
Abstract
The porphyrias are a heterogeneous group of metabolic diseases resulting from a variable catalytic defect of one of the eight enzymes involved in the heme biosynthesis pathway; they are mostly inherited diseases, but in some circumstances the metabolic disturbance may be acquired. The specific patterns of tissue overproduction (and hence accumulation and excretion) of toxic heme precursors, associated with each enzymatic deficiency, are responsible for the characteristic biochemical and clinical features of each of these diseases. Moreover, even in the presence of a specific inherited enzymatic defect, many different environmental factors (such as drugs, calorie restriction, hormones, sunlight exposition, infections, etc.) often play a key role in triggering the clinical expression of the various forms of porphyrias. The porphyrias are often misdiagnosed diseases, due their multiform clinical manifestations, able to mimic many other more common diseases. For this reason, many different specialists, such as surgeons, psychiatrists, gastroenterologists, neurologists, emergency physicians and dermatologists may be variably involved in the diagnostic process, especially for the forms presenting with acute and life-threatening clinical features. According to the clinical features, the porphyrias can be classified into neuropsychiatric (characterized by neurovisceral crises involving autonomic and central nervous system but also the liver and the kidney with possible consequences in terms of neurological, psychic, cardiac, respiratory, liver and kidney functions), dermatological (mostly presenting with cutaneous lesions due to photosensitivity), and mixed forms. From a strictly clinical point of view, porphyrias presenting with neurovisceral attacks are also referred as acute porphyrias: they are the object of the present review. An accurate diagnosis of acute porphyria requires knowledge and the use of correct diagnostic tools, and it is mandatory to provide a more appropriate therapeutic approach and prevent the use of potentially unsafe drugs, able to severely precipitate these diseases, especially in the presence of life-threatening symptoms. To date, availability of a relatively stable haem preparation (haem arginate) has significantly improved the treatment outcome of acute porphyric attacks, so the knowledge about the diagnosis and the management of these diseases may be relevant for physicians working in internal medicine, neurology and emergency units.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
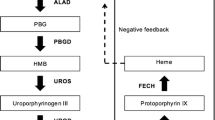
The term porphyrias refers to a heterogeneous group of metabolic diseases resulting from a variable catalytic defect of one of the seven enzymes involved in the haem biosynthesis pathway (Fig. 1). The specific patterns of tissue overproduction (and hence accumulation and excretion) of toxic haem precursors, associated with each enzymatic deficiency, are responsible for the characteristic biochemical and clinical features of each disease (Table 1) [1–3].

Metabolic pathway of heme biosynthesis
The porphyrias are mostly inherited diseases (Table 1), mainly in an autosomal dominant manner (often with incomplete penetrance), even if autosomal recessive and more complex patterns of inheritance are demonstrated [3]. In some circumstances (porphyria cutanea tarda, lead poisoning), the metabolic disturbance may be acquired, as an effect of factors able to induce a variably reversible enzymatic inhibition [4]. Moreover, even in the presence of a specific inherited enzymatic defect, many different environmental factors (such as drugs, calorie restriction, hormones, sunlight exposition, infections, etc.) often play a key role in triggering the clinical expression of the various forms of porphyrias [1, 3–6]. Similarly, the interaction with other genes (“modifying genes”) has been demonstrated to be crucial on influencing the variable penetrance of these diseases [7, 8].
Although porphyrias are rare conditions, they have a pan-ethnic diffusion with a widely variable prevalence (0.5–10 per 100.000 in different populations) from country to country and on the type of porphyria [3]. To date, no data are available about their prevalence in Italy.
The porphyrias are often misdiagnosed diseases due to their multiform clinical manifestations, able to mimic many other diseases. For this reason, many different specialists, such as surgeons, psychiatrists, gastroenterologists, neurologists, emergency physicians, or dermatologists, may be variably involved in the diagnostic process, especially for the forms presenting with acute and life-threatening symptoms (see below).
As clinical features alone are not sufficiently specific either to confirm a diagnosis or to distinguish between the different forms, a correct interpretation of the appropriate biochemical tests is mandatory for accurately diagnosing and managing these diseases [2, 9–12].
General classification of porphyrias
The seven main types of porphyrias (Table 1) are presently classified as erythropoietic [congenital erythropoietic porphyria (CEP) and erythropoietic protoporphyria (EPP)] or hepatic (all the other forms) in type, depending on the primary organ in which excess production of porphyrins or other precursors takes place [2, 3].
Erythropoietic porphyrias and three hepatic porphyrias, namely, porphyria cutanea tarda (PCT), hereditary coproporphyria (HC) and variegate porphyria (VP) present clinically with variable grade of cutaneous symptoms consequent to photosensitivity due to accumulation of porphyrins, (which are highly photoactive molecules) in plasma and skin. Four hepatic porphyrias, namely, acute intermittent porphyria (AIP), hereditary coproporphyria (HCP), variegate porphyria (VP) and the very rare ALA-dehydrase deficit porphyria may present clinically with recurrent severe acute neurovisceral crises, due to an accumulation in tissues and plasma of non-porphyrin heme precursors [13].
