
Abstract
The distribution of arsenic between calcium ferrite slag and liquid silver (wt pct As in slag/ wt pct As in liquid silver) with 22 wt pct CaO and between iron silicate slag with 24 wt pct SiO2 and calcium iron silicate slags was measured at 1573 K (1300 °C) under a controlled CO-CO2-Ar atmosphere. For the calcium ferrite slags, a broad range of oxygen partial pressure (10–11 to 0.21 atm) was covered, whereas for the silicate slags, the oxygen partial pressure was varied from 10–9 to 3.1 × 10–7 atm. The measured relations between the distribution ratio of As and the oxygen partial pressure indicates that the oxidation state of arsenic in these slags is predominantly As3+ or AsO1.5. The measured distribution ratio of arsenic between the calcium ferrite slag and the liquid silver was about an order of magnitude higher than that of the iron silicate slag. In addition, an increasing concentration of SiO2 in the calcium-ferrite-based melts resulted in decreases in the distribution of arsenic into the slag. Through the use of measured equilibrium data on the arsenic content of the metal and slag in conjunction with the composition dependent on the activity of arsenic in the metal, the activity of AsO1.5 in the slags was deduced. These activity data on AsO1.5 show a negative deviation from the ideal behavior in these slags.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Low levels of toxic elements such as arsenic, antimony, bismuth, cadmium, mercury, selenium, and tellurium commonly are found in base metal ores. Clean, coarsely grained ore bodies are becoming depleted with the ore bodies of the future becoming more complex, finer grained, and containing increasing amounts of toxic elements. Worldwide, the industry mines and processes hundreds of millions of tons of base metal ores each year; thus, the accumulated mass of minor elements introduced into the biosphere is large and one could expect a significant environmental impact to result. The current knowledge of the levels of the minor elements mined and brought into the biosphere each year is limited and insufficient for accounting purposes. However, research at the Commonwealth Scientific and Industrial Research Organisation (CSIRO) is aimed at gathering the required data for developing predictive models to account for the dispersion of minor elements in various solid, liquid, and gaseous streams. The present study provides a component of the work carried out to enable the development of predictive tools for studying the deportment of minor and trace elements between different phases during the processing of base metal ores.
The accurate accounting of arsenic in copper smelting processes is a challenging task, and mathematical tools often are sought to assist in bridging gaps in data and accounting for uncertainties resulting for nonrepresentative sampling and characterization of inhomogeneous samples from the commercial processes. Predictive models also are necessary to assist the industry in understanding the effects that process variables have on the deportment of minor elements such as arsenic between phases. This understanding can be used to develop alternative practices for controlling the deportment of minor elements between different product streams and for the safe storage/disposal of such toxic elements.
The thermodynamics of arsenic (As), such as its equilibrium distribution between slag and copper, its oxidation state in the slag, and its activity coefficient in the various slags, have been the subject of a numerous studies[1–9] over the last 30 years. A summary of the experimental conditions and partial results from these publications are provided in Table I with some key findings being summarized subsequently.
Nagmori et al.[1] measured the distribution of As between the liquid copper alloys and the iron silicate slags at oxygen potentials (\( P_{{{\text{O}}_{ 2} }} \)) in the range of 10–11 to 10–6 atm at 1473 K (1200 °C). They proposed the following equilibrium:
for which the equilibrium constant is expressed as follows:
The distribution coefficient \( L_{\text{As}}^{S/C} \) is defined as follows:
Rearranging Eq. [3] yields the following:
where [ ] refers to the arsenic in the metal, () refers to the oxidic arsenic in the slag, n T is the total mole number of constituents in 100 g of each phase when all constituents are expressed with a mononuclear metal atom base such as FeO1.5, CuO0.5, etc. Provided that n T and the activity coefficients are kept constant, the plot of \( \log L_{\text{As}}^{S/C} \) against \( \log P_{{{\text{O}}_{2} }} \)could give a linear relationship with a slope of ν/2, which suggests the dissolved species in the slag. However, the \( \gamma_{{{\text{AsO}}_{1.5} }} \) in the slag may not be constant, and it could change with oxygen partial pressure because of the change of slag chemistry (Fe3+/Fe2+ ratio). The dependency of the \( \log \gamma_{{{\text{AsO}}v}} \) on oxygen partial pressure (Eq. [5]) may not be strong because oxygen partial pressure only changes the Fe3+/Fe2+ ratio, whereas other major components are kept at the same level. Equation [5] is expressed as follows:
Considering the effect of the oxygen partial pressure on the activity coefficient, Eq. [4] can be rearranged as follows:
The “a” term in Eq. [6] is expected to be much smaller than v/2, the slope of \( L_{\text{As}}^{S/C} \) still would be close to v/2, which indicates the oxidation state of As in slag.
