
Opinion Statement
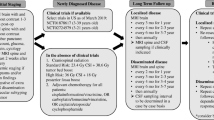
Approximately 70 % of newly diagnosed children with medulloblastoma (MB) will be classified as “standard risk”: their tumor is localized to the posterior fossa, they undergo a near or gross total resection, the tumor does not meet the criteria for large cell/anaplastic histology, and there is no evidence of neuroaxis dissemination by brain/spine MRI and lumbar puncture for cytopathology. Following surgical recovery, they are treated with craniospinal radiation therapy with a boost to the posterior fossa or tumor bed. Adjuvant therapy for approximately 1 year follows anchored by the use of alkylators, platinators, and microtubule inhibitors. This approach to standard risk MB works; greater than 80 % of patients will be cured, and such approaches are arguably the standard of care worldwide for such children. Despite this success, some children with standard risk features will relapse and die of recurrent disease despite aggressive salvage therapy. Moreover, current treatment, even when curative causes life-long morbidity in those who survive, and the consequences are age dependent. For the 20-year-old patient, damage to the cerebellum from surgery conveys greater risk than craniospinal radiation; however, for the 3-year-old patient, the opposite is true. The challenge for the neuro-oncologist today is how to identify accurately patients who need less therapy as well as those for whom current therapy is inadequate. As molecular diagnostics comes of age in brain tumors, the question becomes how to best implement novel methods of risk stratification. Are we able to obtain specific information about the tumor’s biology in an increasingly rapid and reliable way, and utilize these findings in the upfront management of these tumors? Precision medicine should allow us to tailor therapy to the specific drivers of each patient’s tumor. Regardless of how new approaches are implemented, it is likely that we will no longer be able to have a single standard approach to standard risk medulloblastoma in the near future.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Medulloblastoma (MB) is the most common malignant brain tumor of childhood, making up 20 % of all primary central nervous system tumor diagnoses in patients younger than age 19 years. Incidence peaks between ages 5 and 9 years, but patients of all ages can be affected [1]. Most patients present with symptoms of hydrocephalus due to obstruction by tumor at the level of the fourth ventricle or cerebral aqueduct, and ataxia.
Medulloblastoma was first described as being distinct from glioma by Bailey and Cushing in 1925 [2]. It is a primitive neural ectodermal tumor (PNET), consisting of small, round, blue cells arising in the posterior fossa. The World Health Organization in the 2007 revision [3] classified medulloblastoma into five main subtypes: classic; large cell; anaplastic (the two of which often are grouped together under the moniker large-cell anaplastic, LCA); desmoplastic nodular (DMB); and medulloblastoma with extensive nodularity (MBEN). DMB and MBEN are variants primarily of infants and have an excellent prognosis. LCA MB has a high proliferative index and is generally associated with poorer outcomes [3–5].
The advent of molecular diagnostics has allowed MB to be classified into four distinct subgroups based on the presence of characteristic molecular gene signatures: wingless (WNT), sonic hedgehog (SHH), Group 3, and Group 4 [6••, 7••]. Several retrospective analyses indicate that these categories confer different clinical outcomes. Those in the WNT group have an excellent prognosis, whereas those in Group 3, which are characterized by MYC amplification, fare very poorly. SHH and Group 4 have intermediate prognoses [6••, 7••, 8•, 9]. There is some overlap between traditional histology and the molecular genetics; however, neither is exclusive [9]. Group 3 tumors tend to have LCA histology, but conversely some WNT pathway tumors can be LCA as well. DMB almost always have SHH mutations, but these also can be present in classic and LCA histology tumors [6••, 7••, 8•, 9].
Despite the body of literature describing the molecular subgroups, these findings have yet to translate directly to the clinical realm. Standard of care treatment remains maximal up front surgical resection followed by radiation and chemotherapy. The intensity of adjuvant therapy depends on the presence of risk factors at diagnosis. Standard risk patients are those who are older than 3 years and without metastatic disease or evidence of anaplasia. They also must have undergone a near gross total resection with <1.5 mm2 of residual tumor remaining [10••, 11]. Those meeting these criteria have an overall survival close to 85 % [10••, 11, 12, 13•]. Any of the above risk factors will categorize patients as “high risk,” necessitating intensified therapy and an overall survival of approximately 60 % [14]. Infant patients present a particular challenge due to the significant neurocognitive effects associated with radiation therapy. Radiation-avoiding strategies often are attempted in children <3 years of age, or in the case of DMB most can be cured without radiation [15, 16]. The remainder of this article will focus specifically on standard risk patients, who constitute the vast majority of patients with MB.