According to these clinical features, the porphyrias can also be broadly classified into neuropsychiatric (neurovisceral crises), dermatological, and mixed forms. From a strictly clinical point of view, porphyrias presenting with neurovisceral attacks are also referred to as the acute porphyrias [2, 3, 13] (Tables 1, 2). These forms will be the object of the present review.
The acute porphyrias
Classification: four inherited and one acquired form
Four of the seven inherited varieties of porphyrias may present clinically as recurrent attacks of neurovisceral symptoms (neurovisceral crisis) and are collectively known as “the acute porphyrias” [13, 14]. The most common of these disorders is acute intermittent porphyria (AIP) [1], with a worldwide prevalence within the range of 1 ± 10/100.000 [3]. Acute attacks of AIP are clinically more severe, even though they are formally indistinguishable from those of the less common conditions: variegate porphyria (VP) [15] and hereditary coproporphyria (HC) [16], and of the extremely rare homozygous ALAD-deficient porphyria (ALAD-P) [17]. Quite similar acute clinical manifestations may also be seen in lead poisoning (a condition also referred as plumboporphyria), which can be considered a classical example of an acquired disturbance of haem synthesis (Table 1) [18]. Clinical and biochemical features of the above-mentioned acute porphyrias are summarized in Table 2.
Clinical features
The clinical syndrome of the acute porphyric attack and its precipitant factors
The cardinal sign of an acute porphyria is the porphyric attack, whose clinical features are greatly variable (Table 3) even if other symptoms may occasionally occur, the most common complaint is severe abdominal pain, usually excruciating, mimicking an “acute abdomen” and prompting immediate attention. It is generally accompanied by nausea and vomiting, and by neurological and psychiatric symptoms (ranging from depression and apathy to extreme agitation or psychosis with hallucinations). Back pain extending to or involving the proximal limbs is also frequently present, together with signs of vegetative dysfunction (hypertension with postural hypotension, tachycardia and constipation) [1, 2, 13].
An acute attack may be preceded by a period of minor behavioral changes such as anxiety, irritability, restlessness and insomnia, and may proceed rapidly to symptoms of severe autonomic and acute motor and sensory neuropathy. Muscular weakness, particularly a proximal motor neuropathy, is quite common. It can progress to general paralysis leading to respiratory impairment and death from cardiorespiratory arrest, resembling a Guillain−Barré syndrome. Mild sensory changes frequently accompany the predominantly motor neuropathy often in a “bathing trunk” distribution [14].
Hyponatremia and hypomagnesemia may occur as a result of dehydration, nephrotoxicity, or occasionally inappropriate antidiuretic hormone secretion [19]. These water/electrolyte disturbances may contribute to neurological and psychiatric symptoms of the porphyric acute crisis.
The extreme variability in the clinical picture of the porphyric attack is the reason for its denomination of “little imitator” (the great imitators being syphilis and hysteria) [2, 20]; it is also the reason why many patients remain frequently clinically ill-defined: descriptions of patients subjected to abdominal surgeries without any pathological finding, and of patients admitted to mental hospitals for long periods due to inaccurate diagnosis are reported, as well [20, 21].
The clinical manifestation of an acute porphyria is extremely rare before puberty (except in ALAD-P, where neurovisceral crises may arise early in the childhood) with a peak age of presentation in the early 30s, and are four to five times more common in women than in men [2, 3, 22]. The disease remains latent throughout life in some people, (it has been estimated that only 10–15% of gene carriers experience the clinical syndrome), even in the presence of precipitating factors [23]. A third of patients have no family history, such conditions probably having remained latent or unidentified for several generations. The frequency and severity of the attacks vary widely: some people experience frequent and sometimes life-threatening attacks, even in the apparent absence of exogenous precipitant factors [1].
In most cases an acute porphyric attack results from having been triggered by some “porphyrinogenic challenge” (exogenous precipitating factors), such as a recent introduction of a new drug, or changes in sex hormone balance (as during menses or hormonal therapies), local or general anesthesia, sedative use (especially barbiturates), alcohol misuse or consumption of illicit substances (amphetamines, cocaine and derivatives), prolonged fasting or attempts at weight reduction by diet restriction, current mental or physical stress, or acute viral or bacterial infection.
Attacks may occur during pregnancy, when estrogen concentrations are high, and the patient should be advised to avoid pregnancy until remission has been present for at least 2 years; nevertheless in most cases pregnancy is symptom-free. On the contrary, particular care should be taken at the delivery and post-delivery periods: they represent important precipitant factors (stress, blood losses, starvation, etc.) and a high rate of porphyric attacks have been described throughout. If acute attacks do occur during pregnancy or during delivery or in the immediate post-delivery period, they should be treated as is customary (see below) [24].
