
Abstract
The bone is an essential organ for locomotion and protection of the body, as well as hematopoiesis and mineral homeostasis. In order to exert these functions throughout life, bone tissue undergoes a repeating cycle of osteoclastic bone resorption and osteoblastic bone formation. The osteoclast is a large, multinucleated cell that is differentiated from monocyte/macrophage lineage cells by macrophage colony-stimulating factor (M-CSF) and receptor activator of nuclear factor-κB ligand (RANKL). RANKL transduces its signal through the signaling receptor, RANK. RANKL/RANK signaling activates NFATc1, the master regulator of osteoclastogenesis, to induce osteoclastogenic gene expression. Many types of cells express RANKL to support osteoclastogenesis depending on the biological context and the dysregulation of RANKL signaling leads to bone diseases such as osteoporosis and osteopetrosis. This review outlines the findings on osteoclast and RANKL/RANK signaling that have accumulated to date.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Bone is a unique organ in vertebrates acquired during the course of evolution. Its development is considered to be related to the migration of animals out of the sea onto land. Terrestrial animals need to withstand the more powerful effects of gravity on the ground. Moreover, they need to store minerals, which aquatic animals take up from sea water. Bone tissue consisting of hydroxyapatite has evolved in terrestrial animals to meet these demands (Ahlberg et al. 2005, 2008).
Bone tissue undergoes a continuous cycle of osteoclastic bone resorption and osteoblastic bone formation in order to be able to function properly over a lifetime. The balance between bone resorption and formation is influenced by mechanical loading on the musculoskeletal system, as well as interaction with other biological systems such as the endocrine, nervous and immune systems (Okamoto et al. 2017). With the integrative activity of this complex regulatory machinery, the shape, volume and density of the bone are maintained. There is vigorous bone formation in the growth period to increase the body size (Bassett and Williams 2016). In response to mechanical demands, bone remodeling tips toward formation, whereas unloading leads to decreased bone mineral density (Turner et al. 2009). In case of bone fracture healing, osteoblasts synthesize bone matrix for the fixation of the bone fragments, whereas osteoclasts resorb small bone fragments in the period just after fracture and immature bone matrix in the late phase of the healing (Einhorn and Gerstenfeld 2015; Ono and Takayanagi 2017).
The osteoclast is a large, multinucleated cell with an ability to degrade bone tissue by secreting H+, Cl−, cathepsin K (CtsK) and matrix metalloproteinases (MMPs) in the resorption area. These cells differentiate from monocyte/macrophage lineage cells. M-CSF stimulation of myeloid cells expressing the corresponding receptor, c-Fms, supports the survival of these progenitors and induces the expression of RANK, the essential receptor for osteoclastogenesis (Edwards and Mundy 2011). Stimulation of this receptor by RANKL drives the downstream signal, leading to osteoclastogenesis. After the discovery of RANKL and NFATc1, numerous studies on the pathway of RANKL/RANK signaling have been carried out and revealed the complex mechanisms underlying osteoclastogenesis (Edwards and Mundy 2011; Okamoto et al. 2017).
In this review, we outline the accumulated findings on the mechanism underlying the differentiation and function of osteoclasts, with a particular focus on RANKL/RANK signaling.
Osteoclast differentiation from progenitor cells
All hematopoietic lineage cells, including osteoclasts, originate from hematopoietic stem cells (HSCs) in the bone marrow. HSCs undergo self-renewal and differentiation into each hematopoietic cell type. HSCs do not directly become mature cells; instead, they give rise to several types of oligopotent progenitor cells that further develop into lineage-restricted progenitor cells, forming a hierarchical differentiation tree (Seita and Weissman 2010) (summarized in Fig. 1). HSCs lose their self-renewal capacity and become multipotent progenitors (MPPs), which still retain pluripotency. MPPs differentiate into oligopotent progenitors, common myeloid progenitors (CMPs), megakaryocyte–erythrocyte progenitors (MEPs) and common lymphoid progenitors (CLPs). Each stage of hematopoietic cell differentiation has recently been defined by the expression patterns of cell surface markers: HSCs, Lin−Sca-1+c-Kit+CD34−; MPPs, Lin−Sca-1+c-Kit+CD34+; CMPs, Lin−Sca-1lo/−c-Kit+CD34+IL-7R−FcγRlo; MEPs, Lin−Sca-1−c-Kit+CD34−IL-7R−FcγR−; and CLPs, Lin−Sca-1loc-KitloIL-7R+CD27+Flk2+(Seita and Weissman 2010) (Fig. 1).

Osteoclast progenitor cells. Lin−Sca-1+c-Kit+CD34−hematopoietic stem cells (HSCs) have a capacity for self-renewal and pluripotent ability to differentiate into all hematopoietic cell types. HSCs lose the self-renewal capacity upon differentiation and give rise to oligopotent progenitor cells. Among these progenitors, common myeloid progenitor cells (CMPs) are the origin of osteoclasts. During osteoclastogenesis, osteoclast progenitors express c-Fms and RANK, receptors for M-CSF and RANKL, respectively. Stimulation by these cytokines generates osteoclasts
Osteoclast progenitor cells originate from CMPs. It was reported in an early study that the c-Kit+CD11blo/− population in the bone marrow yields osteoclasts (Arai et al. 1999). The study also showed that c-Kit+CD11blo/−c-Fms+ cells differentiate into macrophage-lineage cells more frequently than the c-Fms− counterpart that has the capacity to produce granulocytes and erythrocytes as well, indicating that c-Fms expression specifies the fate of progenitor cells towards monocyte/macrophage-lineage cells.
c-Kit+CD11blo/−c-Fms+ cells can be divided according to the expression of CD27 and Flt3. The CD27+Flt3+ subset was shown to be a common progenitor of macrophages, dendritic cells (DCs) and osteoclasts, whereas CD27−Flt3− progenitors differentiate into macrophages and osteoclasts, but not DCs (Xiao et al. 2017, 2013). Stimulation of c-Kit+CD11blo/−c-Fms+ cells by M-CSF induces RNAK on these cells, and RANK+ cells lose c-Kit expression (Arai et al. 1999). Together, these findings indicate that osteoclast progenitors undergo a stepwise differentiation and finally become c-Fms+RANK+ progenitors, which are ready for RANKL stimulation.
Some studies have reported the transdifferentiation of immature DCs into osteoclasts. These studies have also pointed out that the transdifferentiation was promoted under inflammatory conditions: T cell stimulation by bacterial componetns (Alnaeeli et al. 2006), IL-1β (Speziani et al. 2007), or tumor necrosis factor (TNF)-α (Speziani et al. 2007). These results suggest that immature DCs may play a role as osteoclast progenitors in inflammatory bone loss, but the physiological relevance of DC-derived osteoclasts at present is not fully understood.
RANKL: the cytokine essential for osteoclastogenesis
Discovery of RANKL
Although osteoclast progenitors, monocytes and macrophages, are found in most body tissues, osteoclasts are found only in bone. Therefore, it was speculated that cells specifically existing in the bone tissue would support osteoclastogenesis (Rodan and Martin 1981). Co-culture experiments of spleen cells and osteoblastic cells were conducted and successfully yielded osteoclasts, suggesting that osteoblasts express an osteoclast differentiation factor (ODF) on their cellular membrane (Suda et al. 1999).
As a result of the quest to identify the binding partners for a factor inhibiting osteoclastogenesis (osteoprotegerin: OPG or osteoclastogenesis inhibitory factor: OCIF), ODF and OPG ligand (OPGL) were found (Lacey et al. 1998; Simonet et al. 1997; Tsuda et al. 1997; Yasuda et al. 1998). ODF and OPGL subsequently turned out to be identical to the molecules reported as RANKL and TRANCE (Anderson et al. 1997; Fuller et al. 1998). The essential signaling receptor for this cytokine was reported as ODF receptor (ODFR), which was found to be identical to RANK (Nakagawa et al. 1998). OPG/OCIF appeared to serve as a decoy receptor for RANKL. Today, these cytokine, signaling receptor and decoy receptor are called RANKL (encoded by Tnfsf11), RANK (encoded by Tnfrsf11a) and OPG (encoded by Tnfrsf11b), respectively.
The structures of RANKL/RANK/OPG
RANKL is a type II transmembrane protein of the TNF superfamily, with its N terminus constituting the intracellular region and C terminus containing the receptor-binding domain (Nelson et al. 2012). Like most TNF-family cytokines, RANKL forms a homotrimer by hydrogen bond and hydrophobic interactions, the former of which is thought to be requisite for the binding selectivity and the latter for the association of the monomers (Lam et al. 2001). An inter-subunit cleft that forms between the subunits functions as the receptor-binding site (Fig. 2) (Liu et al. 2010), the occupation of which using a TNF receptor loop peptide mimic leads to the inhibition of osteoclastogenesis and bone resorption (Aoki et al. 2006; Ozaki et al. 2017).

Molecular structures of RANKL/RANK/OPG. Molecular structure images of the RANKL trimer (left), RANKL–RANK complex (center) and RANKL–OPG complex (right). The pictures in the top row are top view images and ones in the bottom row are side view images. Polymerization of the RANKL monomers forms a cleft between the monomers (arrows). The cleft and adjacent loops (orange lines) interact with the cytokine-binding domains of RANK or OPG (gray lines). RANKL and RANK bind with a stoichiometry of 3:3, whereas RANKL and OPG bind with a stoichiometry of 3:2. The structure files for the RANKL trimer, RANKL–RANK complex and RANKL–OPG complex were obtained from the RCSB PDB web-site repository (http://www.rcsb.org/pdb/home/home.do) (Lam et al. 2001; Liu et al. 2010; Nelson et al. 2012). Molecular graphics were composed using the UCSF Chimera package (Pettersen et al. 2004)
The RANKL expressed on the cell surface can be cleaved by MMPs, generating a soluble form (Hikita et al. 2006; Lum et al. 1999). The soluble form was reported to retain cytokine activity, which is enhanced by polymerization (Lum et al. 1999; Nakashima et al. 2000). Inhibition of RNAKL shedding by the knockdown of Mmp14 in an osteoblast–osteoclast co-culture system resulted in increased osteoclastogenesis, indicating that membrane-bound RANKL is more effective than its soluble form (Hikita et al. 2006). To elucidate the physiological relevance of the membrane-bound and soluble forms of RANKL, a genetically modified mouse strain was recently generated in which the RANKL cleavage site is trimmed and the soluble form cannot be produced (the Tnfsf11ΔS/ΔS mouse) (Nagashima et al. 2017). Using these mice, the soluble form of RANKL was shown to be dispensable for the regulation of micro-fold cells (M cells), cells that transport antigens in the intestine to immune cells (Nagashima et al. 2017). The Tnfsf11ΔS/ΔS mouse and its counterpart, a mouse unable to produce membrane-bound RANKL (if generated), would be promising tools for the elucidation of the roles of the two types of RANKL in osteoclast differentiation, bone development and bone disease.