Interventional procedures
Surgery
-
Management of obstructive hydrocephalus
-
Due to the tumor’s predilection to grow in the midline of the cerebellum, most patients with MB present with signs and symptoms of increased intracranial pressure (ICP) resulting from obstructive hydrocephalus. The majority of patients have a history of headaches and nausea/vomiting for weeks to months before definitive presentation. Young children may exhibit irritability and poor feeding. In severe cases, patients may present with lethargy and altered mental status. Physical exam findings may include Cushing’s triad of bradycardia, hypertension, and widened pulse pressure. Ocular findings, including papilledema and sixth nerve palsies, may be present if the degree of hydrocephalus is severe [17].
-
Depending on the severity of obstructive hydrocephalus, the surgeon may choose to perform a cerebrospinal fluid (CSF) diversion procedure before, or at the time of, definitive surgical resection of the tumor. Emergent management is generally accomplished by an external ventricular drain (EVD). Permanent CSF diversion, when required, is accomplished by an endoscopic third ventriculostomy (ETV) or ventriculoperitoneal (VP) shunt. In an ETV, an opening is made in the floor of the third ventricle allowing cerebrospinal fluid (CSF) to flow directly into the basal cisterns, bypassing the area of obstruction in the posterior fossa. Complications of these procedures are related to entering the ventricular system and primarily include infection [17].
-
-
Maximal surgical resection of the tumor
-
Maximal tumor resection is standard of care for patients with MB. Historically patients with bulk residual disease have fared poorly, although the majority of this literature predates current treatment approaches [18]. Approximately 25 % of patients undergoing resection of MB will develop some degree of cerebellar mutism, also known as posterior fossa syndrome. This can be a devastating complication where patients are unable to produce words (may use sounds), are extremely irritable, and develop hypotonia and ataxia. It usually manifests 24 hours after resection of a posterior fossa tumor and is disproportionately represented in cases of medulloblastoma. The duration of the syndrome is variable lasting weeks to months, but the language difficulties may be lifelong [19•]. Other complications of surgery include disruption of the blood supply, which causes infarction of the surrounding normal brain, and intraoperative bleeding, which causes subdural hematomas.
-
-
Surgical considerations in adults
Surgical management is generally identical to children. The midline location is less common with more lateral tumors approaching the cerebellopontine angle.
Radiation therapy
-
Local control
-
Radiation therapy (XRT) should begin ~30 days following definitive surgery [10••, 20]. Historically, the entirety of the cerebellum was radiated. Increasingly, the tumor bed with an appropriate margin is radiated as supported by published evidence indicating that a conformal boost to the tumor bed can achieve similar local control and reduce exposure of the eighth cranial nerves [21]. Therapy is delivered in daily fractions of 1.8 Gy to a final dose of 54-59.4 Gy. Ignoring infants, there are no children with MB for whom XRT is contraindicated, although non-XRT approaches have been studied [22].
-
-
Treatment of micrometastatic disease
-
Craniospinal irradiation (CSI) is delivered to the entire brain and spine and given concurrently with primary site radiation for the first 13 days of therapy, to a total dose of 23.4 Gy. This dosing regimen is well established for this population [10••, Class IIa]. The most recent Children’s Oncology Group (COG) study was designed to test whether the CSI dose could be reduced to 18 Gy. The study recently closed to accrual and results are pending.
-
Complications of XRT for pediatric patients with standard risk MB are significant. Acute side effects of radiation therapy include anorexia and nausea due to the proximity of the spinal fields to the GI tract. Close attention must be paid to the patient’s weight and nutrition during this treatment. In children, the bone marrow of the vertebral bodies is active and important for the maintenance of normal blood counts, so cytopenias are common during and immediately after radiation. In addition to neutropenic immunosuppression, severe lymphopenia results from CSRT leading to a risk for Pneumocystis jiroveci pneumonia (PJP) for which prophylaxis with sulfamethoxazole or equivalent antibiotic is required [23, Class IIb]. While care is taken to limit radiation exposure to sensitive CNS structures, such as the supratentorial brain, hypothalamic pituitary axis (HPA), cochlea, and eyes/optic nerves/optic chiasm [13•, 24], some exposure is unavoidable. CSI and local control measures are felt to be responsible in part for endocrine abnormalities, hearing loss, and neurodevelopmental decline seen in long-term survivors of medulloblastoma [25–27]. Prepubertal children are especially vulnerable to these effects, because their growth and development are immature. Radiation exposure also leads to risk of secondary malignancy, including meningioma and glioblastoma [13•, 28].
