
Abstract
Considerable studies have evaluated the interaction between Wnt/β-catenin signaling and numerous cellular processes. Emerging findings now demonstrate that Wnt/β-catenin signaling interacts with the life cycle of the Human Immunodeficiency Virus type 1 (HIV-1). Wnt/β-catenin is a restrictive pathway to HIV replication in multiple target cells including peripheral blood mononuclear cells and astrocytes. The molecular interaction between Wnt/β-catenin signaling and HIV has been evaluated in astrocytes because they express robust level of this pathway. The cross talk that occurs between these two components has significant biologic consequences to HIV-mediated neuropathogenesis. This perspective highlights current knowledge regarding the interaction between Wnt/β-catenin signaling and HIV, the interplay between these two pathways as it impacts key features of NeuroAIDS, and provides an assessment of knowledge gaps in the field that could propel our understanding of this interaction to inform novel strategies to exploit Wnt signaling for therapeutic intervention in HIV/NeuroAIDS.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Wnt/β-catenin signaling
Wnt/β-catenin is a highly conserved signal transduction pathway involved in many processes from early embryogenesis to adult multi-organ homeostasis. This pathway plays a role in numerous cellular events including cell differentiation, communication, survival, and proliferation. Although the pathway is rather complex involving many agonists and antagonists, key events in Wnt/β-catenin signal transduction are depicted in Fig. 1. The pathway is initiated by binding of Wnt ligands, which are small secreted glycoproteins, to seven-transmembrane frizzled receptors and low density lipoprotein receptor-related protein 5 and 6 (LRP5/6) co-receptors to transduce a signal leading to the hypophosphorylation and stabilization of β-catenin. β-catenin is a central mediator of this pathway. It associates with cadherins at the cell membrane to regulate cellular adhesion or translocates to the nucleus where it functions as a transcriptional co-activator. As a transcriptional co-activator, β-catenin binds to members of the T cell factor/lymphoid enhancer factor (TCF/LEF) family of transcription factors to regulate gene expression. Members of TCF/LEF family of transcriptional factors include LEF1, TCF-1, TCF-3, and TCF-4 and all have a high mobility group (HMG) domain allowing them to induce a sharp bend in the DNA helix. Without a co-activator such as β-catenin, TCF/LEFs are associated with gene repression. However, in association with β-catenin, TCF/LEF can lead to gene repression or induction. β-catenin displacement of negative regulators of TCF/LEF such as transducin-like enhancer protein (TLE) and histone deacetylases (HDACs), and recruitment of co-factors such as BCL9, Pygopus (PYGO) and CBP/p300 leads to Wnt target gene transcription. On the other hand, β-catenin/TCF binding in conjunction with recruitment of HDACs and other epigenetic modifiers leads to gene repression. Wnt target genes are ever growing and a list is found at the Wnt home page at http://www.stanford.edu/group/nusselab/cgi-bin/wnt/. In the absence of Wnt protein binding, a multi-protein destruction complex composed of Axin, adenomatous polyposis coli (APC), casein kinase 1α (Ck1α), and glycogen synthase kinase 3β (GSK3β) phosphorylates β-catenin on Ser45 by Ck1 and on Thr 41, Ser33 and Ser37 by GSK3β, leading to β-catenin ubiquitination by βTrcp and proteasomal degradation. Wnt-independent activation of β-catenin occurs when either negative regulators of β-catenin are inhibited or when activators of β-catenin are engaged. For example, lithium chloride, at doses not to exceed 1 mM, selectively inhibits both isoforms of GSK3 α and β, resulting in β-catenin activation (Hedgepeth et al. 1997). At higher concentrations (4–5 mM) lithium inhibits inositol monophosphatase (IMPase) and inositol polyphosphatase, leading to a decreased inositol (1,4,5) trisphosphate (IP3) (Williams and Harwood 2000) response.

