
Abstract
We have recently demonstrated that in an autosomal dominant porphyria, erythropoietic protoporphyria (EPP), the coinheritance of a ferrochelatase (FECH) gene defect and of a wild-type low-expressed FECH allele is generally involved in the clinical expression of EPP. This mechanism may provide a model for phenotype modulation by minor variations in the expression of the wild-type allele in the other three autosomal dominant porphyrias that exhibit incomplete penetrance: acute intermittent porphyria (AIP), variegata porphyria (VP) and hereditary coproporphyria (HC), which are caused by partial deficiencies of hydroxy-methyl bilane synthase (HMBS), protoporphyrinogen oxidase (PPOX) and coproporphyrinogen oxidase (CPO), respectively. Given the dominant mode of inheritance of EPP, VP, AIP and HC, we first confirmed that the 200 overtly porphyric subjects (55 EPP, 58 AIP, 56 VP; 31 HC) presented a single mutation restricted to one allele (20 novel mutations and 162 known mutations). We then analysed the available single-nucleotide polymorphisms (SNPs) present at high frequencies in the general population and spreading throughout the FECH, HMBS, PPOX and the CPO genes in four case-control association studies. Finally, we explored the functional consequences of polymorphisms on the abundance of wild-type RNA, and used relative allelic mRNA determinations to find out whether low-expressed HMBS, PPOX and the CPO alleles occur in the general population. We confirm that the wild-type low-expressed allele phenomenon is usually operative in the mechanism of variable penetrance in EPP, but conclude that this is not the case in AIP and VP. For HC, the CPO mRNA determinations strongly suggest that normal CPO alleles with low-expression are present, but whether this low-expression of the wild-type allele could modulate the penetrance of a CPO gene defect in HC families remains to be ascertained.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
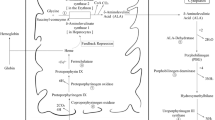
The inherited porphyrias are disorders in which the activity of one of the enzymes involved in the heme biosynthesis pathway is partially deficient (Fig. 1; Sassa et al. 2000; Anderson et al. 2001). They are classified as hepatic or erythropoietic, depending on where most of the porphyrins or their precursors are produced. Acute hepatic porphyrias (AHPs), mainly acute intermittent porphyria (AIP, MIM 176000), variegata porphyria (VP, MIM 176200) and hereditary coproporphyria (HC, MIM 121300), are the acute porphyrias with the highest incidence and clinical severity. AIP, VP and HC are caused by partial deficiencies of hydroxy-methyl-bilane synthase (HMBS; EC 4.3.1.8), protoporphyrinogen oxidase (PPOX; EC 1.3.3.4) and coproporphyrinogen oxidase (CPO; EC 1.3.3.3), respectively (Fig. 1). The clinical features of AHPs consist essentially of neurological symptoms, although VP and HC patients may also present with cutaneous symptoms. AHPs are inherited as autosomic dominant traits with incomplete penetrance. The related molecular defects are characterised by high molecular heterogeneity, with more than 400 mutations reported so far in the three genes (Human Gene Mutation Database: http://www.hgmd.org), without any strong evidence for phenotype/genotype relationship. This incomplete penetrance and variable clinical expression limit the effectiveness of genetic counselling, and all presymptomatic patients have to follow strict rules with regard to medication and lifestyle, even though less than 10% of them are ever likely to develop symptoms (Anderson et al. 2001).