The signaling receptor of RNAKL, RANK, is a type I transmembrane protein and belongs to the TNF receptor (TNFR) superfamily of molecules, all of which consist of four cysteine-rich domains (CRDs). Of these, the middle two domains can directly bind to the corresponding ligands. As is for most of the other TNFR molecules, RANK rotates around the hinge region between CRD2 and CRD3, which contributes to the close contact to RANKL (Liu et al. 2010). Upon binding of RANKL, RANK trimerizes and transduces the signal via adaptor molecules. The RANKL signaling pathway is discussed in the latter part of this manuscript.
The decoy receptor for RANKL, OPG, belongs to the TNFR superfamily as well. It contains four CRDs and bears a hinge region between CRD2 and CRD3 in order to bind to RANKL. Because of conformational difference from RANK in the hinge region, OPG rotates more than RANK and binds to RANKL in a different manner (Fig. 2). Such differences are considered to result in the higher affinity of OPG to RANKL than RANK.
The RANKL/RANK/OPG system in bone
Studies on genetically modified mice have demonstrated the essentiality of RANKL/RANK signaling in bone metabolism. Tnfsf11−/− mice, Tnfrsf11a−/− mice and mice overexpressing the rat Tnfrsf11b gene exhibit severe osteopetrosis (Dougall et al. 1999; Kong et al. 1999; Li et al. 2000; Min et al. 2000). In contrast, Tnfrsf11b−/− mice develop osteoporosis (Bucay et al. 1998; Mizuno et al. 1998). The bone phenotypes of these mice clearly demonstrate the significance of RANKL signaling in osteoclastogenesis. In human patients with bone diseases, mutations in RANKL, RANK and OPG have all been reported. Familial expansile osteolysis (FEO), expansile skeletal hyperphosphatasia (ESH) and the familial form of early-onset Paget’s disease of bone (PDB2) are rare bone diseases with constitutive activation of RANK owing to in-frame duplication of exon 1. Juvenile Paget’s disease (JPD) is another type of Paget’s disease that arises because of mutations in OPG (Walsh and Choi 2014).
FEO is an autosomal dominant disease characterized by deafness and loss of adult dentition in early life due to excessive bone resorption and resulting deformity. From the late juvenile period, osteolytic lesions appear in the tibia, ulna, humerus and femur, with bone thinning, pain and fracture (Whyte and Mumm 2004; Wright et al. 2009). Constitutive activation of RANK in FEO is caused by the mutations 84dup18 and 83dup18.
ESH is an autosomal dominant disease of accelerated bone remodeling, the patients of which suffer deafness, tooth loss, hypercalcemia and widening of the long bones accompanied by pain (Whyte and Mumm 2004; Wright et al. 2009). ESH is caused by a TNFRSF11A mutation, which is similar to that found in FEO (84dup15). However, unlike FEO, the long bones affected by ESH exhibit hyperostosis.
PDB is characterized by progressive osteoclast-mediated osteolysis and the subsequent excessive bone formation that lead to changes in the overall appearance of the bone (Whyte and Mumm 2004; Winn et al. 2017). Despite the fact that the causes of PDB and FEO both lie in the RANKL/RANK/OPG system, the affected bone is different; the skull, spine, pelvis and femur are the common sites affected in PDB (Winn et al. 2017; Wright et al. 2009). Bone deformity leads to fracture, osteoarthritis, nerve entrapment and pain. It is also reported that Pagetic bone frequently undergoes tumor transformation. PDB2 is caused by a mutation in TNFRSF11A (75dup27), while JPD is caused by mutations in TNFRSF11B (D182del, C65R, C87Y, F117L, V199RfsX5 and D323SfsX3).
Osteopetrosis (Albers–Schönberg disease) is another osteoclast-related bone disease characterized by an increased bone mass owing to dysfunction or loss of osteoclasts. The bone marrow in these patients is occupied by bone tissue, resulting in impaired hematopoiesis, which in turn leads to recurrent infections, anemia and hemorrhage. The affected bones easily undergo fracture. Nerves can be compressed by proliferating bone tissue, leading to blindness and deafness (Coudert et al. 2015). According to the inheritance pattern and the clinical features, osteopetrosis is divided into groups: adult-onset autosomal dominant osteopetrosis (ADO)1; ADO2; intermediate autosomal osteopetrosis (IAO); and autosomal recessive osteopetrosis (ARO). Mutations in the molecules related to osteoclast function are known to result in the differing types of osteopetrosis (LDL receptor-related protein 5: LRP5, ATPase A3 subunit: TCIRG1, chloride voltage-gated channel 7: CLCN7, osteoporosis-associated transmembrane protein 1: OSTM1, pleckstrin homology domain containing, family M member 1: PLEKHM1, sorting nexin 10: SNX10 and carbonic anhydrase II: CA2) (Chen et al. 2016; Cleiren et al. 2001; Coudert et al. 2015; Sly et al. 1983). Several different mutations in TNFRSF11A and TNFSF11 (A145delS177, M199K and V277WfsX5), in which the trimerization of RANKL is hampered, have been reported as the causes of ARO (Wright et al. 2009). These together indicate the clinical significance of the RANKL/RANK/OPG system for bone metabolism.
RANKL-independent osteoclastogenesis
The studies introduced above have clearly established the concept that RANKL is an indispensable and exclusive cytokine for osteoclastogenesis. However, a controversy over its exclusivity arose soon after the concept was put forward: factors other than RANKL [TNF-α, A proliferation-inducing ligand (April), B cell activating factor belonging to the tumor necrosis factor family (BAFF), nerve growth factor (NGF), Insulin-like growth factor (IGF-1), IGF-2, lymphotoxin-related inducible ligand that competes for glycoprotein D binding to herpesvirus entry mediator on T cells (LIGHT), transforming growth factor (TGF)-β, IL-6, IL-11, secreted osteoclastogenic factor of activated T cells (SOFAT) and ROS generated by lysyl oxydase (LOX)] were reported to induce osteoclastogenesis (Tanaka 2017). Because these factors are not always reported to induce osteoclastogenesis in the absence of RANKL, the controversy has yet to be brought to a close.
The inconsistency is derived, in part, from experimental difficulties. In most of the culture experiments, cells form murine bone marrow or human peripheral blood were used as the osteoclast progenitors. These cells contain not only osteoclast progenitors but also mesenchymal cells that can express RANKL, thus stimulating osteoclast progenitors. Cell sorting based on antigen–antibody reaction can be an effective means of collecting a “pure” population of osteoclast progenitors. However, the purity of the sorted cells is largely dependent on the affinity and the specificity of the antibodies employed. It is reported that anti-human CD14 antibodies, which are supposed to enrich myeloid cells, bind to mesenchymal cells owing to cross-reactivity (Pilz et al. 2011). The use of such antibodies for collecting osteoclast progenitors results in the contamination of mesenchymal cells that express RANKL, resulting in an ambiguity over whether the factors above induce osteoclastogenesis independently on RANKL (Cox et al. 2015; Tsukasaki et al. 2017).
Thus, in the studies on RANKL-independency, the use of RANK-deficient progenitor cells and the purification of progenitor cells with a truly reliable strategy are absolutely required. Reports have shown that Tnfrsf11a−/− osteoclast progenitor cells are able to differentiate into osteoclasts via TNF-α stimulation under conditions in which progenitors are deficient in the transcription factor RBP-J or co-stimulated by TGF-β (Kim et al. 2005; Zhao et al. 2012). It has been also shown that a local injection of TNF-α induced osteoclastogenesis in the calvariae of Tnfrsf11a−/− mice, but the resulting osteoclasts were not functional (Li et al. 2000; Zhao et al. 2012). These together suggest that TNF-α as well as other cytokines may be able to induce osteoclastogenesis under certain conditions, but cannot completely substitute for RANKL.
Osteoclastogenesis-supporting cells: sources of RANKL in the skeletal system
As described above, osteoblasts were found to support osteoclastogenesis, which has become a dogma ever since (Takahashi et al. 1988; Udagawa et al. 1989). However, some reports have suggested that osteoblasts do not exert effects on osteoclasts as supposed because there is no decrease in the osteoclast number in mouse models in which the osteoblast number or function have declined (Corral et al. 1998; Ogata et al. 2007; Weinstein et al. 2002). These reports have led to a rethinking of the dogma.
RANKL is expressed by several types of cells in bone (osteoblasts, osteocytes, immune cells, etc.), but the expression is higher in bone tissue (osteoblasts and osteocytes) than other cells (Nakashima et al. 2011). Purified osteocytes were shown to more vigorously express RANKL and support osteoclastogenesis than osteoblasts, suggesting that osteocytes have a crucial role in bone remodeling (Nakashima et al. 2011). To investigate this, several lines of genetically modified mice, in which Tnfsf11 was deleted in specific cell types in the bone, were examined (Nakashima et al. 2011; Xiong et al. 2011, 2015). Analyses of neonatal or young mice in their growth period revealed that hypertrophic chondrocytes in the growth plate and osteoblasts, which are targeted by Prrx1-Cre, Sp7-Cre, BGLAP-Cre, Dmp1-Cre or Col10a1-Cre, but not osteocytes, serve as the source of RANKL in bone metabolism (Nakashima et al. 2011; Xiong et al. 2011). In older mice, on the other hand, osteoblasts do not contribute to RANKL expression; instead, osteocytes do. The long bones and spine become osteopetrotic with impaired osteoclastic bone resorption in Tnfsf11f/fDmp1-Cre mice, Tnfsf11f/ΔDmp1-Cre mice and Tnfsf11f/fSost-Cre mice, but not in Tnfsf11f/fSp7-Cre mice (Nakashima et al. 2011; Xiong et al. 2011, 2015). Thus, it is indicated that osteocytes contribute to osteoclastogenesis in the adult, not in the developmental phase of the life course.