The diagnosis of an underlying porphyric condition greatly depends on the awareness of the clinician of the possibility of an acute porphyric state. The probability of the disease being present may be significantly increased when the patient can report that he/she is a member of a porphyria kindred, or is actually a carrier of porphyria, in the best case verified by a personal warning card, issued by the specialist responsible for the diagnosis. This is the strategy applied by the European associations of patients affected by porphyria, and proposed by some European initiatives for protection of these patients against unsuitable (and potentially dangerous) medical management in emergencies [25].
The role of drugs in precipitating porphyric attacks, due to their possible effect on haem metabolism (see below), is well-established, so it is mandatory that any drug treatment for a patient with porphyria is prescribed according to an accurate reference to a drug list [13]. This is particularly important in surgery, many anesthetics being well-known powerful precipitant factor of acute attack [5]. Table 4 resumes the most commonly used drugs considered as unsafe or potentially unsafe for patients affected by acute porphyria. Complete lists of potentially safe and unsafe drugs are continuously upgraded, and they are available on the net, for example, at http://www.drugs-porphyria.org/ and http://www.porphyria-europe.com. Some drugs are strictly forbidden, due to their association with great number of severe attacks, as it may be argued especially by the structure of a former website (made by a collaborative initiative of the Sweden and the Norwegian Porphyria Centers); however, most drugs are considered only potentially dangerous, and the majority of patients often tolerate them. For this last group of drugs, a commonsense assessment of benefit versus risk is needed: an acute attack is in fact less likely to be precipitated in case of latent disease, or if the patient has experienced only a single attack, or if the concentrations of urinary metabolites (porphobilinogen, and particularly aminolevulinic acid, see below) are within the normal range at the time of the prescription [26].
Associated pathologic conditions
Different epidemiological studies have suggested a connection among AIP, hypertension and renal disease. Hypertension has often been described as well in HCP and VP, but the incidence of renal involvement in these conditions remains ill-defined. Treatment of hypertension decreases the risk for renal damage in carriers of a gene for acute porphyria; symptomatic patients should especially be managed with a regular monitoring of blood pressure and renal function [27].
A strong association between acute porphyria and hepatocellular carcinoma (HCC) has also been described: 27% of individuals carrying the W198X mutation of the PBG-D gene are reported to be affected by HCC in a mortality study [28].
Pathophysiology
The pathophysiologic mechanisms underlying the clinical features of acute porphyrias are still poorly understood: the recent development of an animal model of gene knockout AIP will probably allow rapid progress in this area [29]. Possible mechanisms include: (1) damage by free radicals (not completely confirmed) [30], (2) direct neurotoxicity of the accumulating porphyrin precursor δ-aminolevulinic acid (ALA) and (3) haem deficiency in nervous tissue [31]. The biochemical landmark of the porphyric acute attack is the significant accumulation of non-porphyrin precursors δ-aminolevulinic acid (ALA) and porphobilinogen (PBG) in biological fluids, as a consequence of massive excretion from the liver due to induction of haem biosynthesis [2]. The haem synthesis is effected by a sequence of enzymatic steps, as discussed above (Fig. 1): the overall synthetic rate depends mostly on the current activity of the first enzyme of the chain, 5-aminolevulinate synthetase (ALA-S) (a rate-limiting step). Liver ALA-S activity is feedback-governed by the amount of free haem present within the hepatocyte. A decrease of this “regulatory” free haem pool gives rise to ALA-S induction, and hence to an acceleration of haem biosynthesis, as an effect of increased inflow of metabolites into the pathway. A second rate-limiting enzymatic reaction is the third step of haem synthesis [catalyzed by porphobilinogen deaminase (PBG-D)]. In contrast to ALA-S step, which represents a variable control step for the synthetic process, the constantly limited capacity of the PBG-D step functions as an “inbuilt safety valve” against overproduction of toxic porphyrins down the synthetic chain. In the acute porphyrias, the enzymatic activity of PBG-D is significantly compromised: as a consequence of a genetically determined deficiency in AIP; probably as an inhibitory effect by porphyrins produced in surplus in VP and HC. Restoration of normal porphyrin metabolism in an AIP symptomatic patient (who thereafter became attack-free) by liver transplantation, confirms the key role of the liver as a source of non-porphyrin precursors in the circulation [32]. Exogenous and endogenous factors able to induce the activity of the rate-limiting enzyme (ALA-S) (as in the case of drugs or pathological conditions reducing the free haem pool) are thus known to trigger porphyric manifestations. These may be consequent to overproduction of toxic metabolites at the third, enzyme-deficient step of the chain, but also to impaired production of the end-product, haem. It has been demonstrated that ALA is neurotoxic [33]. The blood-brain barrier protects the brain from toxic agents, but certain areas—such as the hypothalamus and limbic area—do not have such protection [14]. Moreover, circulating porphyrins and their precursors can cause vascular injury, leading to impaired permeability, and resulting in reversible focal edema in the brain [14]. The autonomic and peripheral nervous systems are especially vulnerable to this toxic action, due to absence of a specific barrier protection. Vulnerability of neurons to toxic reactions can vary among individuals, and, therefore some patients are more prone to paresis than others: some patients with acute porphyria have constantly elevated levels of porphyrins and their precursors during the symptom-free phase, but even their excretion level increases during an acute attack [23]. ALA has also been suggested as a carcinogenic for the liver [28]. ALA can cross the placenta and possibly cause toxicity to the developing fetal brain [24].