Classification of the major human porphyrias. ALA delta-aminolevulinic acid, PBG porphobilinogen, HEP hepatoerythroporphyria. *Autosomal dominant inheritance has been documented in familial porphyria cutanea and recessive inheritance has been documented in HEP
Inconsistent phenotype/genotype relationships in simple Mendelian disorders have often been attributed to environmental factors or to modifier genes that modulate the clinical expression of a gene defect at a major locus. In AHPs various triggering factors, such as fasting, infectious disease or medication have been identified, but remain insufficient to explain the penetrance of a dominant mutation, suggesting the existence of further susceptibility loci. A new genetic approach suggests that in dominantly inherited disorders, clinical manifestations cannot be simply a matter of haplo-insufficiency, and that additional enzyme deficiencies may be necessary for phenotype expression to occur. In these disorders, slight variations in the expression of the normal allele in trans to the mutated allele may have a major impact on the clinical expression of the disease by lowering gene expression below a critical threshold. The penetrance of a dominant mutation may thus be modulated by functional polymorphisms at the same locus. This mechanism of incomplete penetrance has been suggested in dominant Hirschsprung disease (Borrego et al. 1999), and demonstrated in hereditary elliptocytosis (Alloisio et al. 1991; Wilmotte et al. 1993) and in the predisposition to tumorigenesis in familial adenomatous polyposis (Yan et al. 2002). We have recently demonstrated that in dominant erythropoietic protoporphyria (EPP, characterised by a partial deficit in ferrochelatase activity, FECH, EC4.99.1.1) the clinical penetrance of a FECH gene defect is systematically modulated in the presence of a common wild type, low-expression FECH gene in trans to a dominant mutation. A common intronic single-nucleotide polymorphism (SNP), IVS3−48T/C, is responsible for the low-expression mechanism by modulating the use of a constitutive aberrant acceptor splice site; the aberrantly spliced mRNA is degraded by a nonsense-mediated decay mechanism, producing a lower steady-state level of FECH mRNA (Gouya et al. 2002). In AHPs, enzyme activities are 50% of normal in both overt and presymptomatic patients (Sassa et al. 2000). However, enzyme activities are measured in peripheral blood cells, whereas AHPs are mainly hepatic disorders. Moreover, two of these assays are technically tedious, and may lack sensitivity; the stability of proteins, especially of CPO, is low in lymphocytes so that enzyme decay may occur if blood sampling, lymphocyte isolation and storage take more than 2 h to perform (Da Silva, personal communication). This means that enzyme measurement in blood samples in acute hepatic porphyrias cannot provide any evidence for or against a hypothetical effect on enzyme activity levels from an allele trans to the mutation.
In this study, a new EPP cohort of 55 patients and a control group of 80 unrelated subjects of Caucasian origin, are studied in order both to validate the method and to confirm in a larger cohort that the low expression of a wild-type allelic variant is generally required for EPP to be clinically expressed (Gouya et al. 2002). We then investigated whether this phenomenon is restricted to EPP, or whether it could be also involved in AIP, VP and HC. We have developed two complementary methods to test this hypothesis: (1) in 145 AIP, HC, and VP subjects with identified mutations, we analysed the distribution of all common intragenic SNPs in the HMBS, CPO, and the PPOX genes by case-control association studies; (2) in the control group, we performed relative quantifications of respective HMBS, CPO, and PPOX allele mRNA to explore the putative functional consequences of SNPs on the abundance of wild-type mRNAs.
Material and methods
Patients
Procedures involving human subjects were performed in accordance with the 1983 revision of the Declaration of Helsinki, and informed consent was obtained from all subjects prior to their inclusion in the study. All the porphyric families included in the study had been diagnosed in the Centre Français des Porphyries during the period 1998–2003, and patients from each family were undergoing clinical follow-up at the centre. A total of 200 subjects from unrelated French porphyric families of Caucasian origin were studied. Overt French Caucasian patients were selected on the basis of a typical history of painful neurovisceral episodes for the 58 AIP and 31 HC patients, and/or dermatological symptoms for 56 VP patients; 30 VP patients (53%) had isolated dermatological symptoms. Fifty-five French Caucasian overt EPP patients were selected on the basis of a typical history of skin photosensitivity. In all these patients, porphyria was diagnosed on the basis of low enzyme activity, in the red blood cells for HMBS or in the lymphocytes for CPO, PPOX and FECH, and identification of the gene defect.
