
Abstract
Oil content and fatty acid composition in 444 ground cottonseed kernel samples were analyzed using near infrared reflectance spectroscopy (NIRS). Calibration equations were developed for oil and fatty acid contents with the modified partial least squares (MPLS) regression method. The correlations between NIRS and reference values in external validation were in agreement with the predictions in calibration. Each equation was assessed based on the relative prediction determinant for external validation (RPDv). Equations corresponding to total oil content (RPDv = 11.495) and linoleic acid (RPDv = 5.026) showed high accuracy. For palmitic acid (RPDv = 1.914), myristic acid (RPDv = 1.724) and oleic acid (RPDv = 1.999), the equations were predicted with relatively high accuracy while those for palmitoleic acid (RPDv = 0.686), stearic acid (RPDv = 0.792), linolenic acid (RPDv = 0.475) and 1-eicosenoic acid (RPDv = 0.619) were poorly predicted. The equations for traits with RPDv > 1.5 could be reliably used in screening samples for breeding programs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cottonseed is an important secondary product of the cotton industry. Its main derivatives are the cake and meal which are used for making animal feed, and the oil which is used for both domestic and industrial food preparation. Cottonseed contains between 20 and 23% oil [1] and belongs to the group of unsaturated vegetable oil seeds that include safflower, corn, soybean, rapeseed and sunflower. Fats and oils account for a considerable portion of the calories in human diet and their nutritional and health impacts on the human body is of major concern to consumers [2]. Research findings have shown that excessive consumption of fats and oils contribute significantly to the increased risk of cardiovascular and other diseases [3]. To address these concerns, there have been numerous efforts aimed at developing new profiles in oilseed plants based on the genetic variability for fatty acid composition, to facilitate the selection and breeding of lines with desirable nutritional and health as well as flavor and consistency values [4]. The identification of target traits and profiling often involves the evaluation of large numbers of seed samples and the current standard screening methods for oil content and fatty acid composition seem to be less suitable in this respect since they are labor-intensive, time consuming and could limit the scale of a breeding program in most cases.
A rapid, non-destructive and low cost analytical method would be more preferable for large scale screening. Near-infrared spectroscopy (NIRS) is one of the most reliable multi-trait evaluating techniques in agricultural and food analysis that meets these requirements [5]. It can be used to analyze a large number of seed samples in small quantities in the form of whole grains or powders. With NIRS, the error sources related to laboratory analysis are avoided and the control of NIRS related errors leads to equations that can predict the contents of a sample with high accuracy and precision. NIRS has been applied to fatty acid profiling in oil seeds including rapeseed [6], peanut [7], sesame [8], and shea nut [9]. It has also been used, to study the gossypol content in cottonseed meal [10] and oil content in cottonseed [11]. However, there seems to be limited information on its application to fatty acid profiling in cotton seed. The objective of the current experiment was to use NIRS to develop prediction models for oil content and fatty acid composition in cottonseed kernel that could be applied to breeding programs.
Materials and Methods
Sample Preparation
Cottonseed samples used in this study were harvested from cotton plants grown in Hangzhou in the Zhejiang province and Sanya on Hainan island of China in 2007 and 2008. They included hybrid and breeding materials as well as materials from the cotton germplasm. An original population of 1,179 cottonseed samples was used. Samples meant for oil and fatty acid content measurement were acid de-linted and dried under the sun for 2–3 days. Each cottonseed shell was slightly cracked on the tip with a nutcracker before being manually removed and separated from the kernel. Prior to grinding, dirt and all foreign materials were manually separated from the kernels. Samples of 200 kernels each, were ground with a Universal High-speed Grinder DFT-50 (Linda Machinery Company Ltd, Wenlin, Zhejiang Province, China) six times at 10 s intervals for each time. Each sample was mixed between grindings to minimize caking and separation by particle size.
Reference Methods
The determinations of oil content and fatty acid composition were conducted by the Testing and Supervisory Center of Agricultural Products, MOA (Zhengzhou, Henan Province, China) using Soxhlet extraction and gas liquid chromatography methods, respectively.