Diagnosis
Biochemical assessment: a key role for diagnosis in the acute phases and for the management of patients
When a patient known to be affected by acute porphyria presents with symptoms of a possible acute attack, the obvious question is whether those symptoms are due to its disease: not all symptoms in porphyric patients are due to porphyria; moreover, porphyric patients may be affected by other diseases. The diagnostic problem is obviously even more challenging in the case of a patient not known to have porphyria, when the diagnostic explanation of a porphyric crisis may not be thought of by the physician. Table 4 summarizes some of most common pathological conditions whose differential diagnosis should include an acute porphyria crisis.
In contrast to the ALAD deficiency porphyria, which may start in early infancy, clinical symptoms of PAI, HC, and VP almost never present before puberty [17]. HC and VP also belong to the group of cutaneous porphyrias: in these cases, skin fragility and bullous eruptions may be important (and often the sole) presenting symptoms. Even though AIP may be characterized with more frequent and severe neurovisceral symptoms, the acute neurological presentation does not differ qualitatively among the different forms of acute porphyria, including lead poisoning [13, 14, 16].
Clinical features alone are not sufficiently specific either to confirm a diagnosis or to differentiate between the various forms of acute porphyria, so a prompt (in acute phase of disease) assessment and interpretation of the appropriate laboratory biochemical tests (i.e. determination and quantification of porphyrin and non-porphyrin precursor patterns in biological samples) is mandatory for an accurate diagnosis, and a successful management of these diseases [2, 9–12].
An acute attack of porphyria is invariably associated with an increase of urinary excretion of non-porphyrin precursor [aminolevulinic acid (ALA) and porphobilinogen (PBG)]. So, in the evaluation of a patient suspected to have an acute porphyric attack, a fresh, light-protected urine sample should be sent to a specialist laboratory for adequate assessment of ALA and PBG concentrations (to date, HPLC assays are the most accurate, but rapid, column-chromatographic screening tests are available) [13, 34]. A diagnostic clue may be provided by urine darkening (red tint, varying from port to diluted strawberry sap) on standing, as an effect of PBG polymerization to porphyrins and other pigments (such an effect being typically enhanced by sunlight exposure). The Watson-Schwartz test may be used as a simple assay to test the presence of elevated urinary PBG levels (and probably a simplified kit, suitable for “bedside” assessment, will be made soon available). This test may be considered a “first-line” guide to confirm (or rule-out) a suspicion of AIP, PV and CP diagnosis (all characterized by PBG urinary increase, especially during the acute attack), whereas it is not suitable for ALA-D porphyria or plumboporphyria (both characterized by only ALA urinary increase).
Between attacks, however, urinary concentrations of these precursors (PBG, and particularly ALA) may return to a normal level, making more difficult an accurate diagnosis when the patient is between the acute crises. A typical plasma red fluorescence under UV light (at 626 ± 628 nm) is usually present in VP, and it also is a valuable fluorometric diagnostic tool for family studies [35].
AIP, VP, and HC may be differentiated by analysis of fecal porphyrin patterns (Table 2). The diagnosis of AIP can be further confirmed by the demonstration of reduced red cell activity of porphobilinogen-deaminase (a value <35–40% out of the normal range is considered highly suggestive of AIP); in some cases it is the sole liver enzyme deficient, making it necessary to perform enzymatic studies on liver biopsy samples [36]. Lead intoxication and the extremely rare ALA-dehydrase (ALA-D) deficiency porphyria, are both due to a significant decrease in activity of ALA-D (and an increase in ALA-S activity), with consequent massive increase of urinary ALA in the presence of normal levels of PBG. Both conditions are also characterized by a significant increase in urinary excretion of coproporphyrin III, and increased levels of erythrocyte zinc protoporphyrin. The decreased activity of ALA-D can be restored by thiols and zinc ions in lead intoxication, but not in ALA-D deficiency porphyria, [37]. Moreover, patients usually present with sideroblastic anemia [3]. Isolated ALA urinary increase may arise also from oral ALA ingestion, as in the cases of systemic ALA loading, used during the photodynamic localization and treatment of a variety of malignant lesions.