Nagamori et al.’s experimental results showed that \( L_{\text{As}}^{S/C} \) is virtually constant and independent of the temperature and oxygen potential in the range of 10–6 to 10–11 atm. Therefore, they concluded that As dissolves in the slag in atomic form rather than as an oxide. Kashima et al.[2,3] examined the distribution of As between a silica-saturated iron silicate slag and liquid copper at 1573 K (1300 °C) and at \( P_{{{\text{SO}}_{ 2} }} \) ranging from 10–6 to 0.1 atm. The oxygen partial pressure was estimated by the relations between \( P_{{{\text{O}}_{ 2} }} \), \( P_{{{\text{SO}}_{ 2} }} \), and the matte grade. By plotting the \( L_{\text{As}}^{S/C} \) against the \( \log P_{{{\text{O}}_{ 2} }} \), the results showed that the As dissolved in the slag predominantly as AsO1.5. Lynch and Schwartze[4] equilibrated the Cu-As alloy with a silica-saturated iron silicate slag at temperatures between 1473 K (1200 °C) and 1536 K (1263 °C) and an oxygen partial pressure of 10–10 atm. The As distribution ratio obtained by Lynch and Schwartze showed much scatter. By assuming the As exists in the slag in atomic form, they derived the activity coefficient of As in the slag. However, the Fe3+/Fe2+ ratio in their slag indicates that the CO-CO2 gas–slag system had not reached equilibrium. Dabbs and Lynch[5] used a static dew-point technique to evaluate the solubility of arsenic in the silicate slag at an oxygen partial pressure from 10–12 to 10–8 atm at temperatures of 1458 K (1185 °C) and 1523 K (1250 °C). They found the solubility of arsenic in the slag to be independent of oxygen partial pressure, which indicates that element arsenic exists in the silicate slags. Jimbo et al.[6] found that the dissolution of As in the slag is dependent on the oxygen partial pressure in their experiments. Plots of As solubility in their slags, expressed as \( \log L_{\text{As}}^{S/C} \) vs \( \log P_{{{\text{O}}_{ 2} }} \), suggested that As exists in the slag as AsO and AsO1.5. They also found that when the oxygen partial pressure is strongly reducing, the As solubility in their slag approaches zero, which indicates that As dissolves in the slag only in oxidic form and that no measureable solubility of atomic As exists in such slags at very reducing conditions.
Takeda et al.[7] measured the distribution of As between molten copper and calcium-ferrite-based slags at 1523 K (1250 °C) under varied oxygen partial pressures. They concluded that As exists in the slag as AsO1.5. Their results also showed a much higher distribution ratio and hence the solubility of As in calcium ferrite slags as compared with iron silicate slags. Eerola et al.[8] also studied the distribution behavior of As between molten copper and calcium ferrite slags under copper fire-refining conditions. The \( L_{\text{As}}^{S/C} \) they measured at the oxygen partial pressure ranging from 10–8 to 10–6 atm are in good agreement with the results from Takeda et al.,[7] which suggests that As exists in the slag as AsO1.5 in calcium-ferrite-based slags. When the oxygen partial pressure was increased above 10–6 atm, the \( L_{\text{As}}^{S/C} \) reported by Eerola et al.[8] is higher than that of Takeda et al.,[7] which indicated that AsO2.5 and AsO1.5 could coexist in the slag at such oxygen potentials. Kim and Sohn[9] investigated the effects of CaO, Al2O3, and MgO additions to silica-saturated iron silicate slags on the As distribution ratio between molten copper and slag at 1523 K (1250 °C) as well as the oxygen partial pressure in the range 10–12 to 10–6 atm. They found that the As distribution ratio is independent of oxygen partial pressure, supporting the atomic dissolution hypothesis of As in the slag. They also found that the As distribution ratio was not affected significantly by the additions of basic oxides such as CaO and MgO to the silicate-saturated slags.