-
Radiation therapy is required for long-term survival in most patients with MB and reflects a required cost of treatment. There is a growing debate regarding the energy source, namely whether there is a therapeutic advantage to the use of protons versus photons, although there have been no clinical studies comparing the two modalities. Early reports of proton use shows that clinical responses can be achieved at a similar rate to photons. Many small studies report reduced dosimetry to nontarget tissues and even reduced incidence of secondary malignancy [29, 30]. There is a robust experience using photon-based radiation approaches in MB, and the long-term outcome data for proton-based therapy is just starting to mature. Additionally, there are very few proton beam facilities available for any given geographic area, especially ones that can accommodate pediatric patients. At this time, there is insufficient evidence to support the routine use of proton therapy and photons remain the standard of care for this disease [24, 31, Class IV].
-
-
Radiation therapy considerations in adults
As with children, adults require CSI with a boost to the primary tumor site. Given the reduction in CSI-associated morbidity in adults compared with children, most adults receive 36 Gy CSI as opposed to the 23.4 Gy utilized in children [32, Class III].
Pharmacologic treatment
Chemotherapeutic treatment of residual gross and micrometastatic disease
The goal of chemotherapy in medulloblastoma is to assist in the local control of tumor and the management of micrometastatic disease. As with most chemotherapeutics, these drugs affect rapidly dividing cells, including those of the gastrointestinal tract, hair follicles, and bone marrow. This leads to risk of nausea and vomiting, diarrhea and/or constipation, hair loss, and myelosuppression. The drugs and doses listed are those used in the treatment of standard risk MB patients and are well established in this population [10••, Class IIa]. Table 1 outlines one typical approach to standard-risk MB patients that has been widely adopted. Please note that alternative regimens incorporating additional chemotherapeutic agents or utilizing different dosages and frequencies often are used in patients who are high-risk, infant, or adult.
Cisplatin
- Mechanism of action :
-
Induces cellular apoptosis by cross-linking DNA [33].
- Standard dosage :
-
75 mg/m2/dose IV each cycle for a total of 6 cycles [10••].
- Main drug interactions :
-
Caution should be taken with other nephrotoxic and ototoxic drugs, specifically, aminoglycosides, loop diuretics, and amphotericin.
- Major side effects :
-
Ototoxicity and nephrotoxicity. It is especially important to monitor hearing in these patients as radiation to the posterior fossa can induce hearing loss. An audiogram should be obtained before each course of cisplatin, and the dose should be reduced by 50 % if low-frequency hearing loss is detected [33, 34]. Electrolyte wasting is common and fluids containing sodium, potassium, and magnesium should be administered with the infusion. Serum creatinine and electrolytes, including calcium, magnesium, and phosphorous should be monitored during and after the infusion. Electrolytes should be replaced as needed [33].
- Cost :
-
Inexpensive
Cyclophosphamide
- Mechanism of action :
-
Alkylating agent
- Standard dosage :
-
1,000 mg/m2/dose IV for 2 doses each cycle for a total of 3 cycles [10••].
- Major drug interactions :
-
None
- Major side effects :
-
Hemorrhagic cystitis, which is usually prevented by the concurrent infusion of mesna at 360 mg/m2 with each dose of cyclophosphamide [35, Class Ib]. Nephrotoxicity also may occur, and cyclophosphamide should always be administered with IV fluids. Patients should be counseled regarding risk of infertility. Postpubertal males may want to consider sperm banking [36, Class III].
- Cost :
-
Inexpensive
Lomustine
- Mechanism of action :
-
Alkylating agent
- Standard dosage :
-
75 mg/m2/dose PO each cycle for a total of 6 cycles [10••].
- Major drug interactions :
-
None
- Major side effects :
-
Significant nausea and prolonged myelosuppression, especially with cumulative dosing [37]. There may be fertility risks associated with this drug. The most serious long-term risk with lomustine is a rare but increased risk of secondary malignancy particularly myelodysplastic syndrome and myeloid leukemia [23, 38].
- Cost :
-
Expensive
Vincristine
- Mechanism of action :
-
Microtubule inhibitor that prevents cell division by binding to the tubulin component of microtubules and leading to metaphase arrest [37].
- Standard dosage :
-
1.5 mg/m2 IV to a maximum dose of 2 mg [10••] weekly during CSRT and intermittently during adjuvant therapy
- Major drug interactions :
-
Vincristine is metabolized by the CYP450 system. Inhibitors of this enzyme, such as azole antifungals, should be avoided if possible. Inducers of P450 also should be avoided, including rifampin, phenobarbital, phenytoin, and carbamazepine.