Oil Extraction and Fatty Acid Determination
Oil content in the cottonseed samples was determined using the Soxhlet extraction method [12] for oil content determination in cottonseed. Ground cottonseed kernels (40 g) were weighed into a 43 mm × 123 mm paper thimble (Whatman International Ltd., Maidstone, England) and covered with a piece of cotton wool before placing it in the Soxhlet extractor. Approximately 300 ml of commercial hexane (40% n-hexane, CAS No. 110-54-3 Ningbo Samreal Chemical Co. Ltd. China) was used for extractions. The amount of oil extracted by Soxhlet was considered to be the total amount of oil available in the sample.
Fatty acids were determined according to the trans-methylation procedure [1]. A 50-μL amount of the oil obtained from the Soxhlet method was transferred into a test tube. To form fatty acid methyl esters (FAME), 200 μL of 0.5 N metallic sodium in methanol (0.5 N methanolic base, CAS No7440-23-5, PubMed Supelco, Inc., Bellefonte, PA) was added to the extracted oil and the tube was capped and heated to 70 °C for 10 min with periodic vortex mixing. Upon cooling, 1 mL of brine and 1 mL of hexane were added and the contents were vortex mixed again. After allowing the phases to separate, 1 mL of the organic phase containing methyl esters was used for gas chromatography.
Gas Liquid Chromatography
FAME obtained were analyzed according to FAME standards [13] for fatty acid determination in cottonseed using gas chromatogram (Agilent 6890N, Agilent Technologies Inc., Palo Alto CA, USA) with an SP-2560 flexible fused capillary column (biscyanopropyl polysiloxane Supelco Bellefonte, PA, USA). The carrier gas was helium at a split ratio of 50:1 and with a linear velocity of 18 cm/s. A single injection of 1 μL was made per sample. The temperatures of the injection port and detector were 250 and 280°, respectively with gas flows of 40 mL/min for hydrogen and 450 mL/min for air. The initial oven temperature of 110 °C (which was held for 5 min) was increased at 3 °C/min to 215 °C and held again for 24 min. Methyl esters of myristic, palmitic, palmitoleic, stearic, oleic, linoleic, linolenic and 1-eicosenoic acids (Sigma Chemical Co., St. Louis, DE, USA) were used for internal standardization of the FAME of each sample. HP CHEMSTATION software (Agilent Technologies Inc., Wilmington, D., USA) was used in the identification and quantification of individual fatty acids according to their percentage areas on the calibration curve based on the signals from the detector of the chromatograph.
NIRS Analysis
Spectral Data Collection
Spectral data collection, processing and calibration were conducted with the WinISI II software (v1.5, FOSS NIRSystems, Silver Springs, MD, USA). Spectral measurements were performed using a near infrared scanning monochromator (NIRS System model 5000 NIRS Systems Inc, Silver Spring., MD, USA) equipped with a sample auto-charger. Powdered seed samples (about 3 g/sample) were placed in a small ring cup of 36 mm (inner diameter), and then scanned in the diffused-reflectance mode. Scanning was performed twice on each sample by rotating the ring cup to an angle of 90° before the second scan in order to minimize the effects of particle size. Each spectrum represented the average of 32 scans and was recorded as the logarithm of the reciprocal of reflectance (log (1/R)) [14]. Data were stored at every 2 nm interval in the wavelength range from 1,100 to 2,498 nm to give a total of 700 data points. The NIR spectra of each sample were transformed into principal components (which are linear combinations of the original data) for further chemometric analysis.
Definition of Calibration and Validation Sets
Discriminant analysis was used to define the sample population from which samples for calibration and external validation were selected solely on the basis of spectral data. The CENTER algorithm which is included in the WinISI II software (v1.5, FOSS NIRSystems, Silver Springs, MD, USA) was applied to calculate the distances that define the population [14].This algorithm performs principal component analysis by reducing the original spectra information (Log 1/R values) to a small number of linearly independent variables, thus making it easy to calculate spectral distances [15]. These new variables were used to calculate the center of the sample population and the distance from each sample in the population from that center (expressed as the Mahalanobis distance or Global H or GH), as well as the distance between a sample and its neighbor (expressed as Neighborhood H or NH). The Global H defines the population boundaries of a complete set and the Neighborhood H defines the boundaries of a subset. In the present study, the population was defined through a cutoff point of GH = 3, resulting in a new population of 1,174 samples, after 5 abnormal or extreme samples (considered as outliers) were removed [14]. The SELECT algorithm which is also included in the WinISI II software was applied to select suitable and representative samples for the calibration and validation sets. It arranges spectral distances of the whole sample set (from the shortest to the longest distance from the center of the population), and samples, represented by their corresponding spectra, are randomly selected to form the calibration and validation subsets [16]. In this study, a total subset of 289 samples was constructed by a cutoff of NH = 0.35. After adding another variant subset of 155 samples, the newly defined population with 444 samples was randomly split into two sets. One of every 3 samples was selected for the external validation set (n = 148) and the remainder were kept as the calibration set (n = 296).