As summarized in Table I and as discussed previously, considerable discrepancies seem to persist between the published experimental data on the oxidation state of As in slags. Furthermore, the distribution ratios obtained for similar silicate slags under comparable conditions also shows some discrepancies. The studies discussed as well as other published equilibrium data on the solubility of copper in iron silicate and calcium ferrite slags[10] have shown that copper oxide can have significant solubility in both iron silicate and calcium ferrite slags, and its solubility increases with oxygen partial pressure. Liquid copper alloys have a strong affinity for arsenic[11] and readily could “contaminate” silicate slags through suspension/emulsification. Thus, it is possible that the aforementioned discrepancies are at least partly due to (1) suspended copper-arsenic microdroplets in the slag and (2) interactions between oxide(s) of copper with arsenic dissolved in the slag. Thus, the primary objective of the present study was to resolve the discrepancy in the published studies on the oxidation state and the solubility of arsenic in slags and then to deduce the effect of slag chemistry and its oxidation state on the activity and the activity coefficient of the most stable form of arsenic species in slags. Because silver oxide is fairly unstable[12] at oxygen potentials of interest, its solubility in slags is likely to be low. Second, because of differences in other physicochemical properties (e.g., surface and interfacial tensions) of liquid copper and silver alloys, using silver-arsenic alloys rather than copper-arsenic alloys could reduce/eliminate the contamination of the slag by arsenic containing metal droplets. It therefore was decided to use liquid silver-arsenic alloys in the present study in which the classical metal–slag–gas equilibrium technique was used to determine the effects of oxygen partial pressure and slag chemistry on the arsenic distribution between the slags and alloys using a CO-CO2 gas mixture to control the oxygen potential.
Experimental
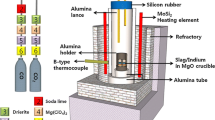
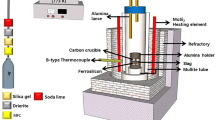
The master CaO-FeOx and SiO2-FeOx slags were prepared by premelting mixtures of reagent grade CaCO3, SiO2, Fe2O3, and prereduced FeOx in a platinum crucible at 1573 K (1300 °C) under an oxygen partial pressure of 10–7 atm. The calcium ferrite slag contained 22 wt pct CaO, and the iron silicate slag contained 24 wt pct SiO2. Master slags of SiO2-CaO-FeOx also were prepared by melting together reagent-grade SiO2, CaCO3, Fe2O3, and FeOx in a platinum crucible at 1573 K (1300 °C) in a muffle furnace without atmospheric control. The master Ag-As alloy was prepared by melting granules of silver and arsenic metal in an alumina crucible at 1273 K (1000 °C) under a flow of nitrogen, which helped to suppress oxide formation. The metal phase at the start of each experiment was fixed at 1 wt pct As by mixing silver granules with an Ag-As master alloy.
A vertical tube furnace fitted with six silicon carbide heating elements was used for the metal–slag–gas equilibration experiments.[13] This furnace was fitted with water-cooled brass end caps to isolate the atmosphere inside the furnace. Reaction gases entered the furnace from the bottom and exited through an alumina tube connected to gas bubblers. The Ar, CO2, and CO gases were regulated by a set of mass flow controllers then were purified by passing through heated copper turnings before being mixed in a column of glass beads and introduced to the tube furnace. A total flow rate of 500 ml/min was maintained for the duration of each experiment. The required CO2/CO ratio for the desired \( P_{{{\text{O}}_{2} }} \) for an experiment was determined by using the following equation from Yazawa and Takeda[14]:
In each experiment, approximately 8 g of the Ag-As (1 wt pct) alloy and 8 g of slag were placed in a magnesia crucible. The crucible was lowered to the hot zone of the vertical tube furnace under an Ar flow in which the temperature, as measured by an R-type (Pt/Pt-13 wt pct Rh) thermocouple, remained constant at 1573 ± 1 K. The thermocouples used were calibrated against the melting point of copper, which have an accuracy of within ± 1 K.
After the required equilibration time, the crucible and its content were removed from the furnace and quickly quenched in the cold water. The slag and metal phases then were removed from the crucible and separated by physical means. Care was taken to avoid contamination of the slag with metallic droplets or with MgO-rich phase from the crucible. Representative samples of the metal and slag then were analyzed by inductively coupled plasma (ICP) techniques after acid digestion. Calcium and silicon in the slag were digested using a borate fusion technique according to an Australian standard.[15] The silver in the slags was digested in concentrated nitric acid to avoid forming insoluble Ag compounds. The metal samples were dissolved in chloride-free nitric acid, using a sealed microwave digestion technique to avoid the loss of volatile species of arsenic from the solution. The solution then was cooled, diluted, and analyzed for arsenic and iron using ICP atomic emission spectrometry. Ferrous oxide and the total iron content of the slag samples were determined with the digestion and titration technique adapted from Young.[16]
The calcium ferrite slags and iron silicate slags investigated were known to be single-phase liquids at the oxygen partial pressures and temperatures of experiments from the works of Takeda et al.[17] and Muan.[18] According to Henao et al.,[19] the calcium iron silicate slag with about 5 wt pct SiO2 and 23 wt pct CaO was saturated with Ca2SiO4 at 1573 K (1300 °C) and an oxygen partial pressure of 10–7 atm, whereas the rest of the calcium iron silicate slags were liquid.