Identification of mutations
None of the patients in this study had been included in previous publications on mutational analysis. DNA was extracted from peripheral blood leukocytes. The specific gene defects were identified by a two-step strategy: mutations were first localized using a screening method, and any that showed an abnormal pattern were then sequenced. Exons 1–15 of the HMBS gene, 1–7 of the CPO gene, 1–13 of the PPOX gene, 1–11 of the FECH gene, along with 50–150 bp of their flanking regions, were amplified using the primers and conditions described elsewhere (Gouya et al. 1996; Puy et al. 1997; Rüfenacht et al. 1998; Rosipal et al. 1999) and were screened for mutations by denaturing gradient gel electrophoresis (DGGE), heteroduplex analysis or dHPLC. Any regions showing abnormal patterns were sequenced to identify mutations and polymorphisms. When no abnormality was detected by screening methods, the gene was sequenced to eliminate the possibility of any other sequence variation, as has previously been reported (Gouya et al. 1996; Puy et al. 1997; Rüfenacht et al. 1998; Rosipal et al. 1999). For DNA sequencing, HMBS, CPO, PPOX and FECH genes were amplified using primers different from those used for the screening analysis (Gouya et al. 1996; Puy et al. 1996, 1997; Rüfenacht et al. 1998; Rosipal et al. 1999).
Nomenclature
Gene mutation nomenclature used in this article follows the recommendations of den Dunnen and Antonarakis (2001). Gene symbols used in this article follow the recommendations of the HUGO Gene Nomenclature Committee (Povey et al. 2001).
Intragenic polymorphism genotyping
The frequencies of 14 known intragenic SNPs distributed over the FECH, HMBS, CPO and PPOX genes were determined by studying overtly porphyric patients and 80 French Caucasian white subjects who did not have porphyria. Among the 14 SNPs, we used all the available non-synonymous coding SNPs (non-syn cSNPs) found frequently in the disease-free population (Table 1; Astrin et al. 1994; Martasek et al. 1994; Puy et al. 1996; Gouya et al. 1999; Whatley et al. 1999; Gouya et al. 2002). In the case of the HMBS gene, Caucasian populations from Western Europe and North America are known to exhibit a marked linkage disequilibrium between four SNPs in intron 1, and one SNP in intron 3 (Yoo et al. 1993). All the HMBS intragenic polymorphic sites studied here gave rise to a co-dominant two-allele polymorphism, and for all of them the distribution of homozygotes and heterozygotes is known to be in a Hardy-Weinberg equilibrium without linkage disequilibrium. The control group was drawn from the Centre d’Etude du Polymorphisme Humain (CEPH, Paris), and polymorphisms frequencies were determined by means of direct sequencing, restriction enzyme digestion, or DGGE as previously described (Puy et al.1997; Rüfenacht et al. 1998; Rosipal et al. 1999; Gouya et al. 2002).
Relative determinations of DNA and mRNA by fluorescent primer extension assay
DNA and total RNA were prepared from lymphoblastoid cell lines, and mRNAs were reverse transcribed using oligo-dT primers. The PCR amplimers and the fluorescent primers used for the HMBS, CPO and PPOX assays are shown in Table 2. FECH primers and the extension assay have already been described elsewhere (Gouya et al. 1999). Extended primers were separated on an Alf DNA automated sequencer (Pharmacia biotech), and the areas under the peaks were calculated using the Fragment Manager program (Pharmacia Biotech). Results are expressed as the peak area ratio (R) of the corresponding allelic mRNA. DNA quantifications were performed as an index when the scatter of the ratios outside the equimolar representation of the two copies of a gene (R=1) reflected the technical limitations of the assay. The results are the mean of 3–9 experiments, as previously described (Gouya et al. 1996).
Statistical analysis
Allele distribution was analysed by the χ2 test. The allele distribution of the 14 polymorphisms in the four genes satisfied the Hardy-Weinberg equilibrium (χ2 test, P>0.5).
Results and discussion
To find out whether a common allele in trans to a dominant mutation could modulate the penetrance in four different human dominant porphyrias, we studied 200 unrelated porphyric patients. We first characterised the specific gene mutation in each patient: 20 novel and 162 previously reported mutations were identified in our porphyric cohort (Table 3). We confirmed the wide heterogeneity of molecular abnormalities in these porphyrias. Moreover, in all recently studied populations, hitherto unreported mutations continue to emerge. This continuing emergence of novel mutations despite the identification of more than 420 mutations, and the low de novo mutation rate demonstrates the usefulness of preliminary identification of the causative mutations in each newly-diagnosed family so that relatives with latent porphyria can be accurately identified (Donnelly et al. 2002; Gregor et al. 2002; Wiman et al. 2002; Yasui et al. 2002). Most mutations were restricted to a single family, and in accordance with the autosomic dominant inheritance, each patient had a single mutation restricted to one allele.