The diagnostic interpretation of these tests is easier during the symptomatic phase (acute crisis) of the disease; in contrast, during remission the tests may partially lose their diagnostic specificity and sensitivity, making more difficult the diagnosis in absence of a specialist’s evaluation and specific genetic tests [38]. Nevertheless, biochemical methods still remain indispensable to monitor the level of activity of the disorder, and to evaluate the risk of development of the organ diseases (liver and kidney), associated with porphyria [11].
Genetic assessment: a key role in diagnosis confirmation, screening and family studies
With the availability of tools for genetic assessment, DNA analysis and carrier detection has become rapid and reliable in almost every case. Besides the diagnostic confirmation for symptomatic patients and the evaluation of the association between genotype and phenotype (i.e. the clinical significance of each single gene mutation), genetic assessment has a key role in family studies (identification of asymptomatic carriers in kindred): asymptomatic carriers may remain completely undiagnosed in absence of genetic screening, due to the low clinical penetrance of the genetic defect (even <25% for AIP), as mentioned above [9, 38, 39]. The risk of developing potentially acute and life-threatening fatal attacks even in these asymptomatic carriers if exposed to possible precipitating factors (including certain medical treatments or causing unnecessary surgical procedures) makes it essential to exclude or confirm the diagnosis of porphyria in all relatives, whenever it has been diagnosed in any family member. Every gene carrier should be informed as soon about the nature of the disorder, and counseled with regard to exposures to avoid, the measures to take if symptoms should appear, and about the probabilities of propagating potential dangerous mutations in new generations.
Thus genetic analysis has an essential role. Nevertheless it still remains difficult due to the high number and the high variability in type of possible gene mutations; [in AIP, for example, to date, over 200 mutations including deletions, insertions, missense, nonsense, and splicing mutations have been identified, and most of these mutations resulted from family-specific analysis] and need to be undertaken by expert laboratories (cfr. http://www.porphyria-europe.com) [40]. To date, the exact locations for all genes responsible for the acute porphyrias have been identified, and a rapid growing knowledge in this area is expected [39].
Therapy
Management of acute attacks
Patients with an acute attack, usually require admission to the hospital in order to initiate prompt specific as well as symptomatic treatments. About 1–2% of acute attacks of porphyria may be fatal (especially as a consequence of respiratory failure, due to acute neuromuscular involvement). One must pay particular attention to potential signs of progressive neuropathy, and if there are any signs of respiratory failure, then aggressively manage the airway. Monitoring of electrolyte balance (for hyponatremia) and renal function must be started as well. During an acute attack only drugs known to be safe in porphyria should be prescribed, and the monitoring of urinary ALA and PBG should be started promptly. A significant rise in their urinary concentrations might in fact suggest a porphyrinogenic action of a drug in use [13, 26, 41]. Identification and elimination of any possible precipitating factor is mandatory: as mentioned above, an acute attack is often precipitated by several factors simultaneously, and no individual guidelines can be given for the use of a single drug [1, 42].
The specific treatment for acute porphyria should be started as soon as possible in order to prevent the possible neurological progression of the crisis, hopefully allowing for a quick remission, and shortening of the hospital stay [1, 41]. At present, glucose and haem arginate administration represent the mainstays of treatment [41]. They both reduce synthesis of ALA, resulting in clinical and biochemical remission, with urinary excretion of ALA and PBG reducing towards a normal range. Glucose solution (up to 2000 cc 20%/die intravenously) should be used in case of mild attacks, and pending administration of haem arginate (but one must be careful of sodium balance!!). According to recent evidence, glucose (such as a good general nutritional status) reduces ALA-S activity and consequently the levels of non-porphyrin precursors by decreasing cell expression of PGC-1 alpha, an important inductor of liver ALA-S [43]. This probably is the reason why starvation represents a well-known precipitant of acute attacks. Therefore it must be prevented and promptly treated: when the patient is vomiting or unable to eat, parenteral nutrition containing a high percentage of glucose is mandatory.
Haem arginate (Normosang®, Orphan Europe, Paris, France) should be administered as soon as possible in case of an acute attack because its ability to reverse an established neuropathy is controversial. It has to be given at a dose of 3 mg/kg/day over 15 min for 3–4 consecutive days by slow intravenous infusion. It acts rapidly (within 1–3 days), and the effect lasts for a week, enough to abolish all acute symptoms. The drug is rather expensive, but it may be available on an urgent need basis from the manufacturing drug company. Haem preparations can cause thrombophlebitis, coagulopathy, and anaphylactic reactions, so they should be administered by a central venous line under careful control; a line washout by saline soon after the drug infusion is advisable [41]. No significant accumulation of iron or haem after several infusions has been reported; nevertheless the monitoring of iron parameters (serum ferritin and transferrin saturation) is advisable in patients undergoing repeated and prolonged infusion. Haem arginate has been used successfully during pregnancy.
