
Abstract
This study determined the effects of miR-338-3p on osteoclast (OC) differentiation and activation. The change levels of miR-338-3p in differentiated OCs were investigated by microRNA microarray assay and quantitative real-time PCR analysis. The effects of miR-338-3p on the differentiation and activation of OCs were determined by tartrate-resistant acid phosphatase staining resorption activity assay and Western blot. Target genes of miR-338-3p were identified by target gene prediction and dual-luciferase reporter gene detection assay as well as Western blot. Results showed that miR-338-3p was markedly downregulated in differentiated OCs. miR-338-3p could inhibit the formation and absorption activity of OCs. Western blot showed that miR-338-3p could influence the change levels of OC differentiation–related proteins. Dual-luciferase reporter gene detection assay and Western blot both showed that miR-338-3p directly targeted IKKβ gene. In conclusion, miR-338-3p may affect the formation and activity of OCs by targeting the IKKβ gene.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Osteoclasts (OC) originate from mononuclear macrophages of hematopoietic stem cells. With the regulation of many factors, OC precursors differentiate on the surface of the bone and fuse into polynuclear OCs. OCs are the only cells with bone resorption ability in the human body; thus, the excessive activity of OC may result in an excessive bone loss (Soo-Jin et al. 2005; Ono and Nakashima 2018). OC bone resorption initiates the cycle of bone remodeling, and human bone mass is maintained, which depends on the dynamic balance of bone formation and bone resorption (Detsch and Boccaccini 2015). In the body, osteoblasts are responsible for the secretion of bone matrix constituents, mineralization of bone matrix, and bone formation (Khandaker et al. 2016). Conversely, OCs are mainly responsible for bone resorption (Ono and Nakashima 2018). Most bone diseases in adults, such as osteoporosis and rheumatoid arthritis, are caused by over-activated OCs (Kim and Kim 2016).
Previous studies indicated that many miRNAs can affect the formation and function of OCs by regulating the target genes or corresponding signal transduction pathways in the process of OC differentiation and activation (Chen et al. 2014; Dole and Delany 2016). However, the regulatory mechanism of most miRNAs on OC differentiation and activation remains to be identified. By using miRNA microarray assay, we found that the expression of miR-338-3p in differentiated OCs was significantly lower compared with undifferentiated OCs (P < 0.01). The results suggested that miR-338-3p might play an important role in OC differentiation. Therefore, miR-338-3p was selected as the research object in this study. miR-338-3p is one of the newly discovered miRNAs in recent years; it is mainly involved in the process of cell differentiation. Several studies have identified the regulatory effects of miR-338-3p on the nervous system, tumor invasion, metastasis, and osteoblast differentiation. For example, miR-338-3p can influence the differentiation of mouse bone marrow stromal cells by targeting Runx2 and Fgfr2 genes to produce osteoblasts (Liu et al. 2014). However, the role of miRNA-338-3p in the regulation of OC formation and function remains unclear. Therefore, in the present study, the mechanisms of miRNA-338-3p in regulating OC formation and function were investigated.
The mmu-miR-338-3p target genes were predicted using the miRWalk 2.0 software. Moreover, the predicted results of miRWalk, miRanda, RNA22, and Targetscan were compared. We found that the key gene of IκB kinase β (IKKβ) (Ikbkb, IKK2) in the NF-κB signal transduction pathways is one of the target genes of mmu-miR-338-3p. The IKK complex was critical to the activation of the NF-κB signaling pathways. OC precursors lacking IKKβ activity exhibited differentiation defects both in vitro and in vivo (Otero et al. 2010). IKKβ plays a dominant role in OC differentiation.
In this study, OCs and their precursors were used as the research objects. microRNA microarray assay was employed to determine the differential expression of miRNAs between differentiated OCs and OC precursors. We selected the significant miRNA to predict and verify its target genes. Thus, the regulatory mechanisms of miRNAs on OC differentiation and activation were elucidated. The results of this study will improve our understanding of the mechanisms of miRNAs regulating the differentiation and activation of OCs and may provide a theoretical basis for the prevention and treatment of osteoporosis caused by abnormal OC activity.