In the present study, the distribution ratio of arsenic between the slag and the liquid silver is based on the bulk analysis of both phases, which are assumed to be the pure liquids. For the slag samples saturated with solid phases, the measured As distribution ratio will deviate from the real value.
Results
The time required for the silver-alloy, calcium ferrite slag and gas to reach equilibrium was determined from two series of experiments. In the first series, pure silver was brought into contact with the master calcium ferrite slag containing 1 wt pct arsenic at 1573 K (1300 °C) and oxygen partial pressure of 10–7 atm. In the second series, the Ag-1 wt pct As alloy was used so that equilibrium could be approached from the opposite direction. The results from these experiments are shown in Figure 1, which depicts the equilibrium between the calcium ferrite slag and metal being approached within 30 hours. Steady Fe3+/Fe2+ values for the slag were recorded, as shown in Figure 2, which suggests that the gas–slag reaction also reached equilibrium in less than 30 hours. The good agreements between the As distribution values from these two series of experiments confirm that the technique produced reproducible results and that a close approach to equilibrium was reached. Similar tests were carried out for determining the time required for the iron silicate slag, metal, and gas to reach equilibrium. As shown in Figures 1 and 2, the iron silicate slag, metal, and gas phases can reach equilibrium within 18 hours.

Variation of the arsenic distribution among the calcium ferrite slag, iron silicate slag, and liquid silver with the reaction time

Fe3+/Fe2+ ratio in slag as a function of time at 1573 K (1300 °C) and 10–7 atm of oxygen partial pressure for the calcium ferrite slag and the iron silicate slag
Through subsequent experiments, the distribution of arsenic between the slag and the metal was measured under different experimental conditions, which include variations in slag chemistry and oxygen partial pressure. The experimental conditions as well as the compositions of the slag and alloy after equilibration are listed in Table II. The master slags were initially MgO-free; however, after equilibration, the calcium ferrite slags contained 2.5 to 3.5 wt pct MgO, and the iron silicate slags contained 4.5 to 6.0 wt pct MgO. No evidence of entrapped silver metal in the slags was observed; the silver level in the slag after the equilibration was always less than 0.04 wt pct. The activity coefficients of AsO1.5 listed in Table II were calculated from Eq. [3] at a given oxygen partial pressure, activity coefficient of As in liquid Ag, and the measured distribution ratio. The calculation of the activity coefficients of AsO1.5 is discussed in more detail in the following section.
Discussion
The variation of the Fe3+/Fe2+ ratio in both the calcium ferrite slag and the iron silicate slag are plotted as a function of \( \log P_{{{\text{O}}_{2} }} \) in Figure 3. As is shown, the Fe3+/Fe2+ ratio in the calcium ferrite slag of this study is in good agreement with that from Takeda et al.,[17] which demonstrates that the slag and gas in this study are in equilibrium. For the iron silicate slag, the slope of the data is close to that of the silica-saturated iron silicate slag given by Michal and Schuhmann,[20] whereas the Fe3+/Fe2+ ratio was approximately doubled by decreasing the SiO2 content to 25 pct. This outcome was supported by Michal and Schuhmann’s[20] finding that the Fe3+/Fe2+ ratio increases with a decrease in SiO2 in the slag. It is worth noting that as a basic oxide, MgO tends to increase the Fe3+/Fe2+ ratio in slags[21,22]; thus, the measured higher Fe3+/Fe2+ ratios in the MgO saturated iron silicate slags is partly caused by MgO.