Recently, tin protoporphyrin, an inhibitor of haem oxygenase, has been observed to prolong remission when administered together with haem arginate; however, its side effects (cutaneous photosensitivity) and potential toxicity prevent its current use, and further confirmation studies are needed [44].
Acute attacks often require symptomatic treatment for hypertension, pain, and seizures (Table 3). Opiates are safe in case of severe pain: pethidine, morphine, or diamorphine are the most often used. Their use should be restricted to the acute phase, and stopped as soon as possible in order to prevent possible and frequently observed drug addiction.
Chlorpromazine may be helpful to favor relaxation and sleep. Propranolol is the first choice for the treatment of symptoms due to sympathetic overstimulation (tachycardia and hypertension).
During an acute attack, seizures are rare, but possible: they usually resolve as the attack subsides. They may be precipitated by hyponatremia, so plasma osmolarity and electrolyte balance should always be checked. In case of hyponatremia, fluid restriction should be prescribed [3, 41]. Rarely, seizures may be the only presenting symptom of AIP. In this case, the treatment should be aimed at the underlying disease, but it is difficult because the most commonly used anticonvulsant drugs are porphyrogenic, and may lead to further exacerbations of the disease. Historically, bromides have been considered as the drug of choice for the control of seizures in patients with acute porphyria. However, more recently, gabapentin and vigabatrine have produced safe and successful seizure control, and are considered at the present as the treatment of choice for these patients [45].
A small number of patients (mainly women) have recurrent and severe attacks with or without apparent precipitants (often menses-related). Induction of menopause by administration of luteinizing hormone releasing hormone agonists has been demonstrated to be successful or at least useful in these situations [46]. Table 6 summarizes the key points for the treatment of acute porphyric attack.
In lead poisoning (plumboporhyria) lead chelation is recommended with oral meso-2,3-dimercaptosuccinic acid (DMSA) in case of mild intoxication (few symptoms and blood lead levels not exceeding 45 μg/dl); in the presence of severe acute toxicity (blood lead levels >60–80 μg/dl) with significant signs and symptoms (anemia, demyelinating peripheral neuropathy, hypertension, interstitial nephritis with renal impairment, acute encephalopathy with convulsions and coma), hospitalization and chelation with edentate calcium disodium (CaEDTA) (via EV or IM) is required, with the addition of dimercaprol to prevent worsening of encephalopathy. Vitamin C is a weak, but natural, lead chelating agent [47].
Prevention of attacks
The above-mentioned precipitating factors (especially alcohol, smoking, and unsafe drugs) should be avoided, as should sudden or prolonged calorie restrictions (low-caloric diets). Patients should get a Medic Alert document, and should be fully informed regarding possible precipitating factors (especially drugs and anesthesia preparation). Prophylactic haem arginate administration on a regular basis for symptomatic patients, (that is patients with history of porphyric attacks) or for asymptomatic patients “at risk” (i.e. with high value of urinary non-porphyrin precursors but no history of porphyric attacks) has been proposed, nevertheless, at the present, specific guide-lines or expert consensus (indications for the timing and the dose to use, as well for the end-point to reach) are still lacking.
Monitoring of associated pathologic conditions
The prognosis of untreated HCC is poor, but early diagnosis coupled with a range of therapeutic options can improve the outcome. It has been observed that the development of a hepatic tumor may be accompanied by increased excretion of porphyrin precursors, or the recurrence of porphyric symptoms that have been absent for several years. The mechanisms by which a process in the liver makes its presence known in an erythrocyte function are obscure, but nevertheless the analysis should be used as a complement to the routine ultrasound investigation of the liver in the yearly hepatocellular carcinoma control recommended for AIP patients above middle age [28].
Forthcoming therapeutical perspectives: is this the future?
Liver transplantation has been proposed as treatment for patients with repeated life-threatening acute attacks resulting in a poor quality of life, requirement of ventilatory support, or a progressive loss of venous access due to hemin infusion. To date, two cases of liver transplantation for AIP have been described: one was completely successful (complete normalization of ALA and PBG values and no recurrence of symptoms) [32, 48]. Similar single observations are available for liver transplantation in PV, and renal transplantation for a severe renal impairment in one patient with AIP. All these data proffer some hope of cure for selected patients with severe forms of these diseases. To date, other therapeutic approaches, such as the infusion of recombinant porphobilinogen deaminase [49] or gene therapy have to be considered as experimental [29]; the latter approach has showed very good results in animal models of AIP, especially after the recent optimization of gene vectors [50].
Conclusions
The acute porphyrias are rare diseases. Nevertheless, their diagnosis is important, and should be considered within the diagnostic process in many different fields of medicine owing to the extreme variability in the presentation of their clinical pictures. An accurate diagnosis requires knowledge and use of the correct diagnostic tools, and it is mandatory to provide optimal therapy, and to prevent the use of unsafe drugs, potentially able to severely precipitate these diseases, especially in the presence of life-threatening symptoms. To date, the availability of a relatively stable haem preparation (haem arginate) has significantly improved the treatment outcome of acute porphyric attacks.