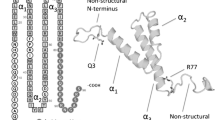
Wnt/β-catenin signal transduction pathway: Wnt protein binding to seven transmembrane Frizzled receptors leads to the recruitment of Axin to the plasma membrane and phosphorylation of its co-receptor, low-density lipoprotein receptor related protein (LRP) 5/6 by casein kinase-1α (CK1α) and glycogen synthase kinase-3β (GSK3β), leading to activation of Dishevelled (Dvl) and destabilization of a β-catenin destruction complex. The destruction complex consists of Axin, adenomatous polyposis coli (APC), CK1α and GSK3β. Hypophosphorylated β-catenin accumulates in the cytoplasm and is able to translocate to the nucleus, where it interacts with TCF/LEF to displace its co-repressors and recruit either positive or negative transcription co-factors to regulate Wnt target genes. Active β-catenin can also bind to cadherins at the cell membrane to regulate cellular adhesion. In the absence of Wnt binding to frizzled and LRP5/6, cytosolic β-catenin is phosphorylated by the destruction complex and undergoes βTrcp-mediated ubiquitination and proteasomal degradation. Image is slightly modified and reprinted from (Henderson and Al-Harthi 2011), copyright © J Neuroimmune Pharmacology [(2011) 6: 247–259, doi:10.1007/s11481-011-9266-7]
The connection between Wnt/β-catenin and HIV
Wnt/β-catenin has emerged as a restriction factor for HIV in a number of cell types, including PBMCs and astrocytes (Wortman et al. 2002; Caroll-Anzinger et al. 2007; Kumar et al. 2008; Henderson et al. 2012a; Narasipura et al. 2012). Many of the studies to evaluate this interaction have been modeled in human astrocytes due to their robust level of Wnt/β-catenin signaling, facilitating molecular studies to probe the interaction between Wnt/β-catenin and HIV.
HIV invades the CNS within weeks of infection and sets the stage for neuroinflammatory events that despite anti-HIV therapy leads to mild/moderate impairment to more severe HIV-associated dementia in a subset of HIV infected individuals (Heaton et al. 2011). This disease spectrum has been collectively termed HIV-associated neurocognitive disorders (HAND). HAND symptoms include decline in memory, learning, and executive function that impairs day to day activity. The pathology of HAND, depending on disease severity, includes reactive astrocytosis, myelin pallor, and perturbations in synaptic and dendritic density that may also include selective neuronal loss. The mechanism of HIV-mediated neurologic disorder is not entirely clear, but is likely driven by both direct (active viral replication) and indirect sequelae to HIV invasion of the brain. Indirect mechanisms include dysregulation of glia, release of viral proteins (gp120, and Tat), and elevation of neurotoxic cytokines/chemokines (e.g TNFα, IL-6) and neurotransmitters (e.g glutamate) from resident brain cells and infiltrating immune cells.
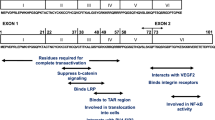
The primary targets for productive HIV replication within the CNS include perivascular (M-2 like) macrophages, microglia, and infiltrating CD4+ T cells. Neurons are not infected by HIV. Astrocytes, which constitute 40–70 % of brain cells and perform vital functions critical for maintenance of blood brain barrier integrity, release of neurotrophic factors, metabolism of toxic neurotransmitters, and immune surveillance by secretion of cytokines/chemokines, are conditionally susceptible to productive HIV replication. β-catenin signaling plays an integral part in regulating HIV in astrocytes. Astrocytes express all four members of the TCF/LEF-1 transcription factor family (TCF-1, TCF-3, TCF-4, and LEF-1) but their relative abundance is probably donor and may even be context-dependent(Narasipura et al. 2012). Primary progenitor-derived astrocytes (PDAs) express higher levels of TCF-3 and LEF1 mRNA than TCF-4 and TCF-1 while the astrocytoma cells lines U87MG and U251 express higher level of TCF-4 (Narasipura et al. 2012). Despite differential expression of TCF/LEF, TCF-4 is a key player in β-catenin-mediated repression of HIV transcription (Wortman et al. 2002; Henderson et al. 2012a; Narasipura et al. 2012). At least four TCF-4 binding sites have been identified within the HIV promoter at −143 to −136 nt; −336 to −329 nt; +66 to +73 nt; and +186 to +195 nt from the