatitis beginning within 1–45 days after starting treatment, is a rare complication of flucloxacillin treatment occurring in 8.5/100,000 of new users [80, 81]. In 2009, Daly and colleagues published an association between HLA-B*57:01 and flucloxacillin-associated DILI. In this GWAS of 51 cases of flucloxacillin DILI and 64 flucloxacillin-tolerant controls, MHC genotyping revealed an association with those carrying the HLA-B*57:01 allele (OR = 80.6, p = 9.0 × 10–19) [82].
Genotyping in Clinical Practice
The application of genetic screening for immunologically mediated drug reactions would have highest utility for those diseases that are prevalent, severe, and associated with HLA markers that show a high positive and 100 % negative predictive value. A user friendly reporting strategy and/or decision support system that is readily available to the healthcare team is an additional advantage. Even if these pre-requisites are met, however, there are many barriers to the translation of a pharmacogenomic test into clinical practice. Independent from the direct translational opportunities of an individual pharmacogenomic test or strategy, pharmacogenomics has provided enormous insights into the mechanisms behind DHS. Currently, the FDA has recommended genetic screening prior to starting abacavir and in high-risk populations prior to starting carbamazepine. Although the product label for allopurinol in Taiwan suggests consideration for HLA-B*58:01 testing and avoidance of allopurinol in those who are positive, the Taiwan Bureau of National Health Insurance (BNHI) is currently not funding HLA-B*58:01 screening prior to allopurinol prescription. Since control of off-label use of carbamazepine and BNHI-funded HLA-B*15:02 screening in Taiwan, allopurinol has surpassed carbamazepine as the most common cause of SJS/TEN. No recommendation has been made by the US FDA for HLA-B*58:01 screening. With the use of genotyping as a screening tool for certain drugs, we are able to predict and prevent severe DHS from occurring. The story of abacavir has provided a “translational roadmap” and highlights the many barriers involved in the process of identifying HLA-associated drug hypersensitivity and implementing clinical screening for specific HLA alleles. In order for HLA genotyping to be useful in the clinical setting, many fundamental criteria must be met. First, the drug should be widely used and have good efficacy, convenience, and tolerability. If there are alternatives with equal efficacy, it would be less useful to define an underlying HLA association rather than choosing an alternative and safer agent. Next, the drug toxicity should be prevalent in the treatment population and associated with significant morbidity, mortality, and/or healthcare costs. Another prerequisite for successful integration of HLA pharmacogenetic testing into routine clinical care is an HLA allele that is strongly associated with an ADR along with a high (ideally 100 %) negative predictive value. The number needed to test to prevent a given DHS disease significantly impacts the cost-effectiveness and feasibility of an HLA screening test, and is dependent on the prevalence of the DHS, the prevalence of the HLA allele in the population of interest, and the positive predictive value (PPV) of the HLA allele for that specific DHS. For instance, even though AHS and flucloxacillin DILI are both strongly associated with HLA-B*57:01, only 30 individuals would need to be screened to prevent one case of AHS, whereas almost 14,000 patients would need to be screened for HLA-B*57:01 to prevent a single case of flucloxacillin DILI, making the latter impractical as a screening strategy [83]. In addition, for widespread clinical translation to occur, the pharmacogenomic association must be established in a large population with a diverse genetic background. For example, the FDA currently recommends HLA-B*15:02 testing only for Asians prior to initiation of carbamazepine therapy [84]. The ability to appropriately diagnose an ADR is another essential feature to the development of HLA screening for a particular drug. Imprecision in phenotyping or imprecise clinical classification may act as another barrier to pharmacogenomics discovery. AHS patch testing was a specific test that was useful to identify the extreme phenotype of true immunologically mediated AHS and overcome the problem of false positive clinical diagnosis [85–87]; however, sufficiently sensitive and specific tests to define a true DHS phenotype does not exist for most immunologically mediated drug reactions [88, 89].