The correct identification of patients mandates an in-depth study of their relatives (by means of molecular biology methods), in order to identify other gene carriers prone to porphyric manifestations, who should be informed about their potential risk if exposed to potential precipitant factors.
References
Kauppinen R (2005) Porphyrias. Lancet 365:241–252
Ventura E, Rocchi E (2001) Le Porfirie. In: Guarini G, Fiorelli G, Malliani A, Violi E, Volpe M (eds) Teodori 2000. Trattato di Medicina Interna.Società Editrice Universo, Roma, pp 2301–2334
Anderson KE, Sassa S, Bishop DF, Desnick RJ (2001) Disorders of heme biosynthesis: X-linked sideroblastic anemia and the porphyrias. In: Scriver CR, Beaudet A, Sly WS, Valle D (eds) The metabolic and molecular basis of inherited diseases. McGraw-Hill, New York, pp 2991–3062
Phillips JD, Bergonia HA, Reilly CA et al (2007) A porphomethene inhibitor of uroporphyrinogen decarboxylase causes porphyria cutanea tarda. Proc Natl Acad Sci USA 104:5079–5084
Rigal JC, Blanloeil Y (2002) Anaesthesia and porphyria. Minerva Anestesiol 68:326–331
Gisbert JP, Garcia-Buey L, Pajares JM, Moreno-Otero R (2003) Prevalence of hepatitis C virus infection in porphyria cutanea tarda: systematic review and meta-analysis. J Hepatol 39:620–627
Badminton MN, Elder GH (2005) Molecular mechanisms of dominant expression in porphyria. J Inherit Metab Dis 28:277–286
Gouya L, Puy H, Robreau AM et al (2004) Modulation of penetrance by the wild-type allele in dominantly inherited erythropoietic protoporphyria and acute hepatic porphyrias. Hum Genet 114:256–262
Kauppinen R (2004) Molecular diagnostics of acute intermittent porphyria. Expert Rev Mol Diagn 4:243–249
Bonkovsky HL, Barnard GF (1998) Diagnosis of porphyric syndromes: a practical approach in the era of molecular biology. Semin Liver Dis 18:57–65
de Rooij WM, Edixhoven A, Wilson JH (2003) Porphyria: a diagnostic approach. In: Kadish KM, Smith KM, Guillard R (eds) The porphyrin handbook. Elsevier, St Louis, pp 211–245
Grob U, Puy H, Jacob K et al (2006) Biochemical compared to molecular diagnosis in acute intermittent porphyria. J Inherit Metab Dis 29:157–161
Elder GH, Hift RJ, Meissner PN (1997) The acute porphyrias. Lancet 349:1613–1617
Solinas C, Vajda FJ (2008) Neurological complications of porphyria. J Clin Neurosci 15:263–268
Kirsch RE, Meissner PN, Hift RJ (1998) Variegate porphyria. Semin Liver Dis 18:33–41
Martasek P (1998) Hereditary coproporphyria. Semin Liver Dis 18:25–32
Sassa S (1998) ALAD porphyria. Semin Liver Dis 18:95–101
Frith D, Yeung K, Thrush S et al (2005) Lead poisoning—a differential diagnosis for abdominal pain. Lancet 366:2146
Usalan C, Erdem Y, Altun B et al (1996) Severe hyponatremia due to SIADH provoked by acute intermittent porphyria. Clin Nephrol 45:418
Crimlisk HL (1997) The little imitator–porphyria: a neuropsychiatric disorder. J Neurol Neurosurg Psychiatry 62:319–328
Tishler PV, Woodward B, O’Connor J et al (1985) High prevalence of intermittent acute porphyria in a psychiatric patient population. Am J Psychiatry 142:1430–1436
Hultdin J, Schmauch A, Wikberg A et al (2003) Acute intermittent porphyria in childhood: a population-based study. Acta Paediatr 92:562–568
Kauppinen R, von und zu Fraunberg M (2002) Molecular and biochemical studies of acute intermittent porphyria in 196 patients and their families. Clin Chem 48:1891–1900
Kanaan C, Veille JC, Lakin M (1989) Pregnancy and acute intermittent porphyria. Obstet Gynecol Surv 44:244–249
Deybach JC, Badminton M, Puy H et al (2006) European porphyria initiative (EPI): a platform to develop a common approach to the management of porphyrias and to promote research in the field. Physiol Res 55(Suppl 2):S67–S73
Thadani H, Deacon A, Peters T (2000) Diagnosis and management of porphyria. BMJ 320:1647–1651
Andersson C, Lithner F (1994) Hypertension and renal disease in patients with acute intermittent porphyria. J Intern Med 236:169–175
Andant C, Puy H, Bogard C et al (2000) Hepatocellular carcinoma in patients with acute hepatic porphyria: frequency of occurrence and related factors. J Hepatol 32:933–939
de Verneuil H, Ged C, Boulechfar S, Moreau-Gaudry F (1995) Porphyrias: animal models and prospects for cellular and gene therapy. J Bioenerg Biomembr 27:239–248