The measured distribution ratio \( L_{\text{As}}^{S/C} \) between the calcium ferrite slags and the iron silicate slags and metal are plotted against the oxygen partial pressure in Figure 4. It is evident that the distribution of As is affected strongly by the oxygen partial pressure over the range studied, which supports the oxidic dissolution of As in the slag. Considering the valence of the As is 3+ or 5+, the As exists in the slag as AsO1.5 or AsO2.5 and the slope of log\( L_{\text{As}}^{S/C} \)should be close to 0.75 or 1.25 according to Eq. [6]. As listed in Table II, the n T of both types of slags in the present study changes slightly with the variation of oxygen partial pressure. Given the low concentration of As in the alloy listed in Table II, the γ As in the alloy also should be constant. Therefore, according to Eq. [6], the slope of the As distribution ratio (Figure 4) would be close to v/2. The lines shown in the figure have a slope of about 0.6, which is close to 0.75 for both iron silicate slags and calcium ferrite slags. This finding indicates that the predominant As species dissolved in both slags was AsO1.5, which agrees with the experimental results from studies in References 7 and 8. Figure 4 also shows that the As distribution ratio between the calcium ferrite slag and liquid silver at an oxygen partial pressure of 10–11 atm deviated from the main tendency. According to Takeda et al.,[17] the calcium ferriet slag with 22 wt pct CaO at 1573 K (1300 °C) and 10–11 atm of oxygen partial pressure is close to the iron γ saturation liquid boundary. Considering the experimental uncertainties, the slag under this condition is likely to be saturated with solid iron γ, which has high affinity for arsenic. As shown in Figure 4, because of the limitation of the experimental technique mentioned before, the bulk analysis of the As in the slag containing solid iron γ leads to the overestimation of the As distribution at 10–11 atm oxygen partial pressure. These results also show that the \( L_{\text{As}}^{S/C} \) of the iron silicate slag is about one magnitude lower than that of the calcium ferrite slag at any given oxygen partial pressure. This finding is also consistent with the results from Takeda et al.,[7] which suggests that the calcium ferrite slag has a greater capacity for acidic oxides such as AsO1.5 than the iron silicate slag. The effect of slag chemistry on the arsenic distribution at a fixed oxygen partial pressure of 10–7 atm at 1573 K (1300 °C) is shown in Figure 5. The As distribution into the slag decreases with an increasing SiO2 content, which suggests that the interaction between the AsO1.5 and basic oxides such as CaO and FeO is stronger than that of SiO2.

Variation of the equilibrium distribution of arsenic between slags and the liquid silver with oxygen partial pressure at 1573 K (1300 °C) from the present work

Variation of the equilibrium distribution of arsenic between slags and the liquid silver with a silica-to-lime ratio in the slag at 1573 K (1300 °C) from the present work
Rearranging Eq. [2] yields the following:
Thus, for a given activity coefficient of As in the metal, concentrations of As in the metal and slag, and the oxygen partial pressure, the activity coefficient of arsenic oxide (\( \gamma_{{{\text{AsO}}_{v} }} \)) can be calculated readily from Eq. [8]. Hino and Azakami[11] used the isopiestic technique to measure the activity of arsenic in the Ag-As alloy at 1423 K (1150 °C). In this technique, pure arsenic was used to equilibrate the arsenic alloy with arsenic vapor. The activity of arsenic then was calculated by considering the equilibrium among the arsenic gas species As, As2, As3, and As4. They reported that the activity coefficient of arsenic at an infinite dilution \( \gamma_{\text{As}}^{\text{O}} \) was about 0.35 at 1423 K (1150 °C). However, Pei,[23] who assessed the activity of arsenic in molten copper, had shown that the thermodynamic data for condensed arsenic and arsenic vapor species from Gokcen’s critical assessment[24] are more accurate than other data such as the data used by Hino and Azakami.