Allele frequencies of intragenic SNPs distributed over the FECH, HMBS, CPO and PPOX genes in the control group and the symptomatic patient group are reported in Table 1. We first analysed the distribution of four intragenic polymorphisms in 55 patients with overt EPP. The patient group exhibited a significantly higher frequency of a specific set of alleles than the control group (Table 1; 4589G, 11286T, 41042T), whereas both groups exhibited a similar distribution for the G32371C polymorphism in exon 7. These findings are related to the strong linkage disequilibrium observed between the functional IVS3−48C allele and the 4589G, 11286T, 41042T alleles (Gouya et al. 2002); the G32371C polymorphism in exon 7 is not affected by this phenomenon. The three other case-control association studies failed to detect any difference in SNP distribution between the control group and the AIP, VP, and HC patient groups, respectively (Table 1).
These data lead to a number of conclusions. First, these findings from a new large EPP cohort are consistent with the role of a low expression of a wild-type allele variant trans to a mutated FECH allele, which is generally required for clinical expression of EPP. Second, by this study, the control group is validated for the subsequent case-control association studies. Third, the absence of common non-syn cSNP in the HMBS gene, and the absence of any association with the three available non-syn cSNPs in the PPOX and CPO genes, suggest that common protein variants may not generally be involved in the pathophysiology of AIP, VP and HC. Finally, these data do not indicate that a common wild-type allele is generally operative in the mechanism of variable penetrance in AIP, VP and HC. However, this absence of significant evidence for such an association could result either from the lack of linkage disequilibrium between the individual SNPs tested and any functional polymorphism, or from the existence of several different functional polymorphisms, each of which makes only a limited individual contribution to penetrance in AHPs. To overcome these two main limitations of the case-control association studies, we performed an independent functional experiment.
Indeed, it has been reported that most of the phenotype variations among closely related organisms are attributable to changes in gene expression rather than to modifications in protein sequence (Brett et al. 2002). Consequently, it can be expected that penetrance in Mendelian disease would often be caused by quantitative modifications in mRNA amounts, rather than by structural alterations of proteins (Brett et al. 2002). Moreover, if the low expression mechanism is generally involved in the penetrance of an autosomal dominant disorder, this would imply that alleles with low expression should be reasonably common in the general population. We therefore analysed the expression level of VP, CPO and HMBS wild-type genes by comparing allele mRNA determinations in 80 control subjects. We have previously demonstrated that the modulating FECH gene is characterised by a lower steady-state level of FECH mRNA (Gouya et al. 1999). In this study, the 10.6% allele frequency of the wild-type, low-expressed FECH allele measured in the 80 unrelated subjects (Fig. 2a; seven low-expressed alleles/66 quantified alleles) is consistent with the 11% we previously reported (Gouya et al. 1996). Of the seven low-expressed FECH alleles, five have low ratios and two have high ratios, related to the absence of linkage disequilibrium between the functional IVS3−48C allele and the G or C 32371 alleles in exon 7 used to quantify mRNA. This finding validates the control group for subsequent mRNA quantifications for the HMBS, PPOX and CPO genes. The relative values of HMBS and PPOX allelic mRNA do not reveal significant variations in the expression level that are any greater than those observed when using genomic DNA as the control (Fig 2c–d). Taken as a whole, our results strongly suggest that a common specific variant does not play any major role in the penetrance of an AIP or VP gene defect. Indeed, both the association studies and mRNA determinations gave convergent results, excluding any important role for a single functional polymorphism trans to a dominant mutation in their molecular pathogenesis.