An alert from the FDA was made in 2007 describing the increased risk of phenytoin- and fosphenytoin-associated SJS/TEN in individuals that carry the HLA-B*15:02 genotype; however, no formal recommendations have been made to implement screening prior to initiation of either drug. In a large, multicenter prospective study from Taiwan, 4,877 carbamazepine-naïve subjects were genotyped for HLA-B*15:02 and those who tested positive were advised to not take carbamazepine. During the study, none of the HLA-B*15:02 negative patients developed SJS/TEN, whereas approximately ten cases would have been predicted based on the incidence of SJS/TEN. Although cross-reactivity with other anticonvulsants was not determined in this clinical study, previous clinical and patch testing studies have suggested that clinical cross-reactivity does exist between carbamazepine SJS/TEN and DIHS/DRESS and structurally related aromatic amine anticonvulsants [90]. It would be prudent to have HLA-B*15:02-positive patients avoid phenytoin, fosphenytoin, oxcarbazepine, eslicarbazepine acetate, phenobarbital, and probably lamotrigine [91]. These studies have significantly improved care to those patients at risk; however, it is worth noting that not all carbamazepine-induced ADRs can be attributed to having the HLA-B*15:02 allele and clinical monitoring is recommended for all patients prescribed this medication [42].
The development of cost-effective laboratory testing that is reproducible, available and user-friendly has been critical in the expansion of genotyping amongst patients prior to the initiation of abacavir and carbamazepine. Although high resolution sequence-based HLA typing is the gold standard methodology for determining an individual HLA genotype, the required expertise and expense largely limit this to research and organ transplantation centers. Most commercial laboratories have developed rapid and cost-effective assays using PCR-based tests. The development of an allele specific quality assurance program and rapid molecular techniques that translated into short turn-around-times and cost-effective testing was key to the widespread clinical translation and feasibility of HLA-B*57:01 screening programs [92, 93]. Flow cytometric approaches using a B17 monoclonal antibody to detect any HLA-B*57 or HLA-B*58 allele prior to performing molecular testing has been proposed as a potentially cost-effective approach with a quick turn-around-time [94]. Identification of SNPs known to be in strong linkage disequilibrium with HLA-B*57:01 such as HCP5 have been performed; however, studies have revealed high but less than 100 % linkage disequilibrium with HLA-B*57:01, meaning that the latter is still the gold standard [95]. Rapid and cost-effective molecular techniques and laboratory approaches similar to those developed for HLA-B*57:01 screening for AHS have now been developed for other alleles such as HLA-B*15:02 HLA-B*58:01 and HLA-A*31:01 [43].
Additional Models for Drug Hypersensitivity
Viruses in the Human Herpesvirus (HHV) family (HHV-6/7, CMV, EBV) have more recently been shown to commonly reactivate during the course of DIHS/DRESS but not abacavir HSR or other SCARs [97]. In many cases, reactivation may occur 2–3 weeks following discontinuation of the implicated drug and symptoms replicate the original clinical manifestations such as fever, rash and/or hepatitis [97]. The altered peptide model has been pivotal to explain how drugs interact with the MHC to cause hypersensitivity and appears to be necessary, but not sufficient, in that it does not explain why a high proportion of patients carrying HLA risk alleles do not develop the reaction in question. For instance, while 55 % of HLA-B*57:01-positive patients treated with abacavir develop AHS, only 1–3 % of HLA-B*15:02-positive patients treated with carbamazepine develop SJS/TEN. Although the specific mechanism is not yet understood, memory T-cell responses to chronic persistent and prevalent viruses such as the HHV that cross-react with an endogenous peptide in certain individuals could explain why only a proportion of individuals carrying a specific risk HLA allele will develop a drug hypersensitivity syndrome.