Materials and Methods
Cells and reagents
The murine monocyte/macrophage cell lines RAW264.7 and 293T were obtained from the American Type Culture Collection (Manassas, VA). Macrophage colony-stimulating factor (M-CSF) and receptor activator of NF-κB ligand (RANKL) were obtained from PeproTech, Inc. (Rocky Hill, NJ). Acid Phosphatase Kit 387-A (tartrate-resistant acid phosphatase (TRAP) staining kit) was acquired from Sigma-Aldrich (St. Louis, MO). OC resorption activity was detected using the Corning Osteo Assay Surface (COAS; Corning Inc., Corning, NY). Dulbecco’s modified eagle medium (DMEM), minimum essential medium alpha (MEM-alpha), and fetal bovine serum (FBS) were purchased from GE Healthcare (Salt Lake City, UT). miRNA-338-3p primer, U6 prime, and All-in-One™ miRNA qRT-PCR Detection Kit were purchased from GeneCopoeia, Inc. (Rockville, MD). Total RNA Kit I was bought from Omega Bio-Tek, Inc. (Norcross, GA). Bicinchoninic acid protein assay kit, nitrocellulose filter membranes, blocking buffer, primary antibody dilution buffer, Western blot wash buffer, and secondary antibody dilution buffer were obtained from (Beyotime Biotechnology, Shanghai, China). Primary anti-c-Fos, anti-phosphorylated (phospho)-c-Fos, anti-c-Jun, anti-phospho-c-Jun, anti-activated T nuclear factor (NFATc1), and anti-IKKβ were all purchased from Cell Signaling Technology, Inc. (Danvers, MA). Anti-β-actin antibody and goat anti-rabbit IgG-horseradish peroxidase (HRP) secondary antibody were bought from Santa Cruz Biotechnology, Inc. (Dallas, TX). Immobilon Western Chemiluminescent HRP Substrate Detection reagent was bought from EMD Millipore (Burlington). Dual-luciferase reporter gene detection system kit was bought from Promega Corporation (Madison). Lipofectamine 2000 transfection reagent was bought from Invitrogen Corporation (Carlsbad, CA).
RAW264.7 cell cultures and M-CSF + RANKL induction
The RAW264.7 cells were incubated in DMEM containing 10% FBS, 2 mM/L l-glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin at 37°C in a humidified atmosphere of 5% CO2. To induce OC differentiation, RAW264.7 cells were resuspended in MEM-alpha and seeded into 96-well plates (3 × 103 cells/mL), COAS (3 × 103 cells/mL), and 6-well plates (1 × 105 cells/mL) for 4 h of cultivation. The medium was changed to MEM-alpha plus 25 ng/mL M-CSF + 30 ng/mL RANKL.
MicroRNA microarray assay
The RNAs of OC precursors (RAW264.7 cells) and differentiated OCs were extracted. MicroRNA microarray assay was performed by KangChen Bio-tech (Shanghai, China), and each sample was biologically repeated three times. The extracted RNA was labeled with miRCURY™ Array Power Labeling kit (Cat #208032-A, Exiqon, Vedbaek, Denmark) after quality inspection. After labeling, the sample was hybridized with miRCURY™ LNA Array (v.19.0) (Exiqon Vedbaek, Denmark) chip according to the manufacturer’s instructions. The chip was scanned using Axon GenePix 4000B chip scanner. GenePix Pro 6.0 was employed to read the chip scan image and extract the signal value of the probe. The same probe takes the median value for merging. The probes that were ≥ 30.0 in all samples were kept, the median value of all chips was standardized, and the differential expression probes were screened. Fold change and P value were used to screen the differentially expressed miRNAs between the two groups. Fold change was used to screen the differentially expressed miRNAs between the two samples. Finally, the differentially expressed miRNAs were clustered, and the cluster diagram was drawn.