[11] It thus was decided to use the data by Gokcen[24] and Pei[23] in the present work to reevaluate the activity of arsenic in liquid silver and to resolve the discrepancies between the data from different authors. The Gibbs energy changes for equilibrium between condensed arsenic and various arsenic vapor species from Gocken[24] are summarized in Table III. These changes were used in the present work to reevaluate the arsenic activity in liquid silver. The recalculated arsenic activities in liquid silver are listed in Table IV. Also listed in Table IV are the original data reported by Hino and Azakami[11] at 1423 K (1150 °C). It is shown that the recalculated arsenic activities are significantly lower than the originally reported values. In Figure 6, the logarithm of the activity coefficient of arsenic is plotted against (1 – X As)2, which shows a linear relation over the measured composition range. Extrapolation of the arsenic activity coefficient to a zero arsenic concentration leads to a value of 0.074 for the arsenic activity coefficient at infinite dilution \( \gamma_{\text{As}}^{\text{O}} \) in liquid silver at 1423 K (1150 °C), which is much lower than the value reported by Hino and Azakami.[11] By applying the regular solution approximation, \( \gamma_{\text{As}}^{\text{O}} \) at 1573 K (1300 °C) was estimated to be 0.095. The activity of arsenic in the alloy from the present work was calculated by using this value of \( \gamma_{\text{As}}^{\text{O}} \). Based on the calculated As activity in the alloy and the distribution ratio of As between the slag and the metal, the activity, and hence the activity coefficient of AsO1.5, was calculated with Eq. [8]. For these calculations, a value of 87,096 was used for the equilibrium constant at 1573 K (1300 °C) for the following reaction[25]:

Takeda et al.[7] measured the distribution of arsenic between the liquid copper and the calcium ferrite slags under varied oxygen partial pressure at 1523 K (1250 °C). The activity coefficients of AsO1.5 in their calcium ferrite slags were calculated based on their measured distribution ratio and the activity coefficient of As in liquid copper. The activity coefficient of AsO1.5 in the silica-saturated iron silicate slags also was calculated by Takeda et al.[7] based on the distribution ratio of arsenic between the iron silicate slag and the liquid copper measured by Kashima et al.[3] at 1573 K (1300 °C). To compare, the AsO1.5 in both the calcium ferrite slag and the iron silicate slag from Takeda et al.[7] and Kashima et al.[3] were recalculated by using the \( \gamma_{\text{As}}^{{}} \) in liquid copper, which was reevaluated by Pei.[23] The \( \gamma_{\text{As}}^{\text{O}} \) used by Takeda et al. was set to be 0.004 at 1523 K (1250 °C), whereas it is estimated to be 0.0018 at 1423 K (1150 °C) in this study. By applying the regular solution approximation, \( \gamma_{\text{As}}^{\text{O}} \) was estimated to be 0.0028 at 1523 K (1250 °C) and 0.0033 at 1573 K (1300 °C). The recalculated activity coefficients of AsO1.5 in both the calcium ferrite slag and the iron silicate slag from Takeda et al.[7] and Kashima et al.[3] and those of this study are plotted against oxygen partial pressure in Figures 7 and 8, respectively.

Comparison of the activity coefficient of AsO1.5 in the calcium ferrite slag from the present work with that obtained by Takeda et al.[7]

As shown in Figures 7 and 8, the activity coefficients of AsO1.5 in the calcium ferrite slag is about one order of magnitude lower than that of the iron silicate slag. Figure 9 shows that the activity coefficient of AsO1.5 increases sharply with an increasing SiO2 content in the slag, which suggests that the interaction between the SiO2 and AsO1.5 is considerably weaker than that between basic oxides such as CaO, FeO, and AsO1.5. Alternatively, one could explain this behavior in terms of strong interactions between acid oxide such as silica and basic oxides such as CaO and FeO, leading to a considerable decrease in the activities of CaO and FeO, which in return, reduces the interaction between the CaO, FeO, and AsO1.5, hence increasing the activity coefficient of arsenic in the slag.

Effect of slag chemistry on the activity coefficient of AsO1.5 in the slag from the present work
Figures 7 and 8 also show that in both calcium ferrite and iron silicate slags, \( \gamma_{{{\text{AsO}}_{ 1. 5} }}^{{}} \) increases with increasing oxygen partial pressures. The slope of the \( \gamma_{{{\text{AsO}}_{ 1. 5} }}^{{}} \)—a in Eq. [6]—is about 0.18 for the calcium ferrite slag and 0.16 for the iron silicate slag. Considering the experimental uncertainty and because a is much smaller than v/2 of 0.75, the assumption that As dissolves in the slag dominantly as AsO1.5 is reasonable according to Eq. [6]. The dependency of \( \gamma_{{{\text{AsO}}_{ 1. 5} }}^{{}} \) on oxygen partial pressure illustrates the effect of the oxidation state of the slag (Fe3+/Fe2+) on the activities of basic oxides such as CaO and FeO. It is known that for a given CaO content in calcium ferrite slags, an increasing oxygen potential leads to a decrease of the activities of CaO and FeO in the slag,[17] which in return, reduces the interaction between the CaO, FeO, and AsO1.5, hence increasing the activity coefficient of arsenic in the slag.