- Major toxicities :
-
Peripheral neuropathy. Muscle weakness is common. It often preferentially affects the lower extremities leading to foot drop, but any muscle group can be affected. Numbness of the lower extremities can occur as can severe pain in any distribution. Jaw pain is an especially common acute side effect. Loss of deep tendon reflexes is common with continued exposure. Visuospatial side effects can occur that impair coordination [39]. Constipation is common and may become severe leading to intestinal obstruction. Foot drop and other signs of significant muscle weakness should be treated by 50 % dose reduction or holding of vincristine if especially severe [10••]. Notably, because vincristine damages peripheral nerves, it can take 6-12 months from the time therapy is discontinued to resume normal pretreatment function [39].
- Cost :
-
Inexpensive
Chemotherapeutic considerations in adults
Chemotherapy is less well tolerated in adults [40], and it has not yielded the clearly beneficial results that it has in children. No randomized, clinical trials have compared XRT alone versus XRT with adjuvant chemotherapy in adults to determine whether there is an incremental benefit to the addition of chemotherapy. Therefore, chemotherapy often is reserved for high-risk, adult patients [41, Class IV].
Physical therapy
-
Multidisciplinary rehabilitative services are recommended for all patients undergoing therapy for MB. Because most patients present with significant symptoms requiring immediate attention, a preoperative assessment is usually not practicable. Postoperatively all patients should undergo a physical and occupational therapy evaluation. If cerebellar mutism or other language-processing disorder is suspected, speech therapy should be incorporated as well. If possible, a complete neuropsychiatric evaluation to assess the patient’s neurocognitive status should be performed before the initiation of radiation therapy. Recommended rehabilitation services can be performed concomitantly with radiation and chemotherapies. An end-of-therapy assessment should be performed to assess functional status and quality of life measures after undergoing successful treatment. Many patients may need educational support. Individualized Educational Plans (IEP) often are required when patients return to school posttreatment [42].
Emerging therapies
-
Subgroup-Specific Therapies
-
Perhaps the most important discovery in medulloblastoma has been the determination of four distinct molecular subgroups and their association with varying clinical outcomes. The first challenge is to identify subgroups accurately at the time of initial diagnosis in order not to delay implementation of standard of care therapy. Diagnostic evaluations are expected to be completed within 30 days of initial resection, because delays in therapy initiation have been associated with poor outcomes [20]. At present, only a handful of centers possess the capability to subclassify MB samples appropriately. Therefore, alternative methods to identify subgroups must be developed and validated.
-
-
WNT
-
Overview: WNT pathway tumors are characterized by overexpression of beta-catenin and activation of its pro-proliferative downstream targets, such as Cyclin D1 and MYC (1., Panel A). These tumors represent 10-20 % of all cases and have close to 100 % overall survival with standard therapy [6••, 7••, 8•].
-
Identification: WNT subgroup may be most amenable to alternative methods of identification, because 70-80 % of MB demonstrating nuclear beta-catenin staining by immunohistochemical methods has correlated with this gene signature [8•, 43]. Monosomy 6 is a readily detectable genetic alteration that also has been correlated with this subgroup. A recent study showed a complete association between beta-catenin staining and the presence of chromosome 6 abnormalities [44], but earlier research recommended caution using this in isolation, because this genetic alteration was present in non-WNT MB [8•].
-
Approaches: various strategies are being studied, including further reductions in CSI doses, omission of XRT altogether, and chemotherapy dose reductions. Specific pharmacologic therapies targeting the WNT pathway in MB are lacking. However inhibition of Tankyrase [45], cyclo-oxygenase [46], and Porcupine [25] to decrease beta-catenin signaling are under investigation in other tumor models.
-
-
SHH
-
Overview: in the SHH pathway, the patched protein (PTCH) normally inhibits smoothened (SMO). When SMO is overactivated, it targets downstream pro-proliferative transcription factors GLI 1-3 [26] (Fig. 1, Panel B). SHH MB accounts for up to 25 % of cases [6••, 7••]. Patients with mutations in this pathway have an intermediate prognosis likely reflective of which part of the pathway is affected with DMB patients likely representing more Ptch mutations and others GLI 1-3. Overexpression of these transcription factors has been linked to less favorable prognoses within this group [27].