Conclusions and Future Perspective
In the future, many more HLA-associated drug hypersensitivity reactions will be elucidated using pharmacogenomics, providing the potential for screening tests prior to initiation of certain drug therapies. Current barriers to the widespread translation of many of these tests into clinical practice include the lack of 100 % negative predictive value, lack of generalizability across diverse populations, and an impractically high number needed to be tested to prevent one case of a given DHS. Fortunately, the provision of rapid, cost-effective and quality assured HLA allele-specific molecular techniques have meant that laboratory programs and technology are now less of a barrier to translation. Although few of these pharmacogenomic discoveries will feasibly translate into routine clinical practice, further research into the immunogenetics and immunopathogenesis of DHS will improve our understanding of the molecular interactions between drugs and HLA molecules. The abacavir story has created a roadmap for the identification of these reactions and implementation of screening into clinical practice. Further work on abacavir has now provided a new immunopathogenetic model for how many drugs may non-covalently interact with the MHC to elicit an altered peptide repertoire and culminate in a DHS phenotype. In the future, increasing insights into the interactions between drugs and the MHC will be key to designing strategies to predict severe reactions before their use in man and to inform drug design to improve drug safety.
References
Papers of particular interest, published recently, have been highlighted as: •Of importance; ••Of major importance
Kongkaew C, Noyce PR, Ashcroft DM. Hospital admissions associated with adverse drug reactions: a systematic review of prospective observational studies. Ann Pharmacother. 2008;42(7):1017–25.
Khan LM. Comparative epidemiology of hospital-acquired adverse drug reactions in adults and children and their impact on cost and hospital stay - a systematic review. Eur J Clin Pharmacol. 2013.
Suh DC et al. Clinical and economic impact of adverse drug reactions in hospitalized patients. Ann Pharmacother. 2000;34(12):1373–9.
Pirmohamed M et al. Adverse drug reactions as cause of admission to hospital: prospective analysis of 18 820 patients. BMJ. 2004;329(7456):15–9.
Pavlos R, Mallal S, Phillips E. HLA and pharmacogenetics of drug hypersensitivity. Pharmacogenomics. 2012;13(11):1285–306.
Schnyder B et al. Direct, MHC-dependent presentation of the drug sulfamethoxazole to human alphabeta T cell clones. J Clin Invest. 1997;100(1):136–41.
Zanni MP et al. HLA-restricted, processing- and metabolism-independent pathway of drug recognition by human alpha beta T lymphocytes. J Clin Invest. 1998;102(8):1591–8.
Zanni MP et al. Allele-unrestricted presentation of lidocaine by HLA-DR molecules to specific alphabeta + T cell clones. Int Immunol. 1998;10(4):507–15.
Pichler WJ. Delayed drug hypersensitivity reactions. Ann Intern Med. 2003;139(8):683–93.
Bharadwaj M et al. Drug hypersensitivity and human leukocyte antigens of the major histocompatibility complex. Annu Rev Pharmacol Toxicol. 2012;52:401–31.
Norcross MA et al. Abacavir induces loading of novel self-peptides into HLA-B*57: 01: an autoimmune model for HLA-associated drug hypersensitivity. AIDS. 2012;26(11):F21–9. Using abacavir-treated HLA-B*57:01 B-cell lines, the authors demonstrated additional evidence that abacavir alters the quantity and quality of self-peptide loading into HLA-B*57:01.
Wei CY et al. Direct interaction between HLA-B and carbamazepine activates T cells in patients with Stevens-Johnson syndrome. J Allergy Clin Immunol. 2012;129(6):1562–9 e5. This article demonstrates a direct interaction of HLA molecules with drugs and provides a potential mechanism for HLA-associated drug hypersensitivity reactions. Supportive of altered peptide repertoire model for carbamazepine.
Illing PT et al. Immune self-reactivity triggered by drug-modified HLA-peptide repertoire. Nature. 2012;486(7404):554–8. Study supporting the altered peptide repertoire model for abacavir hypersensitivity. Abacavir associates with HLA-B*57:01 non-covalently and alters the shape and chemistry of the antigen-binding cleft. This leads to an altered repertoire of endogenous peptides binding HLA-B*57:01. The crystal structure of abacavir-MHC-peptide was resolved using an endogenous peptide. Preliminary evidence to support the altered peptide repertoire model for was also presented.
Ostrov DA et al. Drug hypersensitivity caused by alteration of the MHC-presented self-peptide repertoire. Proc Natl Acad Sci U S A. 2012;109(25):9959–64. Provided structural, biochemical and functional evidence for non-covalent interaction between abacavir and HLA-B*57:01 and the altered repertoire model for abacavir. A synthetic peptide was derived that binds to HLA-B*57:01 only in the presence of abacavir. The crystal structure of abacavir-MHC-peptide was resolved using this peptide.