Osteocytes regulate bone remodeling more than only in the steady state. They have also been shown to be responsible for unloading-induced bone resorption and alveolar bone remodeling in orthodontic tooth movement (Shoji-Matsunaga et al. 2017; Xiong et al. 2011), indicating that osteocytes are involved in mechanical stress-induced bone remodeling. This seems reasonable because osteocytes are recognized to be mechanosensitive cells in bone. The osteocytes embedded in the bone lacunae are connected to other lacunae by bone canaliculi, within which osteocytes extend their dendrites. The bone lacunae and canaliculi are filled with tissue fluid and the osteocytes are anchored in the spaces by adhesive molecules. Upon mechanical loading, there occurs not only a structural deformation of bone matrix, but also a streaming of tissue fluid in the lacuno-canalicular system (Bonewald 2014). These stimuli are thought to be sensed by several different cellular components, such as the primary cilia, integrins, cytoskeletal proteins and ion channels, as a result inducing mechano-responsive gene activation (Nguyen and Jacobs 2013; Takano-Yamamoto 2014). However, the precise mechanism underlying the osteocyte response is largely unknown. Further studies are required to properly understand mechanical stress-induced bone remodeling.
RANKL signaling in osteoclast differentiation
The indispensability of RANKL in osteoclastogenesis has prompted scientists and clinicians to study the RANKL signaling pathway in terms of elucidating the whole picture of osteoclastogenesis and establishing the molecular basis for therapeutic strategies in bone disease. The RANKL signaling pathway is summarized in Fig. 3.

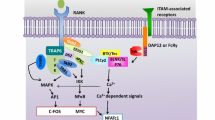
Signal cascade in the process of osteoclastogenesis. RANKL and its receptor RANK transduce a signal via the adaptor molecule TRAF6. TRAF6 recruits TAB2 and TAK1, which in turn activates the NF-κB pathway and MAPK pathway. NF-κB induces c-Fos expression via IKKs. Activation of MAPK pathway results in the activation of the Jun proteins. The c-Fos and Jun proteins associate to form the complex AP-1. The expression of the master regulator of osteoclastogenesis, NFATc1, is driven by AP-1, NF-κB, ATF4, Jdp2, NFATc2 and NFATc1 itself. NFATc1 expression is also enhanced by the demethylation of the NFATc1 promoter by Jmjd3. The activation of NFATc1 is regulated by a co-stimulatory signal pathway. FcRγ, DAP12 and their associating molecules activate Syk, which forms a complex with Btk/Tec and BLNK/SLP76. This complex further activates PLCγ, resulting in calcium signaling. The influx and efflux of Ca2+ is regulated by Ca2+ channels, Tmem64 and Tmem178. The calcium signaling activates calcineurin, which dephosphorylates NFATc1, promoting its entry into the nucleus. Activated NFATc1 promotes its own expression making a autoamplification loop. The calcium signaling also induces c-Fos expression via CAMKIV and CREB. NFATc1, together with other factors including PU.1 and MITF, promotes the expression of osteoclastogenic genes. The expression of Nfatc1 is negatively regulated by LRF, Bcl6, MafB and IRF-8, some of which are inhibited by Blimp1 and Dnmt3a
Adaptor molecules for RANK
RANK adopts a trimetric conformation upon binding to RANKL. The intracellular domain of the RANK trimer is thought to lack signaling domains, but instead, for signal transduction, recruits adaptor molecules: TNF receptor-associated factors (TRAFs), Grb-2-associated binder (Gab) 2 and four and a half LIM domain (FHL) 2 (Bai et al. 2005; Lamothe et al. 2007; Naito et al. 1999; Wada et al. 2005). Among TRAF1/2/3/5/6, which bind to RANK, TRAF6 is essential for osteoclastogenesis, as shown by the analyses of two lines of Traf6−/− mice, which exhibit severe osteopetrosis (Lomaga et al. 1999; Naito et al. 1999). The distinctive role of TRAF6 may be due to the lowest C-terminal homology among the TRAF family molecules (Lomaga et al. 1999). TRAF6 contains an amino-terminal ring finger domain, which has ubiquitin-ligase activity for ubiquitinating TRAF6 itself (Lamothe et al. 2007). This ubiquitination does not serve as a signal for protein degradation, but as a scaffold for TGF-β-activated kinase (TAK) 1-binding protein (TAB) 2 and TAK1(Mizukami et al. 2002). A defect in the kinase activity of TAK1 in osteoclasts has been shown to lead to an impairment in osteoclastic bone resorption (Lamothe et al. 2013; Sumiya et al. 2015). In osteoclastogenesis, TAK1 activates inhibitory κB (IκB) kinases (IKKs) and mitogen-activated protein kinases (MAPKs) for further signaling.
NF-κB signaling in osteoclastogenesis
NF-κB is one of the key transcription factors. It is composed of dimeric transcription molecules: Rel proteins (RelA, RelB and c-Rel), NF-κB1 (p50 and its precursor p105) and NF-κB2 (p52 and its precursor p100). This transcription factor is inactive in the cytosol in the steady state and translocates to the nucleus upon activation, which is dependent on the classical or the alternative pathways (Ghosh and Karin 2002). In the classical pathway, the IKK complex that consists of IKKα, IKKβ and IKKγ (also known as NF-κB essential modulator: NEMO) phosphorylates IκBs, the inactivators of NF-κB. The phosphorylation of the IκB molecules leads to their ubiquitination and subsequent degradation, freeing NF-κB. In the alternative pathway, the homodimer of IKKα cleaves p100 to generate p52. Activated NF-κB dimers induce c-Fos and nuclear factor of activated T cell c1 (NFATc1) expression (Yamashita et al. 2007). (The functions of c-Fos and NFATc1 are described below.) NF-κB has been shown to be essential for osteoclastogenesis, as Nfkb1−/−Nfkb2−/− mice (mice deficient in both p50 and p52) develop osteopetrosis (Franzoso et al. 1997). Likewise, it is reported that Ikbkbf/fMx1-Cre mice, which are deficient in IKKβ in osteoclast progenitors, display an osteopetrotic phenotype, indicating the significance of IKK–NF-κB signaling in osteoclastogenesis (Ruocco et al. 2005).
MAPK signaling in osteoclastogenesis
TAK1 serves as a MAPK kinase kinase (MAPKKK) to phosphorylate the MAPK kinases (MAPKKs) that further phosphorylate the MAPKs, including p38 and c-Jun N-terminal kinase (JNK). Among the MAPKKs, MKK3 was shown to be involved in p38 phosphorylation during osteoclastogenesis (Boyle et al. 2014). The bones of Map2k3−/− mice, which are deficient in MKK3, are osteopetrotic with a tendency towards reduced osteoclasts (Boyle et al. 2014). p38 consists of four subtypes: p38α, p38β, p38γ and p38δ. p38α is most abundantly expressed in osteoclasts and the knockdown of this signaling molecule results in impaired osteoclastogenesis (Bohm et al. 2009). Analyses of younger aged mice in which p38 is deleted specifically in monocytes and macrophages (Mapk14f/fLysM-Cre mice) revealed increased bone mass along with a decrease in osteoclastic bone resorption (Cong et al. 2017).
JNK1 is a MAPK activated by RANKL. JNK1-deficient bone marrow cells (Mapk8−/− BMMs) fail to differentiate into osteoclasts, suggesting its involvement in osteoclastogenesis (David et al. 2002). In these cells, activation of a Jun protein, c-Jun was impaired. MKK7 is a MAPKK that activates JNK1. JNK1 phosphorylation is inhibited by the overexpression of a dominant negative form of MKK7, suggesting a possible role for MKK7 in osteoclastogenesis (Yamamoto et al. 2002). Another member of the Jun proteins, JunB, also has a role in osteoclastogenesis, as Junbf/fLysM-Cre mice exhibit an osteopetrotic bone phenotype (Kenner et al. 2004). There are several other MAPKs and related kinases the functions of which in osteoclastogenesis remain unclear. Therefore, studies using genetically modified mice are required for the clarification of the functions of these molecules (Okamoto et al. 2017).
NFATc1, the master transcription factor in osteoclastogenesis
NF-κB and MAPK signals are not RANKL-specific pathways but RANKL almost exclusively induce osteoclastogenesis, which indicated the existence of osteoclastogenesis-specific transcription factors. In search for such factors, NFATc1 was discovered (Takayanagi et al. 2002). Deficiency in Nfatc1 results in the complete loss of osteoclastic bone resorption, indicating its essentiality in osteoclastogenesis (Aliprantis et al. 2008; Asagiri et al. 2005; Takayanagi et al. 2002).
Downstream of NF-κB and MAPK signaling, c-Fos and Jun proteins are induced as described above. These proteins dimerize to form activator protein-1 (AP-1). NF-κB, AP-1, NFATc2, ATF4 and Jdp2 are recruited to the promoter region of the Nfatc1 gene after RANKL stimulation, together inducing NFATc1 (Asagiri et al. 2005; Cao et al. 2010; Maruyama et al. 2012; Matsuo et al. 2004). The expression of this transcription factor is greatly upregulated after RANKL stimulation by autoamplification machinery (Asagiri et al. 2005; Takayanagi et al. 2002). NFATc1 expression is epigenetically regulated as well. Histone modification profiling in the Nfatc1 promoter region showed an obvious change during osteoclastogenesis, from bivalent H3K4me3/H3K27me3 to monovalent H3K4me3 (Yasui et al. 2011). A histone demethylase, Jmjd3, has been shown to be upregulated and recruited near the Nfatc1 promoter region after RANKL stimulation and enhance osteoclastogenesis (Yasui et al. 2011).
The induced NFATc1 in turn drives a number of osteoclastogenic genes including Dcstamp (dendritic cell-specific transmembrane protein: DC-STAMP), Atp6v0d2 (v-type proton ATPase subunit d2), Oscar, Itgb3 (integrin β3), Ocstamp (osteoclast stimulatory transmembrane protein: OC-STAMP), Acp5 (tartrate-resistant acid phosphatase: TRAP), Calcr (calcitonin receptor) and Ctsk, together with other transcription factors such as PU.1 and microphthalmia transcription factor (MITF) (Kim et al. 2008; Matsumoto et al. 2004; Matsuo et al. 2004; Miyamoto et al. 2012).
Co-stimulatory signaling and calcium signaling in osteoclastogenesis
In T cells, NFATc1 has been known to localize in the cytosol in the steady state in a hyperphosphorylated, latent form that needs to be dephosphorylated by calcineurin for translocation into the nucleus (Hirotani et al. 2004). Likewise, during osteoclastogenesis, nucleus-directed mobilization of NFATc1 was observed and the activation of NFATc1 was shown to be sufficient to induce osteoclastogenesis (Hirotani et al. 2004; Takayanagi et al. 2002).