Spectral Signal Pre-Treatment and Calibration
Calibrations were performed to obtain the regression models for oil and fatty acid contents between spectral data and laboratory reference values using the modified partial least squares (MPLS) regression method. In order to avoid over-fitting of each model, a minimum number of MPLS terms were selected from each model to perform internal cross-validation [17].The pre-treatment of spectral data was done with the CENTER algorithm. Data pre-treatment algorithms reduce or remove unwanted sources of variability in the sample data. They include a mathematical treatment that uses the raw spectra, or their first or second derivatives (to remove background differences whiles enhancing spectral differences); combined with gap sizes in data points over which the derivative is calculated [18]; and a smoothing algorithm that reduces random noise in the spectral data [19]. For example in 2,8,8,1, the first number indicates the order of derivative function (two is the second derivative of log 1/R); the second number is the gap (length in nm); the third number represents the number of data points (segment length) used in the first smoothing and the fourth number is the number of data points in the second smoothing which is normally set at 1 for no second smoothing [17]. Different spectral signal pre-treatment methods were tried for both calibration and internal cross-validation on two randomly selected fatty acids, and the best method was chosen based on the optimum results obtained.
Derivation and smoothing were combined with scatter correction algorithms which help to reduce differences in the spectra related to physical characteristics such as particle size and path length variation among samples [20]. The scatter correction algorithms that were assessed in this study in order to choose the most suitable were: multiple scatter correction (MSC), standard normal variate and detrending (SNV + D), standard normal variate (SNV), detrending (DET), standard multiple scatter correction (S-MSC), weighted multiple scatter correction (W-MSC) and inverse-multiple scatter correction (I-MSC). Two passes of outlier elimination were performed. Pre-treated spectral data was used to develop calibration models for oil content and the corresponding fatty acids. Calibration models were assessed using statistics that included the standard error of calibration (SEC), the coefficient of determination in calibration (R 2), the standard error of cross-validation (SECV) and the coefficient of determination in cross-validation (1-VR) [17]. Optimum calibration equations were obtained based on the highest R 2 or 1-VR and the lowest SEC or SECV values. The SD/SECV ratio which is also referred to as the relative predictive determinant for calibration (RPDc) was used to evaluate the performance of the calibration equations [21].
External Validation
The accuracy and precision of the calibration equations obtained was monitored with the WinISI software using the external validation set [21]. The indicators for external validation were: standard error of prediction (SEP); coefficient of determination in external validation (r 2) and the relative predictive determinant for external validation (RPD), which is the ratio of the standard deviation (SD) for the validation samples to the SEP. The RPD statistic provides a basis for standardizing the SEP [22]. The slope which is the linear regression of the reference values against predicted values as well as bias which constitute the average difference between the reference and predicted values, were also calculated for each model. A slope value close to 1 and a bias close to 0 contribute to the predictive accuracy of a calibration model.
Results and Discussion
Variability for Oil Content and Fatty Acid Composition
The results in Table 1 show the relative values for the means, standard deviations and the range of oil and individual fatty acids in the calibration and validation sets. The diverse sources of the samples as well as the differences in the years and environments of cultivation contributed to the high variability in the sample population. The concentration and scale of variability in oil and fatty acid contents of the samples used in this study are similar to those reported by other authors [1, 23, 24]. There was, however, a reduction in the number of samples for linolenic acid and 1-eicosenoic acid in the calibration and validation sets because some samples had very low contents and could not be detected. The values for oil and fatty acid contents ranged from 0.140 to 61.950% in the calibration set and from 0.120 to 62.270% in the validation set, indicating obvious differences within this population. The widest ranges were observed for oil content (OC), linoleic acid (C18-2), palmitic acid (C16-0) and oleic acid (C18-1); followed by linolenic acid (C18-3), stearic acid (C18-0), myristic acid (C14-0) and palmitoleic acid (C16-1); and the narrowest range was observed for 1-eicosenoic acid (C20-1) both in the calibration and validation sets. Variability within the population as shown by the standard deviation values ranged between 0.025 and 3.274% in the calibration set and between 0.024 and 3.162% in the validation set. The highest variability was observed for oil content, followed by linoleic, oleic, palmitic, stearic and linolenic acids, while palmitoleic, myristic and 1-eicosenoic acids had the lowest values for variability.