Fig. 1 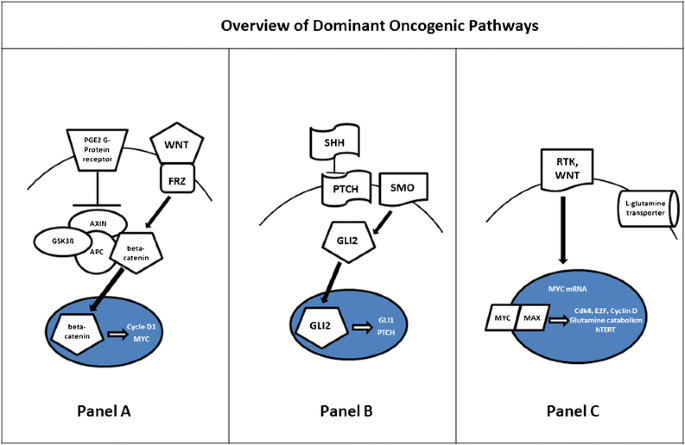
A WNT pathway. The extracellular ligand WNT binds to the receptor Frizzled (FRZ), leading to inhibition of the GSK3β, AXIN, and APC-mediated phosphorylation and degradation of betacatenin in the cytoplasm. Free beta-catenin can then translocate to the nucleus where it activates expression of pro-proliferative genes, including CYCLIN D1 and MYC. B SHH pathway. In canonical Hedgehog signaling, SHHbinds to its receptor PTCH, relieving PTCH inhibition of SMO. This allows SMO to initiate activation of GLI2, which can then translocate to the nucleus and drive transcription of target genes, such as GLI1. C MYC pathway. Signaling through receptor tyrosine kinases, enhanced WNT pathway activation, and gene amplification all lead to increased levels of MYC. MYC protein translocates to the nucleus, where it heterodimerizes with the E-box protein MAX.
-
Identification: although alternative diagnostic approaches, such as FISH, have been proposed [8•], at present digital multiplex PCR is still the primary modality used for confirmation of this subgroup.
-
Approaches: multiple, small molecule inhibitors in the form of synthetic cyclopamine derivatives exist against SMO. Several of these molecules have shown activity against MB in a variety of clinical trials. Unfortunately, SHH tumors frequently demonstrate escape via specific mutations that prevent binding to SMO or amplify downstream effectors, such as GLI2 [47] (Fig. 1, Panel B). Other agents have been identified (e.g., arsenic trioxide) that target these downstream effectors [48, 49] but have yet to be tested in clinical trials in MB.
-
-
Group 3
-
Overview: characterized by MYC amplification, they have the poorest outcomes amongst the subgroups, although specific, overall survival estimates vary. This subgroup accounts for approximately 30 % of MB cases [6••, 7••, 8•, 9]. Downstream targets of MYC are multiple and heavily represented by genes regulating growth and cellular metabolism [50] (Fig. 1, Panel C).
-
Identification: FISH is under investigation as a reproducible and reliable way to identify these patients in real time [8•].
-
Approach: small molecule inhibitors for this subgroup also are under investigation. Telomerase inhibitors dually target the telomerase enzyme as well as the MYC promoter, which has shown promise against MB in vitro [51]. JQ1 is a bromodomain inhibitor that impairs MYC driven transcription in other MYC-driven tumors [52]. Epigenetic modulation with histone deacetylase inhibitors, such as the well-known antiepileptic, valproic acid also is under investigation as a clinical trial in relapsed disease [53].
-
-
Group 4
-
Overview: The remainder of medulloblastoma falls into the heterogeneous Group 4 for which a dominant oncogene has not been identified.
-
Identification: limited to advanced molecular techniques.
-
Approaches: this subgroup does not have a dominant oncogenic pathway defined making targeted therapies extremely challenging. Recent studies indicate it may be associated with NFkappaB activation [6••, 48, 54].
-
-
Immunologic Therapies
-
Immunologic-based therapies have been extremely challenging in MB and other PNETs, and progress in this area has been hindered by a lack of immunologically competent models. However, evidence exists that the host immune response likely plays some role in disease pathogenesis.
-
-
Adoptive T-cell therapies/vaccines
-
Pediatric, vaccine-based approaches have been attempted with limited success. In Belgium, a Phase I study of a pulsed dendritic cell vaccine included 5 MB/PNET patients, none of whom survived beyond 6 months [55]. This lack of success may be partially attributed to a lack of tumor-associated antigens identified in MB. Oba-Shinjo et al. tried to identify the frequency of cancer testis antigens in MB. These are normally expressed in the adult testis but aberrantly found in a variety of human cancers. They have been shown to elicit host antitumor responses in other cancers. Although this study detected high levels of mRNA for cancer testis antigens in MB, corresponding protein expression was not identified [56]. Other studies have shown low levels of HER2, another common cancer antigen, expressed in MB. One study utilized T-cells with chimeric antigen receptors (CARs) against HER2 and demonstrated effective tumor cell killing in vitro and in vivo [57]. Despite many challenges to T-cell–based therapies, a new MB vaccine trial will evaluate the feasibility of combining dendritic cells pulsed with tumor lysate in combination with ex vivo expanded tumor-specific T-cells.
-
-
Adoptive natural killer (NK) cell therapy
-
NK cells can be cytolytic against tumor cells when the NKG2D receptor is activated. Several clinical trials are evaluating the use of autologous and allogeneic infusion of NK cells for the treatment of extracranial cancers. NK cells have shown activity against MB cell lines in vitro [58], and NKG2D has been found on human MB tumor samples [59].