Immunoglobulin-like receptors (IgLRs) are a group of proteins composed of an extracellular region, which has an immunoglobulin-like domain and an intracellular region, which associates with immunoreceptor tyrosine-based activation motif (ITAM)- or immunoreceptor tyrosine-based inhibitory motif (ITIM)-harboring adaptor proteins. IgLRs and their adaptors are expressed on osteoclasts as well as immune cells. Fc receptor common γ subunit (FcRγ) and DNAX-activating protein (DAP) 12 are ITAM-harboring adaptors expressed in osteoclasts that have been shown to have an essential role in osteoclastogenesis through NFATc1 induction (Koga et al. 2004). Paired immunoglobulin-like receptor-A (PIR-A), osteoclast-associated receptor (OSCAR) and Fcγ receptors (FcγRs) are reported to associate with FcRγ; whereas triggering receptor expressed in myeloid cells (TREM)-2, signal-regulatory protein (SIRP) β1, sialic acid-binding Ig-like lectin (Siglec)-15 and myeloid DAP12-associating lectin (MDL)-1 associate with DAP12 (Joyce-Shaikh et al. 2010; Kameda et al. 2013; Koga et al. 2004; Negishi-Koga et al. 2015).
The semaphorins have a variety of functions in organogenesis, immune responses and tumorigenesis. They are divided into 8 subfamilies and most of them use plexins as their receptors. It was demonstrated that Semaphorin (Sema) 6D and its receptor plexin-A1 (PlxnA1), which associate with TREM-2–DAP12, promote osteoclastogenesis (Takegahara et al. 2006). Sema3A and its receptor neuropilin-1 (Nrp1) inhibit osteoclastogenesis by competing with Sema6D for Plxn1, thus protecting bone from excessive resorption (Hayashi et al. 2012).
Phosphorylation of the ITAM motif of FcRγ and DAP12 leads to the activation of spleen tyrosine kinase (Syk), which promotes the formation of the Bruton agammaglobulinemia tyrosine kinase (Btk)/Tec–B cell linker protein (BLNK)/SH2 domain-containing leukocyte protein of 76 kDa (SLP-76) complex (Shinohara et al. 2008). This complex activates phospholipase Cγ (PLCγ), which mediates the hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2) in the cellular membrane, mediating inositol 1,4,5-triphosphate (IP3) release (Shinohara et al. 2008). IP3 binds to its receptor IP3R to induce the release of Ca2+ out of the endoplasmic reticulum (ER). Sarco/endoplasmic reticulum Ca2+ ATPase (SERCA) 2 reuptakes the released Ca2+ into ER. Such influx and efflux of Ca2+ leads to a repetitive fluctuation of [Ca2+] in the cytosol, i.e. calcium oscillation (Yang et al. 2009). Transmembrane protein (Tmem) 64 was shown to associate with SERCA2 and to be essential for its function (Kim et al. 2013). Tmem178 is an NFATc1- and PLCγ2-dependent molecule and suppresses the excessive efflux of Ca2+ (Decker et al. 2015). Calcium oscillation activates calcineurin, which dephosphorylates NFATc1, enabling it to enter the nucleus, so as its expression becomes amplified. Calcium signaling is also reported to induce the c-Fos via Ca2+/calmodulin-dependent kinase (CAMK) IV-cAMP response element-binding protein (CREB) pathway (Sato et al. 2006).
Mice deficient in the factors above exhibit impaired calcium oscillation and subsequent abnormal osteoclastic bone resorption: Tyrobp−/− mice (deficient in DAP12) display mild osteopetrosis and Tyrobp−/− cells have impaired calcium oscillation (Kaifu et al. 2003; Koga et al. 2004; Negishi-Koga et al. 2015); Tyrobp−/−Fcer1g−/− mice (deficient in DAP12 and FcRγ) are osteopetrotic and Tyrobp−/−Fcer1g−/− cells have a defect in calcium oscillation (Koga et al. 2004; Sato et al. 2006); Fcgr3−/− mice (deficient in FcγRIII) are osteopenic and the amplitude of calcium oscillation is higher in Fcgr3−/− cells (Negishi-Koga et al. 2015); Tec−/−Btk−/− mice are osteopetrotic and calcium oscillation in Tec−/−Btk−/− cells is attenuated (Shinohara et al. 2008); Atp2a2+/− mice (haploinsufficient in SERCA2) exhibited osteopetrosis and no calcium oscillation in Atp2a2+/− cells (Yang et al. 2009); Tmem64−/− mice are osteopetrotic and Tmem64−/− cells are defective in calcium oscillation (Kim et al. 2013); and Tmem178−/− mice are osteopenic, with a high level and slow efflux of Ca2+ in Tmem178−/−cells (Decker et al. 2015).
Negative regulation of osteoclastogenesis
NFATc1 expression and activity are not only enhanced, but also suppressed by transcription factors so that osteoclastic bone resorption does not become excessive. Interferon regulatory factor (IRF)-8 is one such molecule. In Irf8−/− cells, the expression of Nfatc1 after RANKL stimulation is high and osteoclastic bone resorption is enhanced in Irf8−/− mice, indicating its suppressive effect on osteoclastogenesis (Zhao et al. 2009). The expression of Irf8 was shown to be high in osteoclast progenitor cells and to gradually decrease during osteoclastogenesis, which is mediated by DNA methyltransferase (Dnmt) 3A through methylation of the 3′-flanking region of the gene (Nishikawa et al. 2015; Zhao et al. 2009).
MafB is a bZIP motif-containing transcription factor that has been known to stimulate macrophage differentiation. Mafb expression was reported to be downregulated by RANKL stimulation, which is under the control of p38 or JNK MAPKs. MafB was shown to suppress the expression of Nfatc1 and Oscar by inhibiting c-Fos, MITF and NFATc1, leading to the suppression of osteoclastogenesis (Kim et al. 2007).
B cell lymphoma (Bcl) 6 is a transcription factor with a BTB/POZ domain in the N-terminus and a zinc finger domain in the C-terminus. Like Irf8 and Mafb, the expression of Bcl6 decreases during osteoclastogenesis. Bcl6 was revealed to suppress osteoclastogenesis as Bcl6−/− cells underwent enhanced osteoclastogenesis and Bcl6−/− mice developed an osteoporotic phenotype due to the increased osteoclastic bone resorption. The mechanism underlying this suppression is explained by the repression of the transcription of Nfatc1, Dcstamp and Ctsk (Miyauchi et al. 2010).
B lymphocyte-induced maturation protein (Blimp) 1 is a transcriptional repressor, the expression of which is upregulated by NFATc1 (Nishikawa et al. 2010). Osteoclast-specific Blimp1-deficient mice (Prdm1f/ΔCtskCre/+ mice) were reported to be osteopetrotic because of reduced osteoclastic bone resorption (Miyauchi et al. 2010; Nishikawa et al. 2010). Blimp1 was shown to suppress the expression of Irf8, Mafb and Bcl6 by binding to their promoter regions (Miyauchi et al. 2010; Nishikawa et al. 2010).
Leukemia/lymphoma-related factor (LRF) is a transcriptional repressor belonging to the POZ/BTB and Krüppel (POK) family and is involved in various biological processes. Zbtb7a (LRF) expression was shown to be very low before and highly elevated after RANKL stimulation (Kukita et al. 2011; Tsuji-Takechi et al. 2012). LRF is a unique transcription factor in that it regulates osteoclastogenesis in a stage-specific manner. Overexpression of LRF in osteoclast progenitors in the early phase results in suppression and Zbtb7af/ΔMx1-Cre mice have osteopenia, suggesting an inhibitory function in osteoclastogenesis. On the other hand, Zbtb7af/ΔCtskCre/+ mice were osteopetrotic, indicating a promoting function (Tsuji-Takechi et al. 2012).
The studies discussed here have done much to uncover the mechanisms underlying osteoclastogenesis, in which RANKL/RANK signaling plays a central role. The discovery of new mechanisms and molecules reflects the technological advances that have occurred in biology, such as ones in epigenetics. Although key molecules for osteoclastogenesis have been elucidated, there still remain processes, in which the underlying mechanisms are not well understood. The continuing search for novel factors involved in osteoclastogenesis should lead to the establishment of therapeutic strategy for bone diseases.
Osteoclastic bone resorption
As osteoclastogenesis proceeds, osteoclast progenitors fuse so as to generate a large, multinucleated cell. Such enlargement of a cell enables it to widely seal the bone surface, increasing bone resorption efficiency. Inside the sealed zone, bone tissue is degraded by H+, Cl− and catalytic enzymes. Inorganic components of the bone are resorbed under the acidic environment established by HCl. Organic components are degraded by various enzymes including CtsK and MMP-9. This section reviews how osteoclasts become multinucleated and resorb bone matrix (summarized in Fig. 4).

Osteoclastic bone resorption. The integrins expressed on osteoclasts are responsible for the adherence of these cells to the bone surface. Inside osteoclasts, integrins activate c-Src, leading to the formation of a complex with Syk, DAP12, SLP76 and VAV3. This complex activates the Rho-GTPase Rac. Rac induces actin ring formation and isolates the resorption pit. Osteoclasts secrete H+ and Cl− into the isolated pit via ion channels. H+ is produced by the dehydrogenation of a carbonic acid. Cl− is imported into osteoclasts in exchange for HCO3−. Acidification of the resorption pit leads to the decalcification of the bone, as well as the activation of CtsK. CtsK and MMP-9 degrade the organic components, including collagens, whereas TRAP is likely a phenotypic marker
Cellular fusion in osteoclast formation
The fusion of osteoclast progenitors is thought to be crucial for the optimization of the resorption activity. Cellular fusion is achieved by the remodeling of phospholipids, components of the lipid bilayer of the cellular membrane. The phospholipids are divided into five groups: phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylserine (PS), phosphatidylinositol (PI) and sphingomyelin (SM). PC and SM preferentially reside on the outer leaf of the lipid bilayer, whereas PE, PS and PI reside on the inner leaf (Zachowski 1993). Although it is known that the composition and distribution of the phospholipids can change during the course of cellular fusion, our knowledge on the phospholipid dynamics in the osteoclast fusion is currently inadequate (Irie et al. 2017).
One study recently showed that fusing osteoclasts highly express PE, especially on the surface of their filopodia, although PE is normally localized in the inner leaflet. Inhibition of the externalization of PE on osteoclast progenitors resulted in impaired cellular fusion, but the expression of osteoclastogenic genes was relatively preserved (Irie et al. 2017). It was shown that during osteoclast fusion, PE is synthesized and externalized by Acyl-CoA: lysophosphatidylethanolamine acyltransferase 2 (LPEAT2, encoded by Lpeat2) and two ABC transporters (ABCB4, encoded by Abcb4 and ABCG1, encoded by Abcg1), respectively (Irie et al. 2017).