Cutrell AG et al. Updated clinical risk factor analysis of suspected hypersensitivity reactions to abacavir. Ann Pharmacother. 2004;38(12):2171–2.
Shapiro M, Ward KM, Stern JJ. A near-fatal hypersensitivity reaction to abacavir: case report and literature review. AIDS Read. 2001;11(4):222–6.
Symonds W et al. Risk factor analysis of hypersensitivity reactions to abacavir. Clin Ther. 2002;24(4):565–73.
Mallal S et al. Association between presence of HLA-B*5701, HLA-DR7, and HLA-DQ3 and hypersensitivity to HIV-1 reverse-transcriptase inhibitor abacavir. Lancet. 2002;359(9308):727–32.
Hetherington S et al. Genetic variations in HLA-B region and hypersensitivity reactions to abacavir. Lancet. 2002;359(9312):1121–2.
Hughes AR et al. Association of genetic variations in HLA-B region with hypersensitivity to abacavir in some, but not all, populations. Pharmacogenomics. 2004;5(2):203–11.
Saag M et al. High sensitivity of human leukocyte antigen-b*5701 as a marker for immunologically confirmed abacavir hypersensitivity in white and black patients. Clin Infect Dis. 2008;46(7):1111–8.
Mallal S et al. HLA-B*5701 screening for hypersensitivity to abacavir. N Engl J Med. 2008;358(6):568–79.
Schackman BR et al. The cost-effectiveness of HLA-B*5701 genetic screening to guide initial antiretroviral therapy for HIV. AIDS. 2008;22(15):2025–33.
Rauch A et al. Prospective genetic screening decreases the incidence of abacavir hypersensitivity reactions in the Western Australian HIV cohort study. Clin Infect Dis. 2006;43(1):99–102.
Guo Y et al. Studies on abacavir-induced hypersensitivity reaction: a successful example of translation of pharmacogenetics to personalized medicine. Sci China Life Sci. 2013;56(2):119–24.
Chessman D et al. Human leukocyte antigen class I-restricted activation of CD8+ T cells provides the immunogenetic basis of a systemic drug hypersensitivity. Immunity. 2008;28(6):822–32.
Adam J et al. Avidity determines T-cell reactivity in abacavir hypersensitivity. Eur J Immunol. 2012;42(7):1706–16. Paper providing evidence that abacavir interacts with HLA-B*57:01 in a non-covalent and metabolism-independent fashion. Additionally, this study provided evidence that abacavir-reactive T-cell clones are dependent on the drug concentration, TCR avidity and level of HLA-B*57:01 molecules expressed on APCs.
Knowles SR, Dewhurst N, Shear N. Anticonvulsant hypersensitivity syndrome: an update. Expert Opin Drug Saf. 2012;11(5):767–78.
McCormack M et al. HLA-A*3101 and carbamazepine-induced hypersensitivity reactions in Europeans. N Engl J Med. 2011;364(12):1134–43. First GWAS for carbamazepine in Europeans that identified an association with HLA-A*31:01 and individuals of northern European ancestry with various phenotypes of carbamazepine-induced hypersensitivity reactions. Although this study suggested an association between HLA-A*31:01 and carbamazepine SJS/TEN in European populations, not all follow-up studies have supported this.
Chung WH et al. Medical genetics: a marker for Stevens-Johnson syndrome. Nature. 2004;428(6982):486.
Kulkantrakorn K et al. HLA-B*1502 strongly predicts carbamazepine-induced Stevens-Johnson syndrome and toxic epidermal necrolysis in Thai patients with neuropathic pain. Pain Pract. 2012;12(3):202–8.
Mehta TY et al. Association of HLA-B*1502 allele and carbamazepine-induced Stevens-Johnson syndrome among Indians. Indian J Dermatol Venereol Leprol. 2009;75(6):579–82.