Expression levels of miR-338-3p were detected by quantitative real-time PCR assay
According to the experimental design, the cells were cultured and then collected. The total RNAs were extracted, and the cDNAs were inverted. The expression levels of miR-338-3p were detected by quantitative real-time PCR (qRT-PCR) assay and compared with the results of microRNA microarray assay. The miRNA-338-3p expression levels in OC precursors and differentiated OCs were monitored by stem-loop fluorescence quantitative PCR assay.
Transfection of miR-338-3p mimics and inhibitors
miR-338-3p mimics and inhibitors were synthesized by Suzhou GenePharma Co., Ltd. (Suzhou, China). They were transfected into M-CSF + RANKL inducing OC precursors using Lipofectamine 2000 transfection reagent. After 6 h of transfection, the medium was changed to M-CSF + RANKL-induced medium for 96 h of cultivation.
TRAP staining
miR-338-3p mimics and inhibitors were transfected into the M-CSF + RANKL inducing OC precursors employing Lipofectamine 2000 transfection reagent. At the end of cultivation, TRAP staining was performed following the instructions of the TRAP staining kit. In short, cells were fixed by 4% polyformaldehyde solution for 10 min and washed by double-distilled water twice. TRAP solution was added into samples, and cells were stained for 60 min in 37°C. The staining solution was removed, and cells were washed by double-distilled water for twice. Stained TRAP-positive cells were observed under an inverted microscope. Three wells were taken, and ten visual fields were randomly selected to count more than three cell nuclei in each well.
Absorption activity was determined by COAS
miR-338-3p mimics and inhibitors were transfected into the M-CSF + RANKL inducing OC precursors. After cultivation, the medium was discarded, the COAS wells were cleaned twice with double-distilled water, and further ultrasonic washing was conducted for 3 min. Next, 10% hypochlorous acid solution was added into the COAS wells and maintained at room temperature for 5 min. Then, the COAS wells were washed with double-distilled water twice and dried naturally for 3 to 5 h. Ten visual fields were randomly selected for each well and photographed, and the areas of absorption pits were calculated using an image analysis software (version 1.0, Jeda).
Protein preparation and Western blot assay
miR-338-3p mimics and inhibitors were transfected into the M-CSF + RANKL inducing OC precursors. At the end of incubation, cells were collected and lysed in ice for 30 min. After ultrasonic lysis, the samples were centrifuged for 10 min, and then the supernatant was collected. Total protein concentrations of each group were determined for each sample, and equal amounts were separated on 12% SDS-polyacrylamide gels at 110 V for 120 min. The protein samples were transferred to nitrocellulose membranes for 90 min, and the membranes were blocked at room temperature for 90 min. The samples were incubated with primary anti-c-Fos, anti-phosphorylated (phospho)-c-Fos, anti-c-Jun, anti-phospho-c-Jun, anti-NFATc1, anti-β-actin antibody, and anti-IKKβ (1:1000) at 4°C overnight. Then, the nitrocellulose membranes were washed with Tris-buffered saline and Tween 20 solution. The nitrocellulose membranes were incubated with goat anti-rabbit IgG-HRP secondary antibody and goat anti-mouse IgG-HRP secondary antibody (1:5000) at room temperature for 90 min. Enhanced chemiluminescence assay was performed. The gray levels of the protein bands were analyzed using the Image Lab software (version 5.2, Bio-Rad, Hercules, CA).