Certain osteoclast-specific molecules are also known to function in the fusion of osteoclasts. DC-STAMP, one of the osteoclastogenic genes, is essential for cellular fusion of the osteoclast progenitors. Its deficiency results in a failure in osteoclast multinucleation, giving rise to many TRAP+ mononuclear cells both in vitro and in vivo (Kukita et al. 2004; Yagi et al. 2005). Dcstamp−/− TRAP+ mononuclear cells normally express Fos, Nfatc1 and Ctsk. These cells also form a ruffled border and actin ring normally. However, bone resorption activity was lower in the mutant cells (Yagi et al. 2005). These results indicate that DC-STAMP is necessary specifically for the fusion process during osteoclastogenesis and that cellular fusion is crucial for bone resorption.
OC-STAMP is another factor that regulates the fusion of osteoclast progenitors. Ocstamp−/− cells develop into TRAP+ mononuclear cells by RANKL stimulation, similarly to Dcstamp−/− cells, with normal expression in osteoclastogenic genes, normal formation of the ruffled border and sealing zone, and reduced bone resorption activity (Miyamoto et al. 2012). Interestingly, Ocstamp−/− mice do not exhibit significant osteopetrosis, indicating the difference in the contribution of DC-STAMP and OC-STAMP to bone resorption (Miyamoto et al. 2012).
Atp6v0d2 is a subunit of the vacuolar H+-ATPases (V-ATPases) and its expression has been detected in osteoclasts. There were fewer multinuclear osteoclasts differentiated from Atp6v0d2−/− cells by RANKL stimulation, but the number of TRAP+ cells and the expression of osteoclastogenic genes were unaffected (Lee et al. 2006). Although TRAP+ cells were shown to be normally formed in Atp6v0d2−/− mice, osteoclastic bone resorption decreased, suggesting a contribution of Atp6v0d2 to cellular fusion and subsequent bone resorption (Lee et al. 2006). Although these osteoclastogenic molecules are essential for osteoclast fusion, their functional role in phospholipid dynamics remains unclear.
Isolation of the specific site of bone resorption
Osteoclasts adhere to the bone surface via integrin αVβ3, which activates the non-receptor tyrosine kinase c-Src. Activated c-Src then phosphorylates Syk to facilitate the formation of a complex with DAP12 and SLP-76, further activating VAV3 and the Rho-GTPase Rac, leading to actin polymerization (Faccio et al. 2005; Izawa et al. 2012; Zou et al. 2007). Polymerized actin fibers form an actin ring, which is observed as a clear zone under transmission electron microscopy (TEM). The bone surface targeted for resorption is sealed with this structure so that acids and catabolic enzymes do not leak, enabling a fine-tuned targeting of the bone resorption site.
Inside the sealed zone, H+, Cl−, CtsK and MMP-9 are secreted to degrade the inorganic components consisting of hydroxyapatite along with the organic components (mostly collagen). To increase the efficiency of bone resorption, the cellular membrane facing the zone becomes folded, making the ruffled border. It is reported that the osteoclasts of Src−/− mice lack the ruffled border, resulting in a deficiency of bone resorbing activity, suggesting its contribution to bone resorption (Boyce et al. 1992; Soriano et al. 1991). The formation of the ruffled border is closely linked to bone resorption activity because the osteoclasts of mice deficient in synaptotagmin VII, a molecule responsible for exocytosis of enzymes, are unable to form the structure (Zhao et al. 2008).
Ion channels and the degradation of the inorganic components of bone
Osteoclasts secrete H+ via V-ATPases located on the ruffled border. They consist of V1 and V0 domains, which carry out ATP hydrolysis and H+ translocation, respectively (Qin et al. 2012). A deficiency in the subunits of these domains is reported to lead to osteopetrosis. Mice deficient in the a3 subunit of the V0 domain (encoded by the Tcirg1 gene) exhibit an osteopetrotic phenotype, although they do have osteoclasts in the bone (Li et al. 1999; Scimeca et al. 2000). As mentioned in section II-C, mutations in TCIRG1 lead to ARO. A deficiency in the d2 subunit V0 domain (Atp6v0d2−/− mice) resulted in osteopetrosis. However, this seems to be due to a deficiency in cellular fusion than bone resorption activity (see section V-A) (Lee et al. 2006).
ClC-7 and its subunit Ostm1 neutralize the positive charge of the resorption pit by translocating Cl−. Clcn7−/− mice develop a severe osteopetrosis that is consistent with human patients (see section II-C) (Kornak et al. 2001). These mice have TRAP+ osteoclasts but they have few ruffled border and no bone resorption activity (Kornak et al. 2001). Ostm1 was shown to constitute a complex with ClC-7 and is suggested to stabilize it (Lange et al. 2006). Grey-lethal mice (gl/gl mice), which are deficient in Ostm1, as well as human patient with mutations in the orthologous gene exhibit osteopetrosis (see section II-C) (Chalhoub et al. 2003). These findings, taken together, indicate the key role for the ion channel in bone resorption.
CA2 catalyzes a reaction that gives rise to HCO3− and H+ out of carbonic acid. The H+ produced by this reaction is secreted by V-ATPase into the resorption pit. HCO3− is exported by anion exchangers, including SLC4A2, which in turn import Cl− into the cytosol. Mice deficient in CA2 or SLC4A2 were shown to be osteopetrotic (Margolis et al. 2008; Wu et al. 2008). Mutations in CA2 cause ARO, as discussed in section II-C (Sly et al. 1983). Taken together, these findings have indicated that CA2 functions as a supplier of ions for the other functional molecules described here.
Enzymes that degrade the organic components of the bone
Acidification of the resorption site is necessary not only for the dissolution of bone minerals, but also for activating certain enzymes. The cathepsins belong to the class of the cysteine proteinases, which can efficiently degrade the most abundant extracellular matrix (ECM) protein in the bone, type I collagen. CtsK was originally identified as a possible cysteine proteinase from a rabbit osteoclast cDNA library and later found in human bone tissues (Inaoka et al. 1995; Tezuka et al. 1994). The expression of Ctsk is the highest among the known cathepsins in osteoclasts (Ishibashi et al. 2001). Mutations in CTSK have been reported in human patients of an autosomal recessive osteochondrodysplasia, pycnodysostosis (Gelb et al. 1996). Hematopoietic cell-or monocyte/macrophage lineage-specific deletion of Ctsk in mice results in an osteopetrotic phenotype. These mice have an increased number of osteoclasts, but their bone resorption activity is low. In addition, these osteoclasts produced a higher level of sphingosine-1-phosphate (S1P), which increases osteoblastic bone formation (Lotinun et al. 2013).
The MMPs are a group of enzymes that can degrade ECM proteins. MMP-9 is expressed predominantly in osteoclasts and thought to be involved in bone resorption. Mmp9−/− mice were shown to have a deficiency in cartilage resorption during development that was compensated in the later stage, suggesting that MMP-9 plays a role in development (Vu et al. 1998). TRAP begins to be expressed in the osteoclast progenitors just before fusion. Because the functions of TRAP in osteoclasts have not been elucidated well, TRAP is typically thought of more as a phenotypic marker than as a functional molecule.
Imaging of bone resorption
Accumulating evidence has led to a much improved understanding of the functions of osteoclasts. In vitro experiments have enabled a detailed investigation of the mechanisms by which osteoclasts develop and function. However, it has long been known that the osteoclasts generated in the culture dish do not appear as how they are in the body, raising the concern that the cultured osteoclast phenotype does not reflect the actual osteoclasts in the body. Furthermore, the kinetics of these cells cannot be analyzed by in vitro experiments. To address this issue, live imaging techniques have been developed.
The use of animals, soft tissues of which are transparent, such as medaka and zebra fish has provided a simple and effective solution. Using acp5-gfp transgenic or mmp9-rfp transgenic medaka fish, osteoclasts were labeled and the gene expression levels were quantified with the fluorescence (Chatani et al. 2015, 2016). Quantification of mmp9 expression by live imaging suggested that the unloading by spaceflight enhances bone resorption (Chatani et al. 2016). With the use of molecular probes that have the capacity to detect pH variance (described below), more detailed analyses of bone resorption would be achieved.
Intravital imaging is a technique in which spatiotemporal information on tissues, cells and molecules is obtained by detecting fluorescence using two-photon excitation microscopy (TPEM). Bone is an advantageous tissue in this process in that motion artifacts are negligible compared to those that occur in the lung and the heart. However, hydroxyapatite in the bone matrix scatters the excitation light and the light cannot penetrate as deep as soft tissues, making it difficult to observe deep inside. Efforts have been made and now TPEM is applicable to the observation of bone marrow cells for several hours in calvarial bone, the cortical bone of which is thin enough for the analysis (Ishii et al. 2009). The mobilization of osteoclast progenitors and bone resorption by osteoclasts have been successfully visualized using the intravital imaging of the bone marrow (Ishii et al. 2009, 2010; Maeda et al. 2016).
For the purpose of accurate observation, fluorescent proteins or molecular probes with high photostability, intense brightness and a high accessibility toward the targets are required. A molecular probe that is able to make pH variation in the resorption pit visible has been developed (Maeda et al. 2016). The information acquired by such techniques are processed with analytical procedures for the objective and quantitative assessment of osteoclast function. The development of microscopy techniques, design of molecular probes and performance of mathematical analysis require the cooperation of experts from a variety of research fields. With the help of such experts, the elucidation of osteoclast functions will further proceed.
Conclusion
Thus, with the series of studies discussed here, knowledge of the mechanisms underlying osteoclastogenesis has been advanced quite remarkably over approximately the last 20 years. In these studies, comprehensive analyses of transcriptomes/proteomes and genetically modified mice have been employed as powerful tools in the identification of a novel factor related to osteoclasts and in proving its significance in vivo. The accumulated findings might give the impression that most of the crucial processes of osteoclastogenesis have been uncovered, thus discouraging further investigation. However, with the help of novel technologies such as intravital live imaging and mathematical analysis, another edge of the wedge has been delivered and begun to provide further novel insights into osteoclast biology. Findings from such analyses would lead to the development of novel therapeutic strategies for variety of bone diseases. Therefore, from both the scientific and clinical points of view, there remains a room for further studies on the osteoclast and RANK/RANKL biology.