Then SM et al. Frequency of the HLA-B*1502 allele contributing to carbamazepine-induced hypersensitivity reactions in a cohort of Malaysian epilepsy patients. Asian Pac J Allergy Immunol. 2011;29(3):290–3.
Man CB et al. Association between HLA-B*1502 allele and antiepileptic drug-induced cutaneous reactions in Han Chinese. Epilepsia. 2007;48(5):1015–8.
Wang Q et al. Association between HLA-B*1502 allele and carbamazepine-induced severe cutaneous adverse reactions in Han people of southern China mainland. Seizure. 2011;20(6):446–8.
Zhang Y et al. Strong association between HLA-B*1502 and carbamazepine-induced Stevens-Johnson syndrome and toxic epidermal necrolysis in mainland Han Chinese patients. Eur J Clin Pharmacol. 2011;67(9):885–7.
Chen P et al. Carbamazepine-induced toxic effects and HLA-B*1502 screening in Taiwan. N Engl J Med. 2011;364(12):1126–33.
Ko TM et al. Shared and restricted T-cell receptor use is crucial for carbamazepine-induced Stevens-Johnson syndrome. J Allergy Clin Immunol. 2011;128(6):1266–76. Important in vitro study that establishes the essential role of the TCR in the mechanism behind SJS/TEN and explains why some HLA-B*15:02 carriers are tolerant to carbamazepine.
Mizumoto K et al. Case of carbamazepine-induced hypersensitivity syndrome associated with human leukocyte antigen-A*3101. J Dermatol. 2012;39(9):791–2.
Niihara H et al. HLA-A31 strongly associates with carbamazepine-induced adverse drug reactions but not with carbamazepine-induced lymphocyte proliferation in a Japanese population. J Dermatol. 2012;39(7):594–601.
Ozeki T et al. Genome-wide association study identifies HLA-A*3101 allele as a genetic risk factor for carbamazepine-induced cutaneous adverse drug reactions in Japanese population. Hum Mol Genet. 2011;20(5):1034–41. GWAS that showed association with HLA-A*31:01 and carbamazepine-induced immunologically mediated adverse drug reactions in those of Japanese ancestry.
Yip VL et al. HLA genotype and carbamazepine-induced cutaneous adverse drug reactions: a systematic review. Clin Pharmacol Ther. 2012;92(6):757–65.
Amstutz U et al. HLA-A 31:01 and HLA-B 15:02 as genetic markers for carbamazepine hypersensitivity in children. Clin Pharmacol Ther. 2013;94(1):142–9.
Ramasamy SN et al. Allopurinol Hypersensitivity: A Systematic Review of All Published Cases, 1950-2012. Drug Saf. 2013.
Rundles RW, Metz EN, Silberman HR. Allopurinol in the treatment of gout. Ann Intern Med. 1966;64(2):229–58.
Lang Jr PG. Severe hypersensitivity reactions to allopurinol. South Med J. 1979;72(11):1361–8.
Dalbeth N, Stamp L. Allopurinol dosing in renal impairment: walking the tightrope between adequate urate lowering and adverse events. Semin Dial. 2007;20(5):391–5.
Chan SH, Tan T. HLA and allopurinol drug eruption. Dermatologica. 1989;179(1):32–3.
Hung SI et al. HLA-B*5801 allele as a genetic marker for severe cutaneous adverse reactions caused by allopurinol. Proc Natl Acad Sci U S A. 2005;102(11):4134–9.
Dainichi T et al. Stevens-Johnson syndrome, drug-induced hypersensitivity syndrome and toxic epidermal necrolysis caused by allopurinol in patients with a common HLA allele: what causes the diversity? Dermatology. 2007;215(1):86–8.
Kaniwa N et al. HLA-B locus in Japanese patients with anti-epileptics and allopurinol-related Stevens-Johnson syndrome and toxic epidermal necrolysis. Pharmacogenomics. 2008;9(11):1617–22.
Tassaneeyakul W et al. Strong association between HLA-B*5801 and allopurinol-induced Stevens-Johnson syndrome and toxic epidermal necrolysis in a Thai population. Pharmacogenet Genomics. 2009;19(9):704–9.