Relative quantifications of DNA and mRNAs by primer extension assay for the FECH, PPOX, HMBS and CPO genes. Results on the abscissa are expressed as the peak area ratio of the corresponding allelic mRNA or DNA, and are presented in the form of a histogram of the observed distribution at 0.1 intervals. DNA determinations are shown as a control of an equimolar representation of the two copies of a gene. The distribution of DNA and RNA values is similar for the PPOX and HMBS genes (C, D). The distribution of EPP and CPO values (A, B) shows a main subgroup of subjects with equally expressed alleles (solid bar), and a smaller group with a marked disequilibrium in their relative mRNA allelic representation (dashed bar)
Although the epidemiological study does not suggest that the common wild-type modulating allele has an important role in HC, the CPO mRNA determinations clearly demonstrate the presence of common low-expressed CPO genes. HC is much less common than other AHPs. The discrepancy between the case-control association study and CPO mRNA data may be due to the limitations of case control studies (see discussion above), and to the small size of the HC subject group studied. The relative quantifications of CPO allelic mRNAs revealed that the expression of the CPO gene is clearly unbalanced in three subjects (Fig. 2b). This demonstrates the presence of low-expressed CPO genes with an estimated frequency of 5.3% in the control group (three low-expressed alleles/56 quantified alleles, assuming that this ratio reflects the frequency in the group as a whole). DGGE analysis of 500 bp in the 5′UTR region, the entire coding region, the exon/intron boundaries and part of the 3′UTR revealed no mutation. The mechanism leading to low expression remains unknown, and the relationship between the altered allele ratio in 5% of the controls and the penetrance of HC calls for further investigation. Presumably, as previously described (Brett et al. 2002), mutations responsible for the reduced expression could reside deep within introns or far upstream or downstream of the gene. An unknown functional polymorphism, not in linkage disequilibrium with the four SNPs used in the association study, or several different functional polymorphisms may be responsible for this low expression and so contribute to penetrance.
In conclusion, the combined statistical and functional approaches used in this study confirmed the pre-eminence of the low-expressed wild-type allele mechanism in the penetrance of EPP. In AIP and VP this mechanism of modulation of a dominant mutation appears to play only a minor role, and the contributions of other susceptible loci interacting with environmental factors must be investigated. In HC, slight variations in the expression of the normal CPO allele may play a key role in the clinical expression of the disease by situating the amount of gene product above or below the critical threshold. However, further investigations are required, firstly to validate the role of the low-expression mechanism in modulating penetrance both in a larger cohort and in HC families and secondly to characterize the mechanism involved in the low expression of the wild-type allele.
References
Alloisio N, Morle L, Maréchal J, Roux AF, Ducluzeau MT, Guetarni D, Pothier B (1991) Spαv/41: a common spectrin polymorphism at the αIV-αV domain junction. Relevance to the expression level of hereditary elliptocytosis due to α-spectrin variants located in trans. J Clin Invest 87:2169–2177
Anderson KE, Sassa S, Bishop DF, Desnick RJ (2001) The porphyrias. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) The metabolic basis of inherited disease, 8th edn, vol 1. McGraw-Hill, New York, pp 2991–3062
Astrin KH, Desnick RJ (1994) Molecular basis of acute intermittent porphyria: mutations and polymorphisms in the human hydroxymethylbilane synthase gene. Hum Mutat 4:243–252
Borrego S, Saez ME, Ruiz A, Gimm O, Lopez-Alonso M, Antinolo G, Eng C (1999) Specific polymorphisms in the RET proto-oncogene are over-represented in patients with Hirschsprung disease and may represent loci modifying phenotypic expression. J Med Genet 36:771–774
Brett D, Pospisil H, Valcarcel J, Reich J, Bork P (2002) Alternative splicing and genome complexity. Nat Genet 30:29–30
den Dunnen JT, Antonarakis SE (2001) Nomenclature for the description of human sequence variations. Hum Genet 109:121–124