References
Ahlberg PE, Clack JA, Blom H (2005) The axial skeleton of the Devonian tetrapod. Ichthyostega Nat 437:137–140. https://doi.org/10.1038/nature03893
Ahlberg PE, Clack JA, Luksevics E, Blom H, Zupins I (2008) Ventastega curonica and the origin of tetrapod morphology. Nature 453:1199–1204. https://doi.org/10.1038/nature06991
Aliprantis AO et al (2008) NFATc1 in mice represses osteoprotegerin during osteoclastogenesis and dissociates systemic osteopenia from inflammation in cherubism. J Clin Invest 118:3775–3789. https://doi.org/10.1172/JCI35711
Alnaeeli M, Penninger JM, Teng YT (2006) Immune interactions with CD4 + T cells promote the development of functional osteoclasts from murine CD11c + dendritic cells. J Immunol (Baltimore., Md: 1950) 177:3314–3326
Anderson DM et al (1997) A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature 390:175–179. https://doi.org/10.1038/36593
Aoki K et al (2006) A TNF receptor loop peptide mimic blocks RANK ligand-induced signaling, bone resorption, and bone loss. J Clin Invest 116:1525–1534. https://doi.org/10.1172/JCI22513
Arai F et al (1999) Commitment and differentiation of osteoclast precursor cells by the sequential expression of c-Fms and receptor activator of nuclear factor kappaB (RANK) receptors. J Exp Med 190:1741–1754
Asagiri M et al (2005) Autoamplification of NFATc1 expression determines its essential role in bone homeostasis. J Exp Med 202:1261–1269. https://doi.org/10.1084/jem.20051150
Bai S et al (2005) FHL2 inhibits the activated osteoclast in a TRAF6-dependent manner. J Clin Invest 115:2742–2751. https://doi.org/10.1172/JCI24921
Bassett JH, Williams GR (2016) Role of thyroid hormones in skeletal development and bone maintenance. Endocr Rev 37:135–187. https://doi.org/10.1210/er.2015-1106
Bohm C et al. (2009) The alpha-isoform of p38 MAPK specifically regulates arthritic bone loss. J Immunol (Baltimore., Md: 1950) 183:5938–5947 https://doi.org/10.4049/jimmunol.0901026
Bonewald L (2014) Eighth bone quality seminar proceedings 2013. Osteoporos Int 25 Suppl 3:S465–S501. https://doi.org/10.1007/s00198-014-2681-x
Boyce BF, Yoneda T, Lowe C, Soriano P, Mundy GR (1992) Requirement of pp60c-src expression for osteoclasts to form ruffled borders and resorb bone in mice. J Clin Investig 90:1622–1627. https://doi.org/10.1172/JCI116032
Boyle DL et al (2014) Differential roles of MAPK kinases MKK3 and MKK6 in osteoclastogenesis and bone loss. PLoS One 9:e84818. https://doi.org/10.1371/journal.pone.0084818
Bucay N et al (1998) Osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev 12:1260–1268
Cao H et al (2010) Activating transcription factor 4 regulates osteoclast differentiation in mice. J Clin Invest 120:2755–2766. https://doi.org/10.1172/JCI42106
Chalhoub N et al (2003) Grey-lethal mutation induces severe malignant autosomal recessive osteopetrosis in mouse and human. Nat Med 9:399–406. https://doi.org/10.1038/nm842
Chatani M et al (2015) Microgravity promotes osteoclast activity in medaka fish reared at the international space station. Sci Rep 5:14172. https://doi.org/10.1038/srep14172
Chatani M et al (2016) Acute transcriptional up-regulation specific to osteoblasts/osteoclasts in medaka fish immediately after exposure to microgravity. Sci Rep 6:39545. https://doi.org/10.1038/srep39545
Chen X, Zhang K, Hock J, Wang C, Yu X (2016) Enhanced but hypofunctional osteoclastogenesis in an autosomal dominant osteopetrosis type II case carrying a c.1856C > T mutation in CLCN7. Bone Res 4:16035. https://doi.org/10.1038/boneres.2016.35
Cleiren E et al (2001) Albers–Schönberg disease (autosomal dominant osteopetrosis, type II) results from mutations in the ClCN7 chloride channel gene. Hum Mol Genet 10:2861–2867. https://doi.org/10.1093/hmg/10.25.2861
Cong Q et al (2017) p38alpha MAPK regulates proliferation and differentiation of osteoclast progenitors and bone remodeling in an aging-dependent manner. Sci Rep 7:45964. https://doi.org/10.1038/srep45964
Corral DA et al. (1998) Dissociation between bone resorption and bone formation in osteopenic transgenic mice. Proc Natl Acad Sci 95:13835–13840. https://doi.org/10.1073/pnas.95.23.13835
Coudert AE, de Vernejoul MC, Muraca M, Del Fattore A (2015) Osteopetrosis and its relevance for the discovery of new functions associated with the skeleton. Int J Endocrinol 2015:372156. https://doi.org/10.1155/2015/372156
Cox TR et al (2015) The hypoxic cancer secretome induces pre-metastatic bone lesions through lysyl oxidase. Nature 522:106–110. https://doi.org/10.1038/nature14492
David JP, Sabapathy K, Hoffmann O, Idarraga MH, Wagner EF (2002) JNK1 modulates osteoclastogenesis through both c-Jun phosphorylation-dependent and -independent mechanisms. J Cell Sci 115:4317–4325
Decker CE et al (2015) Tmem178 acts in a novel negative feedback loop targeting NFATc1 to regulate bone mass. Proc Natl Acad Sci USA 112:15654–15659. https://doi.org/10.1073/pnas.1511285112
Dougall WC et al (1999) RANK is essential for osteoclast and lymph node development. Genes Dev 13:2412–2424
Edwards JR, Mundy GR (2011) Advances in osteoclast biology: old findings and new insights from mouse models. Nat Rev Rheumatol 7:235–243. https://doi.org/10.1038/nrrheum.2011.23
Einhorn TA, Gerstenfeld LC (2015) Fracture healing: mechanisms and interventions. Nat Rev Rheumatol 11:45–54. https://doi.org/10.1038/nrrheum.2014.164
Faccio R et al (2005) Vav3 regulates osteoclast function and bone mass. Nat Med 11:284–290. https://doi.org/10.1038/nm1194
Franzoso G et al (1997) Requirement for NF-kappaB in osteoclast and B-cell development. Genes Dev 11:3482–3496
Fuller K, Wong B, Fox S, Choi Y, Chambers TJ (1998) TRANCE is necessary and sufficient for osteoblast-mediated activation of bone resorption in osteoclasts. J Exp Med 188:997–1001. https://doi.org/10.1084/jem.188.5.997
Gelb BD, Shi GP, Chapman HA, Desnick RJ (1996) Pycnodysostosis, a lysosomal disease caused by cathepsin K deficiency. Science (New York, NY) 273:1236–1238
Ghosh S, Karin M (2002) Missing pieces in the NF-kappaB puzzle. Cell 109 Suppl:S81–S96
Hayashi M, Nakashima T, Taniguchi M, Kodama T, Kumanogoh A, Takayanagi H (2012) Osteoprotection by semaphorin 3A. Nature 485:69–74. https://doi.org/10.1038/nature11000
Hikita A et al (2006) Negative regulation of osteoclastogenesis by ectodomain shedding of receptor activator of NF-kappaB ligand. J Biol Chem 281:36846–36855. https://doi.org/10.1074/jbc.M606656200
Hirotani H, Tuohy NA, Woo JT, Stern PH, Clipstone NA (2004) The calcineurin/nuclear factor of activated T cells signaling pathway regulates osteoclastogenesis in RAW264.7 cells. J Biol Chem 279:13984–13992. https://doi.org/10.1074/jbc.M213067200
Inaoka T, Bilbe G, Ishibashi O, Tezuka K, Kumegawa M, Kokubo T (1995) Molecular cloning of human cDNA for cathepsin K: novel cysteine proteinase predominantly expressed in bone. Biochem Biophys Res Commun 206:89–96. https://doi.org/10.1006/bbrc.1995.1013
Irie A, Yamamoto K, Miki Y, Murakami M (2017) Phosphatidylethanolamine dynamics are required for osteoclast fusion. Sci Rep 7:46715. https://doi.org/10.1038/srep46715
Ishibashi O, Inui T, Mori Y, Kurokawa T, Kokubo T, Kumegawa M (2001) Quantification of the expression levels of lysosomal cysteine proteinases in purified human osteoclastic cells by competitive RT-PCR. Calcif Tissue Int 68:109–116. https://doi.org/10.1007/bf02678149
Ishii M et al (2009) Sphingosine-1-phosphate mobilizes osteoclast precursors and regulates bone homeostasis. Nature 458:524–528. https://doi.org/10.1038/nature07713
Ishii M, Kikuta J, Shimazu Y, Meier-Schellersheim M, Germain RN (2010) Chemorepulsion by blood S1P regulates osteoclast precursor mobilization and bone remodeling in vivo. J Exp Med 207:2793–2798. https://doi.org/10.1084/jem.20101474
Izawa T, Zou W, Chappel JC, Ashley JW, Feng X, Teitelbaum SL (2012) c-Src links a RANK/alphavbeta3 integrin complex to the osteoclast cytoskeleton. Mol Cell Biol 32:2943–2953. https://doi.org/10.1128/MCB.00077-12
Joyce-Shaikh B et al (2010) Myeloid DAP12-associating lectin (MDL)-1 regulates synovial inflammation and bone erosion associated with autoimmune arthritis. J Exp Med 207:579–589. https://doi.org/10.1084/jem.20090516
Kaifu T et al (2003) Osteopetrosis and thalamic hypomyelinosis with synaptic degeneration in DAP12-deficient mice. J Clin Invest 111:323–332. https://doi.org/10.1172/JCI16923
Kameda Y et al (2013) Siglec-15 regulates osteoclast differentiation by modulating RANKL-induced phosphatidylinositol 3-kinase/Akt and Erk pathways in association with signaling Adaptor DAP12. J Bone Miner Res 28:2463–2475. https://doi.org/10.1002/jbmr.1989
Kenner L et al (2004) Mice lacking JunB are osteopenic due to cell-autonomous osteoblast and osteoclast defects. J Cell Biol 164:613–623. https://doi.org/10.1083/jcb.200308155
Kim N et al (2005) Osteoclast differentiation independent of the TRANCE-RANK-TRAF6 axis. J Exp Med 202:589–595. https://doi.org/10.1084/jem.20050978
Kim K et al (2007) MafB negatively regulates RANKL-mediated osteoclast differentiation. Blood 109:3253–3259. https://doi.org/10.1182/blood-2006-09-048249
Kim K, Lee SH, Ha Kim J, Choi Y, Kim N (2008) NFATc1 induces osteoclast fusion via up-regulation of Atp6v0d2 and the dendritic cell-specific transmembrane protein (DC-STAMP). Mol Endocrinol 22:176–185. https://doi.org/10.1210/me.2007-0237