Jung JW et al. HLA-B58 can help the clinical decision on starting allopurinol in patients with chronic renal insufficiency. Nephrol Dial Transplant. 2011;26(11):3567–72.
Kang HR et al. Positive and negative associations of HLA class I alleles with allopurinol-induced SCARs in Koreans. Pharmacogenet Genomics. 2011;21(5):303–7.
Lonjou C et al. A European study of HLA-B in Stevens-Johnson syndrome and toxic epidermal necrolysis related to five high-risk drugs. Pharmacogenet Genomics. 2008;18(2):99–107.
Genin E et al. Genome-wide association study of Stevens-Johnson Syndrome and Toxic Epidermal Necrolysis in Europe. Orphanet J Rare Dis. 2011;6:52.
Tohkin M et al. A whole-genome association study of major determinants for allopurinol-related Stevens-Johnson syndrome and toxic epidermal necrolysis in Japanese patients. Pharmacogenomics J. 2013;13(1):60–9.
Somkrua R et al. Association of HLA-B*5801 allele and allopurinol-induced Stevens Johnson syndrome and toxic epidermal necrolysis: a systematic review and meta-analysis. BMC Med Genet. 2011;12:118.
Pompeu YA et al. The structural basis of HLA-associated drug hypersensitivity syndromes. Immunol Rev. 2012;250(1):158–66.
Pollard RB, Robinson P, Dransfield K. Safety profile of nevirapine, a nonnucleoside reverse transcriptase inhibitor for the treatment of human immunodeficiency virus infection. Clin Ther. 1998;20(6):1071–92.
Gangar M et al. Frequency of cutaneous reactions on rechallenge with nevirapine and delavirdine. Ann Pharmacother. 2000;34(7–8):839–42.
Martin AM et al. Predisposition to nevirapine hypersensitivity associated with HLA-DRB1*0101 and abrogated by low CD4 T-cell counts. AIDS. 2005;19(1):97–9.
Yuan J et al. Toxicogenomics of nevirapine-associated cutaneous and hepatic adverse events among populations of African, Asian, and European descent. AIDS. 2011;25(10):1271–80. Genetic study investigating different clinical presentations of nevirapine hypersensitivity and reporting several new associations with specific HLA alleles.
Vitezica ZG et al. HLA-DRB1*01 associated with cutaneous hypersensitivity induced by nevirapine and efavirenz. AIDS. 2008;22(4):540–1.
Phillips E et al. Associations between HLA-DRB1*0102, HLA-B*5801, and hepatotoxicity during initiation of nevirapine-containing regimens in South Africa. J Acquir Immune Defic Syndr. 2013;62(2):e55–7. A class II allele DRB1*01:02 which is homologous to DRB1*01:01 is related to nevirapine-associated hepatitis in South African Black populations.
Littera R et al. HLA-dependent hypersensitivity to nevirapine in Sardinian HIV patients. AIDS. 2006;20(12):1621–6.
Gatanaga H et al. HLA-Cw8 primarily associated with hypersensitivity to nevirapine. AIDS. 2007;21(2):264–5.
Gao S et al. HLA-dependent hypersensitivity reaction to nevirapine in Chinese Han HIV-infected patients. AIDS Res Hum Retroviruses. 2012;28(6):540–3.
Chantarangsu S et al. HLA-B*3505 allele is a strong predictor for nevirapine-induced skin adverse drug reactions in HIV-infected Thai patients. Pharmacogenet Genomics. 2009;19(2):139–46.
Chantarangsu S et al. Genome-wide association study identifies variations in 6p21.3 associated with nevirapine-induced rash. Clin Infect Dis. 2011;53(4):341–8.
Likanonsakul S et al. HLA-Cw*04 allele associated with nevirapine-induced rash in HIV-infected Thai patients. AIDS Res Ther. 2009;6:22.
Carr DF et al. Association of human leukocyte antigen alleles and nevirapine hypersensitivity in a Malawian HIV-infected population. Clin Infect Dis. 2013;56(9):1330–9. First study to associate HLA-C*04:01 with nevirapine-associated Stevens-Johnson Syndrome/Toxic epidermal necrolysis in a Black African population.