Dual-luciferase reporter gene detection assay
microRNA was diluted with DEPC-H2O, and the final concentration was 20 μM. In total, 100 μL serum-free DMEM, 10 μL microRNA, and 1.6 μg double fluorescent report vector were successively added into an EP tube and blended completely. Subsequently, 100 μL serum-free DMEM and 4 μL Lipofectamine 2000 were successively added into another EP tube and blended completely. With a 5-min incubation at room temperature, the solutions in the two tubes were mixed together at room temperature for 20 min. 293T cells were fused to 80–90% and washed with PBS. The cells were dissociated and resuspended; they were inoculated into 12-well plates at 5 × 105 cells/well for 24 h of incubation. The medium was discarded, and the above mixture was added into cells gradually. The cells were then incubated at 37°C for 5 h. The transfection medium was removed, and DMEM containing 10% FBS was added for 24 and 48 h of cultivation at 37°C in a humidified atmosphere of 5% CO2. The samples were collected for double luciferase system detection. The cells were washed with cold PBS twice, and 300-μL passive lysis buffer was added into the culture plate. Samples were gently shaken for 15 min at room temperature, and the lysate was transferred to the test plates (100 μL each well) with three repeat wells for each group. A total of 10 μL of LARII reagent (freeze-dried powder luciferase assay substrate dissolved in luciferase assay buffer II solution) was added into each well. The firefly luciferase activity was detected with an enzyme-labeled meter. The test plate was taken out, 10 μL of Stop & Glo reagent was added to each well, and the Renilla activity was detected with an enzyme-labeled meter. The relative expression level was calculated according to the firefly/Renilla ratio.
Statistical analysis
All experiments were repeated in triplicate, and the data were expressed as the mean ± standard error of the mean (SEM). Statistical differences between groups were evaluated by one-way ANOVA followed by Tukey’s post hoc tests using SPSS version 17.0 software. The statistical significance was set at P < 0.05.
Results
miR-338-3p is remarkably downregulated in differentiated OCs
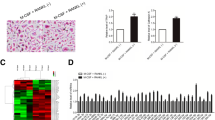
miRNA expression profiles in the OC precursors (RAW264.7 cells) and differentiated OCs were compared by microRNA microarray assay. Results indicated that the expression levels of mmu-miR-199b-5p, mmu-miR-10a-5p, mmu-miR-10b-5p, mmu-miR-24-1-5p, mmu-miR-223-3p, and mmu-miR-338-3p were all downregulated in differentiated OCs. Meanwhile, the expression levels of mmu-miR-291a-5p, mmu-miR-125b-1-3p, and mmu-miR-291b-5p were all increased in differentiated OCs (Fig. 1). In particular, the expression levels of mmu-miR-338-3p were decreased (fold change = 25.4129, P = 0.000541). Further, qRT-PCR assay indicated that the expression levels of mmu-miR-338-3p were reduced significantly in differentiated OCs (P < 0.05) (Fig. 2a). These results were in agreement with the results of microRNA microarray assay. Besides, data indicated that relative mmu-miR-338-3p expression levels were increased significantly by miR-338-3p mimic transfection while decreased significantly by miR-338-3p inhibitor transfection (Fig. 2b, c).

microRNA microarray assay. Compared with undifferentiated cells, the expression levels of mmu-miR-338-3p were significantly decreased (P < 0.01) in differentiated OCs (red: high relative expression, green: low relative expression, gray: missing value). The results are expressed as mean ± SEM. **P < 0.01, *P < 0.05 versus undifferentiated cell groups.

Expression levels of mmu-miR-338-3p were detected by qRT-PCR. (a) Compared with undifferentiated cells, the expression levels of mmu-miR-338-3p in differentiated OCs were significantly decreased (P < 0. 05), which was consistent with the results of microRNA microarray assay. (b, c) Relative mmu-miR-338-3p expression levels were determined. The results are expressed as mean ± SEM. **P < 0.01, *P < 0.05 versus undifferentiated cell groups.
miR-338-3p inhibited the differentiation of OCs
The M-CSF + RANKL-induced OCs were TRAP-positive and multinuclear and had large cell volume. Moreover, the cytoplasm was wine red in color (Fig. 3a). TRAP-positive cell counting showed that compared with the M-CSF + RANKL groups (17.60 ± 2.50), TRAP-positive cells were significantly decreased in the miR-338-3p mimic groups (6.80 ± 1.22) (P < 0.01) and significantly increased in the miR-338-3p inhibitor groups (38.53 ± 2.64) (P < 0.01) (Fig. 3b).