Differences in the means and standard deviations between the calibration set and the validation set were minimal. Except for linolenic acid which had a relatively wider range in the calibration set compared to the validation set, the ranges for the remaining fatty acids in the calibration set were similar to those in the validation set. This indicates that both calibration and external validation sets were selected properly and could represent the total variation in the traits of interest, thus, making the population suitable for NIRS analysis.
Selection of the Spectral Pre-Treatment Method
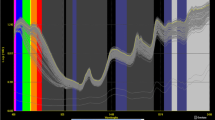
Figure 1a shows the original NIRS spectra of all cotton seed kernels samples used in this study. The original spectra have broad peaks and considerable baseline shifts (vertical offsets) There are also parallel shifts between the spectra caused by scattering of samples [25]. To resolve broad peaks and eliminate baseline and parallel shifts, a number of mathematical treatments combined with scatter correction algorithms were tested on the spectra.

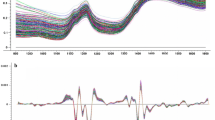
Pre-processing stages of cotton seed kernel spectra. a Original spectra of cotton seed kernel samples in the 1,100 to 2,490 nm wavelength range, b 2,8,8,1 treated spectra of samples in the 1,100 to 2,490 nm wavelength range, c 2,8,8,1 and I-MSC treated spectra of samples in the 1,100 to 2,490 nm wavelength range
In order to ascertain the best mathematical treatment and scatter correction procedure to develop the calibration equations, one saturated fatty acid (palmitic acid) and one unsaturated fatty acid (linoleic acid) were randomly selected to conduct a series of calibrations based on the MPLS regression method using different mathematical treatments combined with scatter correction algorithms. Calibrations were first performed with nine different mathematical treatments (0,0,1,1; 1,4,4,1; 1,8,8,1; 1,12,12,1; 1,16,16,1; 2,4,4,1; 2,8,8,1; 2,12,12,1; 2,16,16,1) without scatter correction on the spectral data of both fatty acids (Table 2).
The RPDc was used to assess the robustness of each equation because it constitutes a standardization of the SECV and can indicate at a glance, the performance of an equation. RPDc values that are more than 3 imply equations with excellent calibration; values between 2 and 3 are considered equations with very reliable predictions; and values between 2 and 1.5 imply limited prediction effects, while values below 1.5 indicate equations with unreliable correlations [22].
Significant effects on the calibration equations for fatty acids were observed when the mathematical treatment 0,0,1,1 (used as control) was compared to the other mathematical treatments. Good calibration models for palmitic acid were developed. They had higher RPDc values with maths treatment 1,4,4,1 (RPDc = 2.156) and 2,8,8,1 (RPDc = 2.222). With regards to linoleic acid, higher calibration effects were obtained by using various math treatments, e.g. 1,8,8,1 (RPDc = 4.834), 1,12,12,1 (RPDc = 4.631), 2,8,8,1 (RPDc = 4.678) and 2,12,12,1 (RPDc = 4.736). Therefore, although the 1,8,8,1 treatment showed the highest RPDc value for linoleic acid among all the treatments, the 2,8,8,1 treatment was selected as the one that produced optimum equations for both palmitic acid and linoleic acid.
In order to reduce parallel shifts due to scattering of samples, the 2,8,8,1 treatment was then used to perform separate calibrations for both fatty acids combined with MSC, SNV + D, SNV, DET, S-MSC, W-MSC and I-MSC as shown in Table 3. Out of all the combinations, the 2,8,8,1 treatment with I-MSC produced the best results for both palmitic acid (RPDc = 2.272) and linoleic acid (RPDc = 6.186). This combination, was thus chosen as the optimum method to fit the spectral data for oil content and the rest of the fatty acids to their corresponding reference values. Figure 1b shows the spectra with 2,8,8,1 mathematical treatment where the spectral differences are more enhanced with more defined absorption peaks, and the baseline shift is corrected. The spectra treated with 2,8,8,1 combined with I-MSC are spaced much closer to each other since scattering has been removed and spectral variation has been greatly reduced as shown in Fig. 1c.