Kim H et al (2013) Tmem64 modulates calcium signaling during RANKL-mediated osteoclast differentiation. Cell Metab 17:249–260. https://doi.org/10.1016/j.cmet.2013.01.002
Koga T et al (2004) Costimulatory signals mediated by the ITAM motif cooperate with RANKL for bone homeostasis. Nature 428:758–763. https://doi.org/10.1038/nature02444
Kong YY et al (1999) OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature 397:315–323. https://doi.org/10.1038/16852
Kornak U et al (2001) Loss of the ClC-7 chloride channel leads to osteopetrosis in mice and man. Cell 104:205–215. https://doi.org/10.1016/S0092-8674(01)00206-9
Kukita T et al (2004) RANKL-induced DC-STAMP is essential for osteoclastogenesis. J Exp Med 200:941–946. https://doi.org/10.1084/jem.20040518
Kukita A et al (2011) The transcription factor FBI-1/OCZF/LRF is expressed in osteoclasts and regulates RANKL-induced osteoclast formation in vitro and in vivo. Arthritis Rheum 63:2744–2754. https://doi.org/10.1002/art.30455
Lacey DL et al (1998) Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell 93:165–176
Lam J, Nelson CA, Ross FP, Teitelbaum SL, Fremont DH (2001) Crystal structure of the TRANCE/RANKL cytokine reveals determinants of receptor–ligand specificity. J Clin Invest 108:971–979. https://doi.org/10.1172/JCI13890
Lamothe B, Webster WK, Gopinathan A, Besse A, Campos AD, Darnay BG (2007) TRAF6 ubiquitin ligase is essential for RANKL signaling and osteoclast differentiation. Biochem Biophys Res Commun 359:1044–1049. https://doi.org/10.1016/j.bbrc.2007.06.017
Lamothe B, Lai Y, Xie M, Schneider MD, Darnay BG (2013) TAK1 is essential for osteoclast differentiation and is an important modulator of cell death by apoptosis and necroptosis. Mol Cell Biol 33:582–595. https://doi.org/10.1128/MCB.01225-12
Lange PF, Wartosch L, Jentsch TJ, Fuhrmann JC (2006) ClC-7 requires Ostm1 as a beta-subunit to support bone resorption and lysosomal function. Nature 440:220–223. https://doi.org/10.1038/nature04535
Lee SH et al (2006) v-ATPase V0 subunit d2-deficient mice exhibit impaired osteoclast fusion and increased bone formation. Nat Med 12:1403–1409. https://doi.org/10.1038/nm1514
Li YP, Chen W, Liang Y, Li E, Stashenko P (1999) Atp6i-deficient mice exhibit severe osteopetrosis due to loss of osteoclast-mediated extracellular acidification. Nat Genet 23:447–451. https://doi.org/10.1038/70563
Li J et al (2000) RANK is the intrinsic hematopoietic cell surface receptor that controls osteoclastogenesis and regulation of bone mass and calcium metabolism. Proc Natl Acad Sci USA 97:1566–1571
Liu C et al. (2010) Structural and functional insights of RANKL-RANK interaction and signaling. J Immunol (Baltimore., Md: 1950) 184:6910–6919 https://doi.org/10.4049/jimmunol.0904033
Lomaga MA et al (1999) TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev 13:1015–1024
Lotinun S et al (2013) Osteoclast-specific cathepsin K deletion stimulates S1P-dependent bone formation. J Clin Invest 123:666–681. https://doi.org/10.1172/JCI64840
Lum L et al (1999) Evidence for a role of a tumor necrosis factor-α (TNF-α)-converting enzyme-like protease in shedding of TRANCE, a TNF family member involved in osteoclastogenesis and dendritic cell survival. J Biol Chem 274:13613–13618. https://doi.org/10.1074/jbc.274.19.13613
Maeda H et al (2016) Real-time intravital imaging of pH variation associated with osteoclast activity. Nat Chem Biol 12:579–585. https://doi.org/10.1038/nchembio.2096
Margolis DS, Szivek JA, Lai LW, Lien YH (2008) Phenotypic characteristics of bone in carbonic anhydrase II-deficient mice. Calcif Tissue Int 82:66–76. https://doi.org/10.1007/s00223-007-9098-x
Maruyama K et al (2012) The transcription factor Jdp2 controls bone homeostasis and antibacterial immunity by regulating osteoclast and neutrophil differentiation. Immunity 37:1024–1036. https://doi.org/10.1016/j.immuni.2012.08.022
Matsumoto M et al (2004) Essential role of p38 mitogen-activated protein kinase in cathepsin K gene expression during osteoclastogenesis through association of NFATc1 and PU.1. J Biol Chem 279:45969–45979. https://doi.org/10.1074/jbc.M408795200
Matsuo K et al (2004) Nuclear factor of activated T-cells (NFAT) rescues osteoclastogenesis in precursors lacking c-Fos. J Biol Chem 279:26475–26480. https://doi.org/10.1074/jbc.M313973200
Min H et al (2000) Osteoprotegerin reverses osteoporosis by inhibiting endosteal osteoclasts and prevents vascular calcification by blocking a process resembling osteoclastogenesis. J Exp Med 192:463–474
Miyamoto H et al (2012) Osteoclast stimulatory transmembrane protein and dendritic cell-specific transmembrane protein cooperatively modulate cell–cell fusion to form osteoclasts and foreign body giant cells. J Bone Miner Res 27:1289–1297. https://doi.org/10.1002/jbmr.1575
Miyauchi Y et al (2010) The Blimp1-Bcl6 axis is critical to regulate osteoclast differentiation and bone homeostasis. J Exp Med 207:751–762. https://doi.org/10.1084/jem.20091957
Mizukami J, Takaesu G, Akatsuka H, Sakurai H, Ninomiya-Tsuji J, Matsumoto K, Sakurai N (2002) Receptor activator of NF- B ligand (RANKL) activates TAK1 mitogen-activated protein kinase kinase kinase through a signaling complex containing RANK, TAB2, and TRAF6. Mol Cell Biol 22:992–1000. https://doi.org/10.1128/mcb.22.4.992-1000.2002
Mizuno A et al (1998) Severe osteoporosis in mice lacking osteoclastogenesis inhibitory factor/osteoprotegerin. Biochem Biophys Res Commun 247:610–615
Nagashima K et al (2017) Identification of subepithelial mesenchymal cells that induce IgA and diversify gut microbiota. Nat Immunol 18:675–682. https://doi.org/10.1038/ni.3732
Naito A et al (1999) Severe osteopetrosis, defective interleukin-1 signalling and lymph node organogenesis in TRAF6-deficient mice. Genes Cells Devot Mol Cell Mech 4:353–362
Nakagawa N et al (1998) RANK is the essential signaling receptor for osteoclast differentiation factor in osteoclastogenesis. Biochem Biophys Res Commun 253:395–400. https://doi.org/10.1006/bbrc.1998.9788
Nakashima T, Kobayashi Y, Yamasaki S, Kawakami A, Eguchi K, Sasaki H, Sakai H (2000) Protein expression and functional difference of membrane-bound and soluble receptor activator of NF-κB ligand: modulation of the expression by osteotropic factors and cytokines. Biochem Biophys Res Commun 275:768–775. https://doi.org/10.1006/bbrc.2000.3379
Nakashima T et al (2011) Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat Med 17:1231–1234. https://doi.org/10.1038/nm.2452
Negishi-Koga T et al (2015) Immune complexes regulate bone metabolism through FcRgamma signalling. Nat Commun 6:6637. https://doi.org/10.1038/ncomms7637
Nelson CA, Warren JT, Wang MW, Teitelbaum SL, Fremont DH (2012) RANKL employs distinct binding modes to engage RANK and the osteoprotegerin decoy receptor. Structure 20:1971–1982. https://doi.org/10.1016/j.str.2012.08.030
Nguyen AM, Jacobs CR (2013) Emerging role of primary cilia as mechanosensors in osteocytes. Bone 54:196–204. https://doi.org/10.1016/j.bone.2012.11.016
Nishikawa K et al (2010) Blimp1-mediated repression of negative regulators is required for osteoclast differentiation. Proc Natl Acad Sci USA 107:3117–3122. https://doi.org/10.1073/pnas.0912779107
Nishikawa K et al (2015) DNA methyltransferase 3a regulates osteoclast differentiation by coupling to an S-adenosylmethionine-producing metabolic pathway. Nat Med 21:281–287. https://doi.org/10.1038/nm.3774
Ogata N, Kawaguchi H, Chung UI, Roth SI, Segre GV (2007) Continuous activation of G alpha q in osteoblasts results in osteopenia through impaired osteoblast differentiation. J Biol Chem 282:35757–35764. https://doi.org/10.1074/jbc.M611902200
Okamoto K et al (2017) Osteoimmunology: the conceptual framework unifying the immune and skeletal systems. Physiol Rev 97:1295–1349. https://doi.org/10.1152/physrev.00036.2016
Ono T, Takayanagi H (2017) Osteoimmunology in bone fracture healing. Curr Osteoporos Rep 15:367–375. https://doi.org/10.1007/s11914-017-0381-0
Ozaki Y et al (2017) Treatment of OPG-deficient mice with WP9QY, a RANKL-binding peptide, recovers alveolar bone loss by suppressing osteoclastogenesis and enhancing osteoblastogenesis. PLoS One 12:e0184904. https://doi.org/10.1371/journal.pone.0184904
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. https://doi.org/10.1002/jcc.20084
Pilz GA et al (2011) Human mesenchymal stromal cells express CD14 cross-reactive epitopes. Cytometry A 79:635–645. https://doi.org/10.1002/cyto.a.21073
Qin A, Cheng TS, Pavlos NJ, Lin Z, Dai KR, Zheng MH (2012) V-ATPases in osteoclasts: structure, function and potential inhibitors of bone resorption. Int J Biochem Cell Biol 44:1422–1435. https://doi.org/10.1016/j.biocel.2012.05.014
Rodan GA, Martin TJ (1981) Role of osteoblasts in hormonal control of bone resorption—a hypothesis. Calcif Tissue Int 33:349–351. https://doi.org/10.1007/bf02409454
Ruocco MG et al (2005) I{kappa}B kinase (IKK){beta}, but not IKK{alpha}, is a critical mediator of osteoclast survival and is required for inflammation-induced bone loss. J Exp Med 201:1677–1687. https://doi.org/10.1084/jem.20042081
Sato K et al (2006) Regulation of osteoclast differentiation and function by the CaMK-CREB pathway. Nat Med 12:1410–1416. https://doi.org/10.1038/nm1515