miR-338-3p influenced OC differentiation in M-CSF + RANKL-stimulated RAW264.7 cells. (a) RAW264.7 cells were seeded into 96-well plates. They were pre-treated with M-CSF + RANKL. After overnight incubation, miR-338-3p mimics and inhibitors were transfected into the M-CSF + RANKL inducing OC precursors. After 6 h of transfection, the medium was changed into M-CSF + RANKL-induced medium. After another 96 h of incubation, TRAP staining was conducted. (b) TRAP-positive multinucleated cells (TRAP+) were counted and compared. The results are expressed as mean ± SEM. **P < 0.01, *P < 0.05 versus M-CSF + RANKL groups. Original magnification × 100.
miR-338-3p inhibited the absorption activity of OC
OCs showed active absorption activity in the M-CSF + RANKL groups and could absorb the bottom of COAS and form obvious resorption lacunae (Fig. 4a). Compared with the M-CSF + RANKL groups (5177.29 ± 6283.96 μm2), the area of absorption pits in the miR-338-3p mimic groups (4175.27 ± 2187.32 μm2) was excessively decreased (P < 0.01), while that in the miR-338-3p inhibitor groups (152,157.65 ± 22,276.90 μm2) was significantly increased (P < 0.01) (Fig. 4b).

miR-338-3p influenced OC absorption activity in M-CSF + RANKL-stimulated RAW264.7 cells. (a) RAW264.7 cells were seeded into COAS and pre-treated with M-CSF + RANKL overnight. Then, miR-338-3p mimics and inhibitors were transfected into the M-CSF + RANKL inducing OC precursors. After 6 h of transfection, the medium was changed into M-CSF + RANKL-induced medium. After another 96 h of incubation, the adherent cells were removed, and the resorption lacunae on the COAS bottom were observed. (b) Pit formation area was calculated and compared. The results are expressed as mean ± SEM. **P < 0.01, *P < 0.05 versus M-CSF + RANKL groups. Original magnification × 100.
miR-338-3p influenced the change levels of proteins and transcription factors related to OC differentiation
Both c-Fos and Jun belong to the AP-1 protein family; these proteins are critical for OC differentiation. Activated c-Fos further activates the downstream transcription factor NFATc1 and eventually induces the differentiation of OC (Ono and Nakashima 2018). The expression levels of NFATc1, p-c-fos, and c-fos were enhanced in the M-CSF + RANKL groups and miR-338-3p inhibitor groups, while the expression levels of NFATc1, p-c-jun, p-c-fos, and c-fos were decreased in the miR-338-3p mimic groups (Fig. 5).

Effects of miR-338-3p on the change levels of proteins and transcription factors related to OC differentiation. RAW264.7 cells were seeded into six-well plates and pre-treated with M-CSF + RANKL overnight. Then, miR-338-3p mimics and inhibitors were transfected into the M-CSF + RANKL inducing OC precursors. After 6 h of transfection, the medium was changed into M-CSF + RANKL-induced medium and incubated for 96 h. After cultivation, total proteins were extracted and an equal amount of proteins was assayed by Western blot.
miR-338-3p directly targeted IKKβ
The RNA-induced silencing complex binds to the 3′ untranslated region (UTR) of the target mRNA and degrades the target mRNA or inhibits its translation (Bartel 2004). miR-338-3p target genes were predicted by miRWalk 2.0 software. One miR-338-3p target gene was located in the coding sequence (CDS) of IKKβ. To investigate whether miR-338-3p directly targets IKKβ, a luciferase reporter construct containing CDS of IKKβ was generated, and two mutations were introduced into the predicted miRNA-binding site (Fig. 6a, b). 293T cells were transfected into miR-338-3p mimics, and luciferase activity was detected. The results showed that the luciferase activity in the IKKβ-3′-UTR WT-miR-338-3p transfection groups was significantly lower than that in the empty vector transfection groups (P < 0. 05); moreover, there was no decrease in luciferase activity in the IKKβ-3′-UTR MUT-miR-338-3p transfection groups (Fig. 6c).