-
-
Immune checkpoint blockade
-
An additional immunologic approach that has been gaining momentum in adult brain tumors is immune checkpoint blockade. This strategy blocks immune checkpoint inhibitors such as PD-1 and CTLA-4 that normally dampen the immune response. Although to date these therapies have not been employed in children, emerging research indicates that like adult gliomas, MB expresses the primary ligand of PD-1, PD-L1 [60].
-

References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Smoll NR, Drummond KJ. The incidence of medulloblastomas and primitive neuroectodermal tumours in adults and children. J Clin Neurosci. 2012;19:1541–4.
Bailey P, Cushing H. Medulloblastoma cerebelli, a common type of mid-cerebellar glioma of childhood. Arch Neurol Psychiatry. 1999;14:192–224.
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK. WHO classification of tumours of the central nervous system. Lyon: IARC; 2007. p. 1–309.
Louis DN, Ohgaki H, Wiestler OD, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114(2):97–109.
Eberhart CG, Kepner JL, Goldthwaite PT, et al. Histopathologic grading of medulloblastomas. Cancer. 2002;94(2):552–60.
Northcott PA, Korshunov A, Witt H, et al. Medulloblastoma comprises four distinct molecular variants. J Clin Oncol. 2011;29(11):1408–14. This paper is the first to describe the molecular subgroups as they are now accepted.
Taylor M, Northcott PA, Korshunov A, et al. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol. 2012;123:464–72. This paper provides a consensus guideline for the clinical significance of the molecular subgroups and also provides a great overview of this topic.
Shih DJ, Northcott PA, Remke M, et al. Cytogenetic prognostication within medulloblastoma subgroups. J Clin Oncol. 2014;32(9):886–96. Proposes a potential way to identify molecular subgroups in real time to be incorporated into patient care.
Ellison DW, Dalton J, Kocak M, et al. Medulloblastoma: clinicopathological correlates of SHH, WNT, and non-SHH/WNT molecular subgroups. Acta Neuropathol. 2011;121:381–96.
Packer RJ, Gajjar A, Vezina G, et al. Phase III study of craniospinal radiation therapy followed by adjuvant chemotherapy for newly diagnosed average-risk medulloblastoma. J Clin Oncol. 2006;24(25):4202–8. Even though this is an older study it provides the backbone for current standard risk therapy in medulloblastoma. Due to smaller sample sizes pediatric trials are slower to complete.
Zeltzer PM, Boyett JM, Finlay JL, et al. Metastasis stage, adjuvant treatment, and residual tumor are prognostic factors for medulloblastoma in children: conclusions from the children’s cancer group 921 randomized phase III study. J Clin Oncol. 1999;17(3):832.
Packer RJ, Sutton LN, Elterman R, et al. Outcome for children with medulloblastoma treated with radiation and cisplatin, CCNU, and vincristine chemotherapy. J Neurosurg. 1994;81:690–8.
Packer RJ, Zhou T, Holmes E, et al. Survival and secondary tumors in children with medulloblastoma receiving radiotherapy and adjuvant chemotherapy: results of children’s oncology group trial A9961. Neuro-Oncology. 2013;15(1):97–103. The first long-term follow-up study to document comparable survival in patients treated with therapy nearly equivalent to the current standard.
Tarbell NJ, Friedman H, Polkinghorn WR, et al. High risk medulloblastoma: a pediatric oncology group randomized trial of chemotherapy before or after radiaton therapy (POG 9031). J Clin Oncol. 2013;31(23):2936–41.
Ashley DM, Merchant TE, Strother D, et al. Induction chemotherapy and conformal radiation therapy for very young children with nonmetastatic medulloblastoma: children’s oncology group study P9934. J Clin Oncol. 2012;30(26):3181–6.
Von Bueren AO, von Hoff K, Torsten P, et al. Treatment of young children with localized medulloblastoma by chemotherapy alone: results of the prospective, multicenter trial HIT 2000 confirming the prognostic impact of histology. Neuro-Oncology. 2011;13(6):669–79.
Gopalakrishnan CV, Dhakoji A, Menon G, et al. Factors predicting the need for cerebrospinal fluid diversion following posterior fossa tumor surgery in children. Pediatr Neurosurg. 2012;48(2):93–101.
Albright AL, Wisoff JH, Zeltzer PM, et al. Effects of medulloblastoma resections on outcome in children: a report from the children’s cancer group. Neurosurgery. 1996;38(2):265–71.