Daly AK, Day CP. Genetic association studies in drug-induced liver injury. Drug Metab Rev. 2012;44(1):116–26.
Hautekeete ML et al. HLA association of amoxicillin-clavulanate–induced hepatitis. Gastroenterology. 1999;117(5):1181–6.
O’Donohue J et al. Co-amoxiclav jaundice: clinical and histological features and HLA class II association. Gut. 2000;47(5):717–20.
Donaldson PT et al. Human leucocyte antigen class II genotype in susceptibility and resistance to co-amoxiclav-induced liver injury. J Hepatol. 2010;53(6):1049–53.
Lucena MI et al. Susceptibility to amoxicillin-clavulanate-induced liver injury is influenced by multiple HLA class I and II alleles. Gastroenterology. 2011;141(1):338–47.
Lucena MI et al. Determinants of the clinical expression of amoxicillin-clavulanate hepatotoxicity: a prospective series from Spain. Hepatology. 2006;44(4):850–6.
Stephens C et al. HLA alleles influence the clinical signature of amoxicillin-clavulanate hepatotoxicity. PLoS One. 2013;8(7):e68111.
Olsson R et al. Liver damage from flucloxacillin, cloxacillin and dicloxacillin. J Hepatol. 1992;15(1–2):154–61.
Russmann S et al. Risk of cholestatic liver disease associated with flucloxacillin and flucloxacillin prescribing habits in the UK: cohort study using data from the UK General Practice Research Database. Br J Clin Pharmacol. 2005;60(1):76–82.
Daly AK et al. HLA-B*5701 genotype is a major determinant of drug-induced liver injury due to flucloxacillin. Nat Genet. 2009;41(7):816–9.
Phillips EJ, Mallal SA. HLA-B*5701 and flucloxacillin associated drug-induced liver disease. AIDS. 2013;27(3):491–2.
Ferrell Jr PB, McLeod HL. Carbamazepine, HLA-B*1502 and risk of Stevens-Johnson syndrome and toxic epidermal necrolysis: US FDA recommendations. Pharmacogenomics. 2008;9(10):1543–6.
Phillips EJ et al. Clinical and immunogenetic correlates of abacavir hypersensitivity. AIDS. 2005;19(9):979–81.
Shear NH et al. A review of drug patch testing and implications for HIV clinicians. AIDS. 2008;22(9):999–1007.
Phillips EJ et al. Utility of patch testing in patients with hypersensitivity syndromes associated with abacavir. AIDS. 2002;16(16):2223–5.
Lee AY, Choi J, Chey WY. Patch testing with carbamazepine and its main metabolite carbamazepine epoxide in cutaneous adverse drug reactions to carbamazepine. Contact Dermatitis. 2003;48(3):137–9.
Buyuktiryaki AB et al. Patch testing is an effective method for the diagnosis of carbamazepine-induced drug reaction, eosinophilia and systemic symptoms (DRESS) syndrome in an 8-year-old girl. Australas J Dermatol. 2012;53(4):274–7.
Lin YT, et al. A patch testing and cross-sensitivity study of carbamazepine-induced severe cutaneous adverse drug reactions. J Eur Acad Dermatol Venereol. 2012.
Phillips EJ, Mallal SA. HLA-B*1502 screening and toxic effects of carbamazepine. N Engl J Med. 2011;365(7):672. author reply 673.
Martin AM et al. Predisposition to abacavir hypersensitivity conferred by HLA-B*5701 and a haplotypic Hsp70-Hom variant. Proc Natl Acad Sci U S A. 2004;101(12):4180–5.
Hammond E et al. External quality assessment of HLA-B*5701 reporting: an international multicentre survey. Antivir Ther. 2007;12(7):1027–32.
Martin AM et al. A sensitive and rapid alternative to HLA typing as a genetic screening test for abacavir hypersensitivity syndrome. Pharmacogenet Genomics. 2006;16(5):353–7.