MiR-338-3p directly targeted IKKβ. (a) miR-338-3p targets in the CDS of IKKβ. (b) IKKβ WT (i.e., IKKβ wild-type sequence was constructed into pmir GLO vector) and IKKβ MUT vectors (i.e., IKKβ mutant sequence was constructed into pmir GLO vector) were constructed. The groups were as follows: mimic-negative control + IKKβ WT, mimic-negative control + IKKβ MUT, mmu-miR-338-3p + IKKβ WT, mmu-miR-338-3p + IKKβ MUT, mimic-negative control + mmu-miR-338-3p-positive control, and mmu-miR-338-3p + mmu-miR-338-3p-positive control with three multiple wells for each group. The results are expressed as mean ± SEM. **P < 0.01, *P < 0.05 versus mimic-negative control groups. (d) RAW264.7 cells were seeded into six-well plates and pre-treated with M-CSF + RANKL overnight. Then, miR-338-3p mimics and inhibitors were transfected into the M-CSF + RANKL inducing OC precursors. After 6 h of transfection, the medium was changed into M-CSF + RANKL-induced medium and incubated for 96 h. After cultivation, total proteins were extracted and an equal amount of proteins was assayed by Western blot.
Western blot showed that compared with the M-CSF + RANKL-induced groups, the expression levels of IKKβ proteins were decreased in the miR-338-3p mimic transfection groups. This finding proved that IKKβ is one of the target genes of miR-338-3p, and miR-338-3p inhibits the expression of IKKβ protein (Fig. 6d).
Discussion
In this study, microRNA microarray assay and qRT-PCR showed that miR-338-3p was significantly downregulated in differentiated OCs. The results indicated that miR-338-3p could inhibit the formation and absorption activity of OCs in vitro; this result is consistent with the previous findings (Zhang et al. 2016). Western blot suggested that miR-338-3p could influence the change levels of OC differentiation–related proteins and transcription factors. Dual-luciferase reporter gene detection assay and Western blot assay showed that miR-338-3p directly targeted IKKβ.
In some cases, the rate of bone reconstruction is higher than that of the physiological state. Given that the period of bone formation is longer than the period of bone resorption, the rate of new bone formation is slower than the rate of absorbing old bone, resulting in more bone loss; therefore, the total bone mass is reduced, which may cause the occurrence of osteoporosis. Inhibition of bone resorption is one of the important strategies for the prevention and treatment of osteoporosis (Xia et al. 2011). At the same time, the bone resorption rate depends on the number and activity of OCs (Horne et al. 2008). The number and activity of OCs are affected by many factors; thus, it is very important to determine the mechanism of these factors.
miRNAs play pivotal roles in bone formation and bone resorption, both of which are related to osteoporosis. Previous studies demonstrated that miRNAs can regulate the differentiation and formation of osteoblasts (Xia et al. 2011). The regulation of miRNAs on OCs and the mechanisms of osteoporosis caused by abnormal OC activity need to be elucidated.