Calibration
Calibration equations developed for the contents of oil and the remaining fatty acids using the 2,8,8,1 mathematical treatment and the I-MSC scatter correction method are shown in Table 4. The coefficients of determination (R 2), standard errors of cross-validation (SECV), standard errors of calibration (SEC) and coefficients of determination in cross-validation (1-VR) were determined for all equations [19]. The equation for total oil content showed high R 2 (0.996) and 1-VR (0.995) with low SEC (0.197) and SECV (0.233) and is considered to be the best equation with the highest RPDc value (14.06). Oleic acid also had a high correlation with high R 2 (0.891) and 1-VR (0.817), low SEC (0.462) and SECV (0.602) and a relatively high RPDc 2.329. Myristic acid had an R 2 of 0.778, a 1-VR of 0.738, an SEC of 0.049, an SECV of 0.054 and an acceptable RPDc of 1.937. Palmitoleic, stearic, linolenic and 1-eicosenoic acids had very low R 2 (0.481, 0.485,0.233 and 0.526, respectively) and 1-VR (0.409, 0.453, 0.112 and 0.501, respectively), and although they also had low SEC (0.027, 0.129, 0.041 and 0.012, respectively) and SECV (0.029, 0.133, 0.044 and 0.013, respectively), their RPDc (1.305, 1.346, 1.062 and 1.391, respectively) were below the limit for reliable predictability [22].
External Validation
With external validation, the predictive abilities of all calibration equations were assessed (Table 5). As in calibration and cross-validation, optimum equations were selected based on low standard errors of prediction (SEP) and high coefficients of determination for external validation (r 2) [17]. Except for the equations for palmitic and oleic acids which had relatively high error values, all other equations had low SEPs. High coefficients of determination were obtained for the equations for oil content (r 2 = 0.993) and linoleic acid (r 2 = 0.963) whiles those for myristic, palmitic and oleic acids (r 2 = 0.753, 0.777 and 0.795, respectively) were relatively high. The lowest values were obtained for palmitoleic, stearic, linolenic, and 1-eicosenoic acids (r 2 = 0.516, 0.472, 0.101 and 0.224, respectively). All equations had biases close to ‘0’ and slopes around ‘1’. The highest RPDvs were obtained from the equations for oil content (11.495) and linoleic acid (5.026). RPDv values corresponding to myristic acid (1.724), palmitic acid (1.914) and oleic acid (1.999) were relatively high while those for palmitoleic acid (0.686), stearic acid (0.792), linolenic acid (0.475) and 1-eicosenoic acid (0.619) were low. All external validation results were in agreement with the assessment statistics for calibration and internal cross-validation with minimal differences.
As shown on the regression plots of reference versus predicted (NIRS) values for oil and fatty acid contents (Fig. 2) not all external validation results obtained in the prediction of individual constituents were accurate. The regression plots for oil content showed a high accuracy in the estimation of this trait with an r 2 of 0.993 and an SEP of 0.273. This was followed by the regression plot for linoleic acid (r 2 = 0.963, SEP = 0.470). There was however less accuracy in the prediction of the remaining fatty acids including myristic acid (r 2 = 0.753) and palmitic acid (r 2 = 0.777). According to the guidelines for the interpretation of RPDv which are similar to those for RPDc [22], the equations for total oil content and linoleic acid are good enough to be used for quality assurance and research applications as well sample screening for breeding programs. Those for myristic, palmitic and oleic acids can be used only for screening whiles those for stearic, palmitoleic and 1-eicosenoic acids are unusable for now.