Melis R et al. Copy number variation and incomplete linkage disequilibrium interfere with the HCP5 genotyping assay for abacavir hypersensitivity. Genet Test Mol Biomarkers. 2012;16(9):1111–4.
Zhang FR et al. HLA-B*13:01 and the dapsone hypersensitivity syndrome. N Engl J Med. 2013;369(17):1620–8.
Pavlos R, Mallal S, Ostrov D, Pompeu Y, Phillips E. Fever, rash and systemic symptoms: understanding the role of virus and HLA in cutaneous drug allergy. J Allerg Clin Immunol In Practice. 2014;2(1):21–33.
Kim SH et al. Carbamazepine-induced severe cutaneous adverse reactions and HLA genotypes in Koreans. Epilepsy Res. 2011;97(1–2):190–7.
Kaniwa N et al. HLA-B*1511 is a risk factor for carbamazepine-induced Stevens-Johnson syndrome and toxic epidermal necrolysis in Japanese patients. Epilepsia. 2010;51(12):2461–5.
Ikeda H et al. HLA class I markers in Japanese patients with carbamazepine-induced cutaneous adverse reactions. Epilepsia. 2010;51(2):297–300.
Hung SI et al. Common risk allele in aromatic antiepileptic-drug induced Stevens-Johnson syndrome and toxic epidermal necrolysis in Han Chinese. Pharmacogenomics. 2010;11(3):349–56.
Lin LC et al. Oxcarbazepine-induced Stevens-Johnson syndrome: a case report. Kaohsiung J Med Sci. 2009;25(2):82–6.
Cheung YK et al. HLA-B alleles associated with severe cutaneous reactions to antiepileptic drugs in Han Chinese. Epilepsia. 2013;54(7):1307–14.
Locharernkul C et al. Carbamazepine and phenytoin induced Stevens-Johnson syndrome is associated with HLA-B*1502 allele in Thai population. Epilepsia. 2008;49(12):2087–91.
Kaniwa N et al. Specific HLA types are associated with antiepileptic drug-induced Stevens-Johnson syndrome and toxic epidermal necrolysis in Japanese subjects. Pharmacogenomics. 2013;14(15):1821–31.
Kim SH et al. HLA-B*5901 is strongly associated with methazolamide-induced Stevens-Johnson syndrome/toxic epidermal necrolysis. Pharmacogenomics. 2010;11(6):879–84.
Alfirevic A et al. HLA-B locus in Caucasian patients with carbamazepine hypersensitivity. Pharmacogenomics. 2006;7(6):813–8.
Singer JB et al. A genome-wide study identifies HLA alleles associated with lumiracoxib-related liver injury. Nat Genet. 2010;42(8):711–4.
Kindmark A et al. Genome-wide pharmacogenetic investigation of a hepatic adverse event without clinical signs of immunopathology suggests an underlying immune pathogenesis. Pharmacogenomics J. 2008;8(3):186–95.
Spraggs CF et al. HLA-DQA1*02:01 is a major risk factor for lapatinib-induced hepatotoxicity in women with advanced breast cancer. J Clin Oncol. 2011;29(6):667–73.
Compliance with Ethics Guidelines
Conflict of Interest
Elizabeth Phillips has served on boards for Merck International Pty, Merck Pty Ltd., Tibotec Pty. Ltd, and ViiV Australia; has received honoraria from ViiV Australia; is a co-director in III Pty Ltd., which holds a patent for HLA-B*57:01; has received royalties from UpToDate; has received payment for development of educational presentations (including service on speakers bureaus) from Merck Pty Ltd. and ViiV Australia; and has had travel/accommodations expenses covered/reimbursed by Gilead Australia.
Eric Karlin declares that he has no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Anaphylaxis and Drug Allergy
Rights and permissions
About this article
Cite this article
Karlin, E., Phillips, E. Genotyping for Severe Drug Hypersensitivity. Curr Allergy Asthma Rep 14, 418 (2014). https://doi.org/10.1007/s11882-013-0418-0
Published:
DOI: https://doi.org/10.1007/s11882-013-0418-0