As shown in Figure 7, within the experiment uncertainty, good agreement exists between values of the \( \gamma_{{{\text{AsO}}_{ 1. 5} }}^{{}} \) in calcium ferrite slags from the present study and from Takeda’s work[7] at oxygen partial pressures below 10–7 atm. According to Takeda’s work,[14] the Cu content in the slag reaches 3 wt pct at an oxygen partial pressure of 10–7 atm. This agreement indicates that low level of copper oxide in the slags used by Takeda et al.[7] has a relatively small effect on the thermodynamics of arsenic in such slags. At an oxygen partial pressure of 10–6 atm, the Cu content in the calcium ferrite slag reaches 6 wt pct.[14] This change of slag chemistry may be responsible for the different values of \( \gamma_{{{\text{AsO}}_{ 1. 5} }}^{{}} \) between the two studies. Another possible reason for these different values could be a higher degree of uncertainty in the determination of low levels of As in the silver alloys at an oxygen partial pressure of 10–6 atm. However, for the iron silicate slags, Figure 8 shows a significant difference from the results of Kashima et al.[3] The silica content in the slag of the present study was about 24 wt pct, whereas that of Kashima et al.[3] reaches saturation. The reason for this difference from Kashami et al.’s data is not clear to the present authors. One possible source of error in Kashima’s study is that the matte, slag, and liquid copper were equilibrated under a gas mixture of SO2-N2. The oxygen partial pressure was estimated by using Eq. [11], which was derived from reaction [10] at a high-matte-grade area in their study. Reaction [10] and Eq. [11] are expressed as follows:
In the low-matte-grade area, the oxygen partial pressure was estimated by using the relations between the \( \log P_{{{\text{O}}_{2} }} \) and the matte grade. This estimation also may contribute to the scatter of their data. However, this is not likely to cause such a large systematic low value of \( \gamma_{{{\text{AsO}}_{ 1. 5} }}^{{}} \). Another possible source of error in their study is the entrapment of liquid copper in their slags, which would lead to a high value of arsenic in the slag and hence a low value of \( \gamma_{{{\text{AsO}}_{ 1. 5} }}^{{}} \) in their slags. As mentioned earlier, the silver levels in the slags from the present work were checked carefully, and the entrapment of silver metal in the slags was not evident. The equilibrium distribution ratios of As among the iron silicate slag, the calcium ferrite slag, and the copper matte under varied SO2 partial pressures at 1573 K (1300 °C) and 1523 K (1250 °C) were determined by Roghani et al.[26,27] The As distribution ratio of the calcium ferrite slag is about 40 times of that for the silica-saturated iron silicate slag at the same matte grade.[26,27] This suggests that the \( \gamma_{{{\text{AsO}}_{ 1. 5} }}^{{}} \) in the iron silicate slag and the calcium ferrite slag can be related as follows:
Based on the \( \gamma_{{{\text{AsO}}_{ 1. 5} }}^{{}} \) in the calcium ferrite slag from this study and from Takeda’s work,[7] the \( \gamma_{{{\text{AsO}}_{ 1. 5} }}^{{}} \) in the silica-saturated iron silicate slag at 1573 K (1300 °C) was estimated by Eq. [12], which was plotted in Figure 8 as a dashed line. The estimated \( \gamma_{{{\text{AsO}}_{ 1. 5} }}^{{}} \) is higher than that of iron silicate slags containing 24 wt pct SiO2, which supports the experimental finding of this study that the increases of SiO2 content in the slag will increase the \( \gamma_{{{\text{AsO}}_{ 1. 5} }}^{{}} \).
Conclusions
A study of the equilibrium distribution of arsenic between CaO-FeOx-SiO2 slags and liquid silver at 1573 K (1300 °C) under varied oxygen partial pressures showed that arsenic distribution is affected strongly by the oxygen partial pressure and that the arsenic exists in slags predominantly as AsO1.5. The distribution ratio between the calcium ferrite slag and the metal was about an order of magnitude higher than that of the iron silicate slag, which indicates that the calcium ferrite slag has a much high arsenic capacity than the iron silicate slag. The distribution ratio decreased with an increasing SiO2 content in slag.
Based on the assessed thermodynamic data of arsenic gas species by Gocken,[24] the arsenic activities in liquid silver were recalculated from the literature work. By using the recalculated activity coefficient of arsenic in infinitely dilute solutions, the activity and, hence, the activity coefficient of AsO1.5 in the slag was calculated from the measured distribution ratio. The increases of the activity coefficient of AsO1.5 with an increasing SiO2 content in the slag suggests that the interaction between the AsO1.5 and the basic oxides such as CaO and FeO are much stronger than that with SiO2.