Donnelly JG, Detombe S, Hindmarsh JT (2002) Single-strand conformational polymorphism and denaturing gradient gel electrophoresis in screening for variegate porphyria: identification of two new mutations. Ann Clin Lab Sci 32:107–113
Gouya L, Deybach JC, Lamoril J, Da Silva V, Beaumont C, Grandchamp B, Nordmann Y (1996) Modulation of the phenotype in dominant erythropoietic protoporphyria by a low expression of the normal ferrochelatase allele. Am J Hum Genet 58:292–299
Gouya L, Puy H, Lamoril J, Da Silva V, Grandchamp B, Nordmann Y, Deybach JC (1999) Inheritance in erythropoietic protoporphyria: a common wild-type ferrochelatase allelic variant with low expression accounts for clinical manifestation. Blood 93:2105–2110
Gouya L, Puy H, Robréau AM, Bourgeois M, Lamoril J, Da Silva V, Grandchamp B, Deybach JC (2002) The penetrance of dominant erythropoietic protoporphyria is modulated by expression of wild-type FECH. Nat Genet 30:27–28
Gregor A, Schneider-Yin X, Szlendak U, Wettstein A, Lipniacka A, Rufenacht UB, Minder EI (2002) Molecular study of the hydroxymethylbilane synthase gene (HMBS) among Polish patients with acute intermittent porphyria. Hum Mutat 19:310
Martasek P, Nordmann Y, Grandchamp B (1994) Homozygous hereditary coproporphyria caused by an arginine to tryptophan substitution in coproporphyrinogen oxidase and common intragenic polymorphisms. Hum Mol Genet 3:477–480
Povey S, Lovering R, Bruford E, Wright M, Lush M, Wain H (2001) The HUGO Gene Nomenclature Committee (HGNC). Hum Genet 109:678–680
Puy H, Robréau AM, Rosipal R, Nordmann Y, Deybach JC (1996) Protoporphyrinogen oxidase : complete genomic sequence and polymorphisms in the human gene. Biochem Biophys Res Commun 226:226–230
Puy H, Deybach JC, Lamoril J, Robréau AM, Da Silva V, Gouya L, Grandchamp B, Nordmann Y (1997) Molecular epidemiology and diagnosis of PBG deaminase gene defects in acute intermittent porphyria. Am J Hum Genet 60:1373–1383
Rosipal R, Lamoril J, Puy H, Da Silva V, Gouya L, De Rooij FW, Te Velde K, Nordmann Y, Martasek P, Deybach JC (1999) Systematic analysis of coproporphyrinogen oxidase gene defects in hereditary coproporphyria and mutation update. Hum Mutat 13:44–53
Rüfenacht UB, Gouya L, Schneider-Yin X, Puy H, Schäfer BW, Aquaron R, Nordmann Y, Minder EI, Deybach JC (1998) Systematic analysis of molecular defects in the ferrochelatase gene from patients with erythropoietic protoporphyria. Am J Hum Genet 62:1341–1352
Sassa S, Kappas A (2000) Molecular aspects of the inherited porphyrias. J Intern Med 247:169–178
Whatley SD, Puy H, Morgan RR, Robréau AM, Roberts AG, Nordmann Y, Elder GH, Deybach JC (1999) Variegate porphyria in Western Europe: identification of PPOX gene mutations in 104 families, extent of allelic heterogeneity, and absence of correlation between phenotype and type of mutation. Am J Hum Genet 65:984–994
Wilmotte R, Marechal J, Morle L, Baklouti F, Philippe N, Kastally R, Kotula L, Delaunay J, Alloisio N (1993) Low expression allele αlely of red cell spectrin is associated with mutations in exon 40 (αv/41 polymorphism) and intron 45 and with partial skipping of exon 46. J Clin Invest 91:2091–2096
Wiman A, Floderus Y, Harper P (2002) Two novel mutations and coexistence of the 991C>T and the 1339C>T mutation on a single allele in the coproporphyrinogen oxidase gene in Swedish patients with hereditary coproporphyria. J Hum Genet 47:407–412
Yan H, Dobbie Z, Gruber SB, Markowitz S, Romans K, Giardiello FM, Kinzler KW, Vogelstein B (2002) Small changes in expression affect predisposition to tumorigenesis. Nat Genet 30:25–26
Yasui Y, Muranaka S, Tahara T, Shimizu R, Watanabe S, Horie Y, Nanba E, Uezato H, Takamiyagi A, Taketani S, Akagi R (2002) A new ferrochelatase mutation combined with low expression alleles in a Japanese patient with erythropoietic protoporphyria. Clin Sci 102:501-506
Yoo HW, Warner CA, Chen CH, Desnick RJ (1993) Hydroxymethylbilane synthase: complete genomic sequence and amplifiable polymorphisms in the human gene. Genomics 15:21–29
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Gouya, L., Puy, H., Robreau, AM. et al. Modulation of penetrance by the wild-type allele in dominantly inherited erythropoietic protoporphyria and acute hepatic porphyrias. Hum Genet 114, 256–262 (2004). https://doi.org/10.1007/s00439-003-1059-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-003-1059-5