Scimeca JC et al (2000) The gene encoding the mouse homologue of the human osteoclast-specific 116-kDa V-ATPase subunit bears a deletion in osteosclerotic (oc/oc) mutants. Bone 26:207–213. https://doi.org/10.1016/S8756-3282(99)00278-1
Seita J, Weissman IL (2010) Hematopoietic stem cell: self-renewal versus differentiation. Wiley Interdiscip Rev Syst Biol Med 2:640–653. https://doi.org/10.1002/wsbm.86
Shinohara M et al (2008) Tyrosine kinases Btk and Tec regulate osteoclast differentiation by linking RANK and ITAM signals. Cell 132:794–806. https://doi.org/10.1016/j.cell.2007.12.037
Shoji-Matsunaga A, Ono T, Hayashi M, Takayanagi H, Moriyama K, Nakashima T (2017) Osteocyte regulation of orthodontic force-mediated tooth movement via RANKL expression. Sci Rep 7:8753. https://doi.org/10.1038/s41598-017-09326-7
Simonet WS et al (1997) Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell 89:309–319
Sly WS, Hewett-Emmett D, Whyte MP, Yu YS, Tashian RE (1983) Carbonic anhydrase II deficiency identified as the primary defect in the autosomal recessive syndrome of osteopetrosis with renal tubular acidosis and cerebral calcification. Proc Natl Acad Sci USA 80:2752–2756
Soriano P, Montgomery C, Geske R, Bradley A (1991) Targeted disruption of the c-src proto-oncogene leads to osteopetrosis in mice. Cell 64:693–702
Speziani C et al (2007) Murine dendritic cell transdifferentiation into osteoclasts is differentially regulated by innate and adaptive cytokines. Eur J Immunol 37:747–757. https://doi.org/10.1002/eji.200636534
Suda T, Takahashi N, Udagawa N, Jimi E, Gillespie MT, Martin TJ (1999) Modulation of osteoclast differentiation and function by the new members of the tumor necrosis factor receptor and ligand families. Endocr Rev 20:345–357. https://doi.org/10.1210/edrv.20.3.0367
Sumiya E et al (2015) Phosphoproteomic analysis of kinase-deficient mice reveals multiple TAK1 targets in osteoclast differentiation. Biochem Biophys Res Commun 463:1284–1290. https://doi.org/10.1016/j.bbrc.2015.06.105
Takahashi N et al (1988) Osteoblastic cells are involved in osteoclast formation. Endocrinology 123:2600–2602. https://doi.org/10.1210/endo-123-5-2600
Takano-Yamamoto T (2014) Osteocyte function under compressive mechanical force. Jpn Dental Sci Rev 50:29–39. https://doi.org/10.1016/j.jdsr.2013.10.004
Takayanagi H et al (2002) Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell 3:889–901
Takegahara N et al (2006) Plexin-A1 and its interaction with DAP12 in immune responses and bone homeostasis. Nat Cell Biol 8:615–622. https://doi.org/10.1038/ncb1416
Tanaka S (2017) RANKL-independent osteoclastogenesis: a long-standing controversy. J Bone Miner Res 32:431–433. https://doi.org/10.1002/jbmr.3092
Tezuka K et al (1994) Molecular cloning of a possible cysteine proteinase predominantly expressed in osteoclasts. J Biol Chem 269:1106–1109
Tsuda E, Goto M, Mochizuki S-i, Yano K, Kobayashi F, Morinaga T, Higashio K (1997) Isolation of a novel cytokine from human fibroblasts that specifically inhibits osteoclastogenesis. Biochem Biophys Res Commun 234:137–142. https://doi.org/10.1006/bbrc.1997.6603
Tsuji-Takechi K et al (2012) Stage-specific functions of leukemia/lymphoma-related factor (LRF) in the transcriptional control of osteoclast development. Proc Natl Acad Sci USA 109:2561–2566. https://doi.org/10.1073/pnas.1116042109
Tsukasaki M et al (2017) LOX fails to substitute for RANKL in osteoclastogenesis. J Bone Miner Res 32:434–439. https://doi.org/10.1002/jbmr.2990
Turner CH et al. (2009) Mechanobiology of the Skeleton. Sci Signal 2:3
Udagawa N et al (1989) The bone marrow-derived stromal cell lines MC3T3-G2/PA6 and ST2 support osteoclast-like cell differentiation in cocultures with mouse spleen. Cells Endocrinol 125:1805–1813. https://doi.org/10.1210/endo-125-4-1805
Vu TH et al (1998) MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell 93:411–422. https://doi.org/10.1016/S0092-8674(00)81169-1
Wada T, Nakashima T, Oliveira-dos-Santos AJ, Gasser J, Hara H, Schett G, Penninger JM (2005) The molecular scaffold Gab2 is a crucial component of RANK signaling and osteoclastogenesis. Nat Med 11:394–399. https://doi.org/10.1038/nm1203
Walsh MC, Choi Y (2014) Biology of the RANKL-RANK-OPG system in immunity bone beyond. Front Immunol 5:511. https://doi.org/10.3389/fimmu.2014.00511
Weinstein RS et al (2002) Promotion of osteoclast survival and antagonism of bisphosphonate-induced osteoclast apoptosis by glucocorticoids. J Clin Invest 109:1041–1048. https://doi.org/10.1172/jci14538
Whyte MP, Mumm S (2004) Heritable disorders of the RANKL/OPG/RANK signaling pathway. J Musculoskelet Neuronal Interact 4:254–267
Winn N, Lalam R, Cassar-Pullicino V (2017) Imaging of Paget’s disease of bone. Wien Med Wochenschr 167:9–17. https://doi.org/10.1007/s10354-016-0517-3
Wright HL, McCarthy HS, Middleton J, Marshall MJ (2009) RANK, RANKL and osteoprotegerin in bone biology and disease. Curr Rev Musculoskelet Med 2:56–64. https://doi.org/10.1007/s12178-009-9046-7
Wu J, Glimcher LH, Aliprantis AO (2008) HCO3-/Cl- anion exchanger SLC4A2 is required for proper osteoclast differentiation and function. Proc Natl Acad Sci USA 105:16934–16939. https://doi.org/10.1073/pnas.0808763105
Xiao Y et al (2013) Osteoclast precursors in murine bone marrow express CD27 and are impeded in osteoclast development by CD70 on activated immune cells. Proc Natl Acad Sci USA 110:12385–12390. https://doi.org/10.1073/pnas.1216082110
Xiao Y, Palomero J, Grabowska J, Wang L, de Rink I, van Helvert L, Borst J (2017) Macrophages and osteoclasts stem from a bipotent progenitor downstream of a macrophage/osteoclast/dendritic cell progenitor Blood Advances. 1:1993–2006 https://doi.org/10.1182/bloodadvances.2017008540
Xiong J, Onal M, Jilka RL, Weinstein RS, Manolagas SC, O’Brien CA (2011) Matrix-embedded cells control osteoclast formation. Nat Med 17:1235–1241. https://doi.org/10.1038/nm.2448
Xiong J et al (2015) Osteocytes, not osteoblasts or lining cells, are the main source of the RANKL required for osteoclast formation in remodeling Bone. PLoS One 10:e0138189. https://doi.org/10.1371/journal.pone.0138189
Yagi M et al (2005) DC-STAMP is essential for cell-cell fusion in osteoclasts and foreign body giant cells. J Exp Med 202:345–351. https://doi.org/10.1084/jem.20050645
Yamamoto A et al (2002) Possible involvement of IkappaB kinase 2 and MKK7 in osteoclastogenesis induced by receptor activator of nuclear factor kappaB ligand. J Bone Miner Res 17:612–621. https://doi.org/10.1359/jbmr.2002.17.4.612
Yamashita T et al (2007) NF-kappaB p50 and p52 regulate receptor activator of NF-kappaB ligand (RANKL) and tumor necrosis factor-induced osteoclast precursor differentiation by activating c-Fos and NFATc1. J Biol Chem 282:18245–18253. https://doi.org/10.1074/jbc.M610701200
Yang YM et al (2009) Alteration of RANKL-induced osteoclastogenesis in primary cultured osteoclasts from SERCA2+/− mice. J Bone Miner Res 24:1763–1769. https://doi.org/10.1359/jbmr.090420
Yasuda H et al (1998) Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci USA 95:3597–3602
Yasui T, Hirose J, Tsutsumi S, Nakamura K, Aburatani H, Tanaka S (2011) Epigenetic regulation of osteoclast differentiation: possible involvement of Jmjd3 in the histone demethylation of Nfatc1. J Bone Miner Res 26:2665–2671. https://doi.org/10.1002/jbmr.464
Zachowski A (1993) Phospholipids in animal eukaryotic membranes: transverse asymmetry and movement. Biochem J 294:1–14
Zhao H, Ito Y, Chappel J, Andrews NW, Teitelbaum SL, Ross FP (2008) Synaptotagmin VII regulates bone remodeling by modulating osteoclast and osteoblast secretion. Dev Cell 14:914–925. https://doi.org/10.1016/j.devcel.2008.03.022
Zhao B et al (2009) Interferon regulatory factor-8 regulates bone metabolism by suppressing osteoclastogenesis. Nat Med 15:1066–1071. https://doi.org/10.1038/nm.2007
Zhao B, Grimes SN, Li S, Hu X, Ivashkiv LB (2012) TNF-induced osteoclastogenesis and inflammatory bone resorption are inhibited by transcription factor RBP-J. J Exp Med 209:319–334. https://doi.org/10.1084/jem.20111566
Zou W et al (2007) Syk, c-Src, the alphavbeta3 integrin, and ITAM immunoreceptors, in concert, regulate osteoclastic bone resorption. J Cell Biol 176:877–888. https://doi.org/10.1083/jcb.200611083
Acknowledgements
This work was supported by Ichiro Kanehara Foundation, Lotte Research Promotion Grant, Takeda Science Foundation, The Uehara Memorial Foundation, Japan Agency for Medical Research and Development (AMED, award number: JP17gm0810003), Japan Society for the Promotion of Science (JSPS), Naito Foundation, Astellas Foundation for Research on Metabolic Disorders, Sumitomo Foundation, Asahi Glass Foundation, Daiichi Sankyo Foundation of Life Science, Secom Science and Technology Foundation (SSTF), Mochida Memorial Foundation for Medical and Pharmaceutical Research, Terumo Foundation and Matsui Life Social Welfare Foundation.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Ono, T., Nakashima, T. Recent advances in osteoclast biology. Histochem Cell Biol 149, 325–341 (2018). https://doi.org/10.1007/s00418-018-1636-2
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00418-018-1636-2