RANKL is a member of the tumor necrosis factor (TNF) family. It is secreted by osteoblasts and can be combined with receptor activator of NF-κB (RANK) on the surface of OCs. The differentiation and activation of OCs are regulated via the RANKL/RANK signaling pathways. In OC precursors, RANKL/RANK can activate downstream key transcription factors related to OC differentiation and activation, such as c-Fos and NFATc1, and induce the expression of OC differentiation–related genes (Shigeru et al. 2012). NFATc1 is a master transcription factor in OC differentiation. NFATc1, AP-1 (Fos/Jun), PU.1, and microphthalmia-associated transcription factor construct specific transcriptional complexes. The transcriptional complexes synergistically act on promoter regions of OC-specific genes, which induce the expression of OC attachment, migration, acidification, and degradation of bone matrix–related genes, such as TRAP, calcitonin receptor, cathepsin K, and integrin-β3, and OC-associated receptors (Zhao et al. 2010).
Several miRNAs, such as miR-21, miR-29a, miR-29b, miR-29, miR-223, miR-378, miR-146a, miR-503, miR-133a, miR-31, miR-125a, miR-148a, miR-124, miR-422a, and miR-155, can regulate the target genes or corresponding signal transduction pathways in the differentiation and activation of OCs, which further influences the formation or function of OCs (Chen et al. 2014; Tang et al. 2014; Dole and Delany 2016). However, regulatory mechanisms of most miRNAs on OC differentiation and activation still need to be determined. miR-338-3p is a newly discovered miRNA; it is mainly involved in the process of cell differentiation. Previous studies have identified the regulatory effects of miR-338-3p on the nervous system, tumor invasion and metastasis, and osteoblast differentiation. For example, miR-338-3p can influence the differentiation of mouse bone marrow stromal cells into osteoblasts by targeting Runx2 and Fgfr2 genes (Liu et al. 2014). Moreover, study demonstrated that miRNA-338-3p played a negative regulatory role in glucocorticoid-induced osteoclast differentiation (Zhang et al. 2016). Nevertheless, the detailed role of miR-338-3p in the regulation of OC formation and function is far still unclear.
Nfkb1 and Nfkb2 knockout mice were found to be unable to form OCs due to the loss of NF-κB activity, which resulted in osteopetrosis occurrence; therefore, the roles of the NF-κB signaling pathways in bone biology have gradually been recognized (Iotsova et al. 1997). The IKK complex is important to the activation of the NF-κB signaling pathways. The IKK complex consists of two catalytic subunits, namely, IKKα and IKKβ, and one regulatory subunit, namely, IKKγ/NF-κB essential modulator (NEMO) (Yong et al. 2015). There are two NF-κB signaling pathways in cells: the classical NF-κB pathway and the alternative NF-κB pathway. The classical NF-κB pathway is usually activated by the IKK complexes, which are stimulated by TNF, IL-1, and RANKL. In this pathway, the main components of the IKK complex are IKKβ and NEMO/IKKγ, which target and degrade IκBα. In the alternative NF-κB pathway, IKKα is activated by NF-κB, which induces NIK, while IKKα controls the translocation into the nucleus of IκB, p100, p52, and subsequent RelB/p52 complexes (Abu-Amer 2013). Although the IKKα and IKKβ kinases possess homologous sequences, their role in OC formation is very different. OCs lacking IKKα activity exhibit dysdifferentiation in vitro, but the lack of IKKα activity does not affect the skeletal phenotype of experimental animal models. However, OC precursors lacking IKKβ activity show dysdifferentiation both in vitro and in vivo (Otero et al. 2010). Thus, compared with IKKα, IKKβ plays a more important role in the differentiation of OC.
Both dual-luciferase reporter gene detection assay and Western blot assay suggested that miR-338-3p directly targeted the IKKβ gene. The results indicated that miR-338-3p may affect the formation and activity of OCs by targeting IKKβ gene.