Robertson PL, Muraszko KM, Holmes EJ, et al. Incidence and severity of postoperative cerebellar mutism syndrome in children with medulloblastoma: a prospective study by the children’s oncology group. J Neurosurg Pediatr. 2006;105:444–51. The only prospective study on cerebellar mutism.
Kortman RD, Kuhl J, Timmerman B, et al. Postoperative neoadjuvant chemotherapy before radiotherapy as compared to immediate radiotherapy followed by maintenance chemotherapy in the treatment of medulloblastoma in childhood: results of the German prospective randomized trial HIT ’91. Int J Radiat Oncol Biol Phys. 2000;46(2):269–79.
Polkinghorn WR, Dukel IJ, Souweidane MM, et al. Disease control and ototoxicity using intensity-modulated radiation therapy tumor-bed boost for medulloblastoma. Int J Radiat Oncol Biol Phys. 2011;81(3):1–6.
Dhall G, Grodman H, Ji L, et al. Outcome of children less than three years old at diagnosis with non‐metastatic medulloblastoma treated with chemotherapy on the “Head Start” I and II protocols. Pediatr Blood Cancer. 2008;50(6):1169–75.
Groll AH, Ritter J, Müller FM. Guidelines for prevention of pneumocystis Carinii pneumonitis in children and adolescents with cancer. Klin Padiatr. 2001;213:A38–49.
Fossati P, Ricardi U, Orecchia R. Pediatric medulloblastoma: toxicity of current treatment and potential role of proton therapy. Cancer Treat Rev. 2009;35(1):79–96.
Liu J, Pan S, Hsieh MH, et al. Targeting Wnt-driven cancer through the inhibition of porcupine by LGK974. Proc Natl Acad Sci. 2013;110(50):20224–9.
Lin TL, Matsui W. Hedgehog pathway as a drug target: smoothened inhibitors in development. OncoTargets Ther. 2012;5:47–58.
Kool M, Jones DT, Jäger N, et al. Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell. 2014;25(3):393–405.
Kotecha RS, Pascoe EM, Rushing EJ, et al. Meningiomas in children and adolescents: a meta-analysis of individual patient data. Neuropathol Appl Neurobiol. 2014;12(13):1229–39.
Chung CS, Keating N, Yock T, et al. Comparative analysis of second malignancy risk in patients treated with proton therapy versus conventional photon therapy. Int J Radiat Oncol Biol Phys. 2008;72:S8.
Yuh GE, Loredo LN, Yonemoto LT, et al. Reducing toxicity from craniospinal irradiation: using proton beams to treat medulloblastoma in young children. Cancer J. 2004;10(6):386–90.
Wolden SL. Protons for craniospinal radiation: are clinical data important? Int J Radiat Oncol Biol Phys. 2013;87(2):231–2.
Friedrich C, von Bueren AO, von Hoff K, et al. Treatment of adult nonmetastatic medulloblastoma patients according to the paediatric HIT 2000 protocol: a prospective observational multicentre study. Eur J Cancer. 2013;49(4):893–903.
Von Hoff DD, Schilsky R, Reichert CM, et al. Toxic effects of cis-dichlorodiammineplatinum (II) in Man. Cancer Treat Rep. 1979;63:1527–31.
McHaney VA, Thibadoux G, Hayes FA, et al. Hearing loss in children receiving cisplatin chemotherapy. J Pediatr. 1983;102:314–7.
Hows JM, Mehta A, Ward L, et al. Comparison of mesna with forced diuresis to prevent cyclophosphamide induced haemorrhagic cystitis in marrow transplantation: a prospective randomised study. Br J Cancer. 1984;50:753–6.
Kenney LB, Laufer MR, Grant FD, et al. High risk of infertility and long term gonadal damage in males treated with high dose cyclophosphamide for sarcoma during childhood. Cancer. 2001;91:613–21.
Rowinsky EK, Donehower RC. The clinical pharmacology and use of antimicrotubule agents in cancer chemotherapeutics. Pharmacol Ther. 1991;52:35–84.
Green DM, Zevon MA, Reese PA, et al. Second malignant tumors following treatment during childhood and adolescence for cancer. Med Pediatr Oncol. 1994;22(1):1–10.
Legha SS. Vincristine neurotoxicity. Med Toxicol. 1986;1(6):421–7.
Tabori U, Sung L, Hukin J, et al. Canadian pediatric brain tumor consortium. Medulloblastoma in the second decade of life: a specific group with respect to toxicity and management: a Canadian pediatric brain tumor consortium study. Cancer. 2005;103(9):1874–80.
Brandes AA, Ermani M, Amista P, et al. The treatment of adults with medulloblastoma: a prospective study. Int J Radiat Oncol Biol Phys. 2003;57(3):755–61.