Rocchi E, Ventura P, Ronzoni A et al (2004) Pro-oxidant and antioxidant factors in acute intermittent porphyria: family studies. J Inherit Metab Dis 27:251–266
Lindberg RL, Martini R, Baumgartner M et al (1999) Motor neuropathy in porphobilinogen deaminase-deficient mice imitates the peripheral neuropathy of human acute porphyria. J Clin Invest 103:1127–1134
Seth AK, Badminton MN, Mirza D et al (2007) Liver transplantation for porphyria: who, when, and how? Liver Transpl 13:1219–1227
Shanley BC, Neethling AC, Percy VA, Carstens M (1975) Neurochemical aspects of porphyria: studies on the possible neurotoxicity of delta-aminolaevulinic acid. S Afr Med J 49:576–580
Minder EI (1986) Measurement of 5-aminolaevulinic acid by reversed phase HPLC and fluorescence detection. Clin Chim Acta 161:11–18
Hift RJ, Davidson BP, van der Hooft C et al (2004) Plasma fluorescence scanning and fecal porphyrin analysis for the diagnosis of variegate porphyria: precise determination of sensitivity and specificity with detection of protoporphyrinogen oxidase mutations as a reference standard. Clin Chem 50:915–923
Erlandsen EJ, Jorgensen PE, Markussen S, Brock A (2000) Determination of porphobilinogen deaminase activity in human erythrocytes: pertinent factors in obtaining optimal conditions for measurements. Scand J Clin Lab Invest 60:627–634
Campbell BC, Brodie MJ, Thompson GG et al (1977) Alterations in the activity of enzymes of haem biosynthesis in lead poisoning and acute hepatic prophyria. Clin Sci Mol Med 53:335–340
Floderus Y, Sardh E, Moller C et al (2006) Variations in porphobilinogen and 5-aminolevulinic acid concentrations in plasma and urine from asymptomatic carriers of the acute intermittent porphyria gene with increased porphyrin precursor excretion. Clin Chem 52:701–707
Thunell S (2006) (Far) Outside the box: genomic approach to acute porphyria. Physiol Res 55(Suppl 2):S43–S66
Cappellini MD, Martinez di Montemuros F, Di Pierro E, Fiorelli G (2002) Hematologically important mutations: acute intermittent porphyria. Blood Cells Mol Dis 28:5–12
Elder GH, Hift RJ (2001) Treatment of acute porphyria. Hosp Med 62:422–425
Kauppinen R, Mustajoki P (1992) Prognosis of acute porphyria: occurrence of acute attacks, precipitating factors, and associated diseases. Medicine (Baltimore) 71:1–13
Handschin C, Lin J, Rhee J et al (2005) Nutritional regulation of hepatic heme biosynthesis and porphyria through PGC-1alpha. Cell 122:505–515
Schuurmans MM, Hoffmann F, Lindberg RL, Meyer UA (2001) Zinc mesoporphyrin represses induced hepatic 5-aminolevulinic acid synthase and reduces heme oxygenase activity in a mouse model of acute hepatic porphyria. Hepatology 33:1217–1222
Krauss GL, Simmons-O’Brien E, Campbell M (1995) Successful treatment of seizures and porphyria with gabapentin. Neurology 45:594–595
Anderson KE, Spitz IM, Sassa S et al (1984) Prevention of cyclical attacks of acute intermittent porphyria with a long-acting agonist of luteinizing hormone-releasing hormone. N Engl J Med 311:643–645
Cohen SM (2001) Lead poisoning: a summary of treatment and prevention. Pediatr Nurs 27(125/126):129–130
Pimstone NR (2005) Roles and pitfalls of transplantation in human porphyria. Liver Transpl 11:1460–1462
Sardh E, Rejkjaer L, Andersson DE, Harper P (2007) Safety, pharmacokinetics and pharmocodynamics of recombinant human porphobilinogen deaminase in healthy subjects and asymptomatic carriers of the acute intermittent porphyria gene who have increased porphyrin precursor excretion. Clin Pharmacokinet 46:335–349
Yasuda M, Domaradzki ME, Armentano D et al (2007) Acute intermittent porphyria: vector optimization for gene therapy. J Gene Med 9:806–811
Conflict of interest statement
The authors declare that they have no conflict of interest related to the publication of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ventura, P., Cappellini, M.D. & Rocchi, E. The acute porphyrias: a diagnostic and therapeutic challenge in internal and emergency medicine. Intern Emerg Med 4, 297–308 (2009). https://doi.org/10.1007/s11739-009-0261-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11739-009-0261-4