References
Abu-Amer Y (2013) NF-kappaB signaling and bone resorption. Osteoporos Int 24:2377–2386
Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116:281–297
Chen C, Cheng P, Xie H, Zhou HD, Wu XP, Liao EY, Luo XH (2014) MiR-503 regulates osteoclastogenesis via targeting RANK. J Bone Miner Res 29:338–347
Detsch R, Boccaccini AR (2015) The role of osteoclasts in bone tissue engineering. J Tissue Eng Regen Med 9:1133–1149
Dole NS, Delany AM (2016) MicroRNA variants as genetic determinants of bone mass. Bone 84:57–68
Horne WC, Duong L, Sanjay A, Baron R (2008) Regulating bone resorption: targeting Integrins, calcitonin receptor, and cathepsin K, Principles of Bone Biology, 3rd edn. Academic Press, pp 221–236
Iotsova V, Caamano J, Loy J, Yang Y, Lewin A, Bravo R (1997) Osteopetrosis in mice lacking NF-κB1 and NF-κB2. Nat Med 3:1285–1289
Khandaker M, Riahinezhad S, Sultana F, Vaughan MB, Knight J, Morris TL (2016) Peen treatment on a titanium implant: effect of roughness, osteoblast cell functions, and bonding with bone cement. Int J Nanomed 11:585–595
Kim JH, Kim N (2016) Signaling pathways in osteoclast differentiation. Chonnam Med J 52:12–17
Liu H, Sun Q, Wan C, Li L, Zhang L, Chen Z (2014) MicroRNA-338-3p regulates osteogenic differentiation of mouse bone marrow stromal stem cells by targeting Runx2 and Fgfr2. J Cell Physiol 229:1494–1502
Ono T, Nakashima T (2018) Recent advances in osteoclast biology. Histochem Cell Biol 149:325–341
Otero JE, Dai S, Alhawagri MA, Darwech I, Abu-Amer Y (2010) IKKbeta activation is sufficient for RANK-independent osteoclast differentiation and osteolysis. J Bone Miner Res 25:1282–1294
Shigeru K, Toru Y, Manabu K, Yuki N (2012) Human receptor activator of NF-κB ligand (RANKL) induces osteoclastogenesis of primates in vitro. In Vitro Cell Dev Biol Anim 48:593–598
Soo-Jin K, So-Young K, Hyun-Hee S, Hye-Seon C (2005) Sulforaphane inhibits osteoclastogenesis by inhibiting nuclear factor-κ B. Mol Cells 20:364–370
Tang P, Xiong Q, Ge W, Zhang L (2014) The role of microRNAs in osteoclasts and osteoporosis. RNA Biol 11:1355–1363
Xia Z, Chen C, Chen P, Xie H, Luo X (2011) MicroRNAs and their roles in osteoclast differentiation. Front Med 5:414–419
Yong X, Lihai Z, Yanpan G, Wei G, Peifu T (2015) The Multiple Roles of Microrna-223 in Regulating Bone Metabolism. Molecules 20:19433–19448
Zhang XH, Geng GL, Su B, Liang CP, Wang F, Bao JC (2016) MicroRNA-338-3p inhibits glucocorticoid-induced osteoclast formation through RANKL targeting. Genet Mol Res 15:1–9
Zhao QX, Wang X, Liu Y, He A, Jia R (2010) NFATc1: functions in osteoclasts. Int J Biochem Cell Biol 42:576–579
Funding
This study was supported by the Project of Natural Science Foundation of Anhui Province (1508085QH172), the Natural Science Research Project of Universities in Anhui (KJ2017A225, KJ2018A1011, and KJ2017A237), the Natural Science Foundation of Bengbu Medical College (BYKY1614ZD), and the National Training Programs of Innovation and Entrepreneurship for Undergraduate (201410367029, 201610367004, 201810367019).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
Editor: Tetsuji Okamoto
Rights and permissions
About this article

Cite this article
Niu, D., Gong, Z., Sun, X. et al. miR-338-3p regulates osteoclastogenesis via targeting IKKβ gene. In Vitro Cell.Dev.Biol.-Animal 55, 243–251 (2019). https://doi.org/10.1007/s11626-019-00325-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11626-019-00325-8