Ross SG, Northman L, Morris M, et al. Cerebellar mutism after posterior fossa tumor resection case discussion and recommendations for psychoeducational intervention. J Pediatr Oncol Nurs. 2014;31(2):78–83.
Pizer BL, Clifford SC. The potential impact or tumour biology on improved clinical practice for medulloblastoma: progress towards biologically driven clinical trials. Br J Neurosurg. 2009;23(4):364–75.
Goschzik T, zur Mühlen A, Kristiansen G, et al. Molecular Stratification of Medulloblastoma: comparison of histological and genetic methods to detect Wnt activated tumors. Pediatr Blood Cancer. 2010;54(4):519-25
Huang SM, Mishina YM, Liu S, et al. Tankyrase inhinition stabilizes axin and antagonizes Wnt signalling. Nature. 2009;461(7264):614–20.
Castellone MD, Teramoto H, Williams BO, et al. Prostaglandin E2 promotes colon cancer cell growth through a Gs-axin-beta-catenin signaling axis. Science. 2005;310(5753):1504–10.
Dijkgraaf GJ, Alicke B, Weinmann L, et al. Small molecule inhibition of GDC-0449 refractory smoothened mutants and downstream mechanisms of drug resistance. Cancer Res. 2011;71(2):435–44.
Kim J, Lee JJ, Gardner D, et al. Arsenic antagonizes the hedgehog pathway by preventing ciliary accumulation and reducing stability of the Gli2 transcriptional effector. Proc Natl Acad Sci U S A. 2010;107(30):13432–7.
Beauchamp EM, Ringer L, Bulut G, et al. Arsenic trioxide inhibits human cancer cell growth and tumor development in mice by blocking hedgehog/GLI pathway. J Clin Investig. 2011;121(1):148–60.
Raabe E, Eberhart CG. Therapeutic targeting of developmental signaling pathways in medulloblastoma: hedgehog, notch, Wnt and Myc. Curr Signal Transduct Ther. 2013;8:1–12.
Shalaby T, von Bueren AO, Hurlimann ML, et al. Disabling c-MYC in childhood medulloblastoma and atypical teratoid/rhabdoid tumor cells by the potent G-quadraplex interactive agent S2T1-6OTD. Mol Cancer Ther. 2010;9(1):167–79.
Je D, Issa GC, Lemieux ME, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146(6):904–17.
Li XN, Shu Q, Su JM, et al. Valproic acid induces growth arrest, apoptosis, and senescence in medulloblastomas by increasing histone hyperacetylation and regulating expression of p21Cip1, CDK4, and CMYC. Mol Cancer Ther. 2005;4(12):1912–22.
Northcott PA, Shih DJ, Peacock J, et al. Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature. 2012;488(7409):49–56.
Ardon H, De Vleeschower S, Van Calenbergh F, et al. Adjuvant dendritic cell-based tumour vaccination for children with malignant brain tumours. Cancer Immun 2008;8(7):54519–525.
Oba-Shinjo SM, Caballero OL, Jungbluth AA, et al. Cancer-testis (CT) antigen expression in medulloblastoma. J Clin Oncol. 2005;23(24):5511-9.
Ahmed N, Ratnayake M, Savoldo B, et al. Regression of experimental medulloblastoma following transfer of HER2-specific T cells. Cancer Res. 2007;67:5957–64.
Castriconi R, Dondero A, Negri F, et al. Both CD133+ and CD133- medulloblastoma cell lines express ligands for triggering NK receptors and are susceptible to NK-mediated cytotoxicity. Eur J Immunol. 2007;37:3190–6.
Fernández L, Portugal R, Valentín J, et al. In vitro natural killer cell immunotherapy for medulloblastoma. Front Oncol. 2013;3(94):1–7.
Martin A, Nirschl C, Polanczyk M, et al. MYC amplification status influences tumor immune evasion in medulloblastoma. Neuro-Oncol. 2013;15 suppl 1:15––16 [Abstract].
Mulhern KR, Palmer SL, Merchant TE, et al. Neurocognitive consequences of risk-adapted therapy for childhood medulloblastoma. J Clin Oncol. 2005;23(24):5511–9.
Sklar CA, Constine LS. Chronic neuroendocrinologic sequelae of radiation therapy. Int J Radiat Oncol Biol Phys. 1995;31(5):1113–21.
Compliance with Ethics Guidelines
Conflict of Interest
Allison M. Martin, Eric Raabe, Charles Eberhart, and Kenneth J. Cohen declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Martin, A.M., Raabe, E., Eberhart, C. et al. Management of Pediatric and Adult Patients with Medulloblastoma. Curr. Treat. Options in Oncol. 15, 581–594 (2014). https://doi.org/10.1007/s11864-014-0306-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11864-014-0306-4