Regression plots showing the relationship between oil and fatty acid percentages as determined by the reference method and predicted percentages by NIRS. a Oil content (OC), b myristic acid (C14-0), c palmitic acid (C16-0), d linoleic acid (C18-2). Dotted and solid lines represent 1:1 (target) and regression lines respectively, r 2 is the coefficient of determination between the reference method and the NIRS method
The highest performances in calibration equations for individual contents corresponded to the traits for total oil and linoleic acid contents, both of which had high variability as shown by their standard deviations in the calibration set (3.274 and 2.165, respectively) and external validation set (3.16 and 2.43, respectively) (Table 1). This suggests that the variability for oil and fatty acid composition is a major factor that contributes to achieving excellent calibration equations [26]. In addition to the effect of fatty acid concentration on predictions, the failure to accurately determine certain individual fatty acids could also be due to similarities in their NIR absorption patterns since different fatty acids have the same absorbing molecular group [27]. An improvement in the filtering of spectral data could help reduce this problem. Another option could also be the application of alternative regression methods, although the MPLS methods have provided reliable results in the evaluation of most agricultural products [28–30]. In a comparative study of fatty acids in soybean using linear and non-linear regression methods [31], a superior performance of non-linear methods based on their respective RPD values was observed, although the sensitivity of the non-linear methods to outliers was also acknowledged.
Conclusion
The sample population used in the present study had a wide range of variability due to the differences in origin as well as the years and environments in which they were grown. The comparative analysis of mathematical treatments combined with different scatter correction methods were necessary to improve upon the accuracy of the predictions. The predictive abilities of the calibration equations were in line with their respective assessments in external validation. In the current experiment, oil content and linoleic acid showed high RPDv (11.495 and 5.026, respectively), whiles myristic, palmitic and oleic acids showed acceptable RPDv (1.724, 1.914 and 1.999, respectively). These results suggest that, for now, NIRS could be suitably used as a standard screening method for the contents of these five traits in large scale cotton breeding programs where the use of conventional screening methods may be a limiting factor.
References
Dowd MK, Boykin DL, Meredith WL, Campbell BT, Bourland FM, Gannaway JR, Glass KM, Zhang J (2010) Fatty acid profiles of cottonseed genotypes from the national cotton variety trials. J. Cotton Sci 14:64–73
Sacks FM, Katan M (2002) Randomized clinical trial on the effects of dietary fats and carbohydrates on plasma lipoproteins and cardiovascular diseases. Am J Med 1139(2):13S–24S
Simopoulos AP (1999) Essential fatty acids in health and chronic diseases. Am J Clin Nutr 70(1):5608–5698
Lawhon JT, Cater CM, Mattil KF (1976) The evaluation of the food use potential of sixteen varieties of cottonseed. J Am Oil Chem Soc 54(2):75–80
Wang W, Paliwal J (2007) Near-infrared spectroscopy and imaging in food quality and safety. Sens Instrum Food Qual Saf 1:193–207
Wu JG, Shi CH, Zhang HZ (2006) Study on developing calibration model of fat acid composition in intact rapeseed by near infrared reflectance spectroscopy. Spectrosc Spect Anal 26:259–262
Tilman BL, Gorbet DW, Person G (2006) Prediction oleic and linoleic acid content of single peanut seed using near-infrared reflectance spectroscopy. Crop Sc 46:2121–2126
Sato T, Maw A, Katsuta M (2003) NIR reflectance spectroscopic analysis of the FA composition in sesame (Sesamum indicum L.) seeds. J Am Oil Chem Soc 80:1157–1161
Davrieux F, Allal F, Piombo G, Kelly B, Okulo JB, Thiam M, Diallo OB, Bouvet J-M (2010) Near infrared spectroscopy for high-throughput characterization of shea tree (Viterllaria paradoxa) nut fat profiles. J Agric Food Chem 57:7811–7819
Lordelo MM, Shaaban SA, Dale NM, Calhoun MC, Vendrel PF, Davis AJ (2008) Near infrared reflectance spectroscopy for the determination of free gossypol in cottonseed meal. Appl Poult Res 7:243–248
Kohel RJ (1998) Evaluation of near infrared reflectance for oil content of cottonseed. J Cotton Sci 2:23–26