References
M. Nagamori, P.J. Mackey, and P. Tarassoff: Metall. Trans. B, 1975, vol. 6B, pp. 295-01.
M. Kashima, M. Eguchi, and A. Yazawa: Trans. Jpn. Inst. Met., 1978, vol. 19, pp. 152-58.
M. Kashima, Y. Nishikawa, M. Eguchi, and A. Yazawa: J. Jpn. Inst. Met., 1980, vol. 96, pp. 907-11.
D.C. Lynch and K.W. Schwartze: Can. Metall. Q., 1981, vol. 20, pp. 269-78.
D.M. Dabbs and D.C. Lynch: Advances in Sulfide Smelting, vol. 1, The Metallurgical Society of AIME, Warrendale, PA, 1983, pp. 143-69.
I. Jimbo, S. Goto, and O. Ogawa: Metall. Trans. B, 1984, vol. 15B, pp. 535-41.
Y. Takeda, S. Ishiwata, and A. Yazawa: Trans. Jpn. Inst. Met., 1983, vol. 24, no. 7, pp. 518-28.
H. Eerola, K. Jylha, and P. Taskinen: Trans. Inst. Min. Metall. C, 1984, vol. 93, pp. C193-99.
H.G. Kim and H.Y. Sohn: Metall. Mater. Trans. B, 1998, vol. 29B, pp. 583-90.
M. Somerville: Masters Thesis, University of Melbourne, Melbourne, Australia, 2000.
M. Hino and T. Azakami: Metall. Rev. MMIJ, 1986, vol. 3, no. 1, pp. 61-78.
O. Knacke, O. Kubachewski, and K. Hesselmann: Thermochemical Properties of Inorganic Substances, Springer-Verlag, Berlin, Germany, 1991.
M.D. Johnston, S. Jahansshahi, and F.J. Lincoln: Metall. Mater. Trans. B, 2007, vol. 38B, pp. 433-42.
A. Yazawa and Y. Takeda: Trans. Jpn. Inst. Met., 1982, vol. 23, no. 6, pp. 328-33.
Australian Standards 1038, Part 14.1: Analysis of Coal Ash, Coke Ash and Mineral Matter, 1981.
R.S. Young: Chemical Analysis in Extractive Metallurgy, Charles Griffin and Company Limited, London, UK, 1971, pp. 172-87.
Y. Takeda, S. Nakazawa, and A. Yazawa: Can. Metall. Q., 1980, vol. 19, pp. 297-05.
A. Muan: Trans. AIME, 1955, vol. 203, pp. 965-77.
H.M. Henao, F. Kongoli, and K. Itagaki: Mater. Trans. JIM, 2005, vol. 46, no. 4, pp. 812-19.
E.J. Michal and R. Schuhmann: J. Met., 1952, vol. 4, pp. 723-28.
L. Yang: Ph.D. Dissertation, University of Newcastle, Sydney, Australia, 1994.
S. Sun and S. Jahanshahi: Metall. Mater. Trans. B, 2000, vol. 31B, pp. 937-44.
B. Pei: Scand. J. Metall., 1993, vol. 22, pp. 24-29.
N.A. Gocken: Bull Alloy Phase Diagrams, 1989, vol. 10, pp. 11-22.
I. Ansara and B. Sundman: Computer Handling and Dissemination of Data, ed., P.S. Glaser, Elsevier Science Ltd., Atlanta, GA, 1986, pp. 154-58.
G. Roghani, Y. Takeda, and K. Itagaki: Metall. Mater. Trans. B, 2000, vol. 31B, pp. 705-12.
G. Roghani, J.C. Font, M. Hino, and K. Itagaki: Mater. Trans. JIM, 1996, vol. 37, pp. 1574-79.
Acknowledgments
The authors wish to thank Mr. Roy Hampson and Mr. Rowan Davidson for carrying out the experimental work. Financial support for this work was provided by CSIRO Process Science and Engineering and the former GK Williams Cooperative Research Centre for Extractive Metallurgy.
Author information
Authors and Affiliations
Corresponding author
Additional information
Manuscript submitted July 1, 2009.
Rights and permissions
About this article
Cite this article
Chen, C., Jahanshahi, S. Thermodynamics of Arsenic in FeOx-CaO-SiO2 Slags. Metall Mater Trans B 41, 1166–1174 (2010). https://doi.org/10.1007/s11663-010-9430-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11663-010-9430-0