Wan PJ, Pakarinen DR, Wakelyin PJ (1998) Concerns for the determination of free fatty acid in cottonseed. J Am Oil Chem Soc 75(10):1321–1324
AOCS Method Ce 1-62 (2005) Fatty acid composition by gas chromatography. AOCS Methods American Oil Chemists Society
Shenk JS, Westerhaus MO (1993) Analysis of agricultural and food products by near infrared reflectance spectroscopy. Infrasoft International (ISI), Silver Spring
Shenk JS, Westerhaus M (1991) Population definition, sample selection, and calibration procedure for near infrared reflectance spectroscopy. Crop Sci 31:469–474
Paz P, Sanchez MT, Perez-Marin D, Guerrero JE, Garrido-Varo A (2009) Instantaneous quantitative and qualitative assessment of pear quality using near infrared spectroscopy. Comput Electron Agr 69:24–32
Shenk JS, Westerhaus MO (1996) Calibration the ISI way. In: Davis AMC, Williams PC (eds) Near infrared spectroscopy: the future waves. NIR Publications, Chichester, pp 198–202
Hruschka WR (1987) Data analysis: wavelength selection methods. In: Williams P, Norris K (eds) Near-infrared technology in the agricultural and food industries. American Association of Cereal Chemistry, St Paul, pp 35–55
Savitzky A, Golay MJE (1964) Smoothing and differentiation of data by simplified least squares procedures. Anal Chem 8:1627–1639
Barnes RJ, Dhanoa MS, Lester SJ (1989) Standard normal variate transformation and de-trending for near infrared diffuse reflectance spectra. Appl Spectrocs 43:772–777
Windham WR, Mertens DR, Barton FE (1989) Protocol for NIRS calibration: sample selection and equation development and validation in near infrared reflectance spectroscopy (NIRS): analysis of forage quality. In: Marten GC, Shenk JS, Barton FE (eds) USDA-ARS agriculture handbook 643, Rev edn edn. US Government Print Office, Washington, pp 96–103
Williams P, Norris K (eds) (2001) Near infrared technology in agricultural and food industries, 2nd edn. American Association of Cereal Chemistry, St Paul
Hamilton KA, Pyla PD, Breeze M, Oslon T, Li M, Robinson E, Gallagher SP, Sorbet R, Chen Y (2004) Bollgard II cotton: compositional analysis and feeding studies of cottonseed from insect-protected cotton (Gossypium hirsutum L.) producing the Cry 1Ac and Cry 2Ab2 proteins. J Agric Food Chem 52:6969–6976
Nyanzi SA, Carsternsen B, Schwack W (2005) A comparative study of the fatty acid profiles of Passiflora seed oils from Uganda. J Am Oil Chem Soc 82(1):41–44
Uddin M, Okazaki E, Fukushima H, Turza S, Yumiko Y, Fukuda Y (2006) Nondestructive determination of water and protein in surimi by near-infrared spectroscopy. Food Chem 96:491–495
Velasco L, Goffman D, Becker HC (1999) Development of calibration equations to predict oil content and fatty acid composition in Brassicaceae germplasm by near-infrared reflectance spectroscopy. J Am Oil Chem Soc 76:25–30
Holdman RT, Edmondson PR (1956) Near infrared spectra of fatty acids and some related substances. Anal Chem 28:1533–1537
Patil AG, Oak MD, Taware SP, Tamhankar SA, Rao VS (2010) Nondestructive estimation of fatty acid composition in soybean (Glycine max (L) Merrill) seeds using near-infrared transmittance spectroscopy. Food Chem 12:1210–1217
Wu JG, Shi CH (2004) Prediction of grain weight, brown rice weight and amylase content in single rice grains using near infrared reflectance spectroscopy. Field Crop Res 87:13–21
Sato T, Takahata Y, Noda T, Yanagisawa T, Morishita T, Saki S (1995) Nondestructive determination of fatty acid composition of husked sunflower (Helianthus annuus L.) seeds by near-infrared spectroscopy. J Am Oil Chem Soc 72:1177–1183
Kovalenko IV, Rippke GR, Hurburgh CR (2006) Measurement of soybean fatty acids by near-infrared spectroscopy. Linear and nonlinear calibration methods. J Am Oil Chem Soc 83(5):421–427
Acknowledgments
The project is supported by the National High Technology Research and Development Program of China (2009AA101104), the Fundamental Research Funds for the Central Universities, and 151 Foundation for the Talents of Zhejiang Province.
Author information
Authors and Affiliations
Corresponding authors
About this article
Cite this article
Quampah, A., Huang, Z.R., Wu, J.G. et al. Estimation of Oil Content and Fatty Acid Composition in Cottonseed Kernel Powder Using Near Infrared Reflectance Spectroscopy. J Am Oil Chem Soc 89, 567–575 (2012). https://doi.org/10.1007/s11746-011-1945-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11746-011-1945-2