transcription initiation site (Henderson et al. 2012a). The +186 site is within the gag leader sequence, just outside of the promoter. The −143 site is of a particular interest. While all sites have >70 % homology to the TCF-4 binding sequence, the −143 site has 100 % homology to the TCF-4 core (5′-(A/T)(A/T)CAAAG-3′) and is present in approximately one-third of 500 HIV LTR sequences reported in the Los Alamos gene bank(Henderson et al. 2012a). TCF-4 binds at a higher affinity at −143 than at any other site (Henderson et al. 2012a). Further, β-catenin is tethered on the HIV LTR at the nt-143 site and knockdown of either TCF-4 or β-catenin enhances HIV transcription (Henderson et al. 2012a; Narasipura et al. 2012). Dual knock down of β-catenin and TCF4 does not further enhance HIV LTR activity, indicating that these factors work together to repress HIV transcription. The effect of mutation and/or deletion of the −143 site on HIV LTR activity was more profound when the HIV LTR is expressed stably than transiently, which suggested possible chromatin involvement in this repression. Indeed, it was found that TCF-4 and β-catenin associate with the nuclear matrix binding protein SMAR1 at −143(Henderson et al. 2012a). SMAR 1 binds matrix attachment regions (MARS) to organize the chromatin into loop domains. SMAR1 is associated with transcriptional repression of HIV by pulling the HIV DNA away from transitional ready complex(Sreenath et al. 2010). The association between TCF-4, β-catenin, and SMAR1 at the −143 site pulls the DNA away from transcriptional machinery and is likely to involve other transcription repressive factors such as Sin-3A or HDACs. TCF-4/β-catenin repression of basal LTR activity likely prevents Tat from reaching a threshold level which would allow it to tether on the TAR region of the LTR in association with a positive elongation complex (pTEFb) to accelerate the rate and efficiency of HIV transcription. As such, by preventing higher level of Tat transcription, β-catenin/TCF-4 may reduce the overall level of Tat in the CNS, which otherwise would promote neuronal excitability and neuroinflammatory processes that play a significant role in HAND.
While studies from our lab have emphasized the role of the −143 site in recruiting TCF-4/β-catenin/SMAR1 to repress basal HIV LTR transcription, the mechanism by which they do so maybe multifaceted. As indicated previously, the HIV promoter contains multiple TCF-4 binding sites and these additional TCF-4 binding sites may cooperate with the −143 site to repress HIV transcription. Further, TCF-4 and β-catenin regulate the expression of other transcription factors relevant to HIV promoter activity. Specifically, both β-catenin and TCF-4 inhibit C/EBP β/δ tethering on the HIV LTR, suggesting that β-catenin and TCF-4 cooperate in this repression (Narasipura et al. 2012). TCF-4, independent of β-catenin, also negatively regulates NFκB tethering on HIV LTR (Narasipura et al. 2012). The requirement for β-catenin to antagonize NFκB activity is context dependent. In astrocytes, NFκB suppression is mediated by TCF-4 without the involvement of β-catenin. In other cells, β-catenin plays a role in suppression of NFκB activity (Deng et al. 2002). TCF-4 suppression of NFκB may be mediated by direct interaction between TCF-4 and NFκB or an indirect effect on upstream regulators of NFκB. Both C/EBP and NFκB are inducers of HIV promoter activity and thus β-catenin/TCF-4 inhibition of these inducers could also contribute to the overall mechanism by which key players in the Wnt/β-catenin pathway repress HIV transcription.
Historically, the association between β-catenin and TCF/LEF is thought to lead to target gene induction. However, there is a significant body of literature that indicates that this association can also lead to gene repression (Hoverter and Waterman 2008). In the case of HIV, the association between β-catenin/TCF-4 is a repressive rather than an inducing signal. LEF-1 enhances HIV transcription, while TCF-4 leads to its repression. One can thus envision a scenario where competition may occur between TCF-4/LEF-1 for β-catenin to regulate HIV expression. In essence, β-catenin/TCF-4 complex joins a list of several transcription factors that repress HIV promoter activity that include YY-1, LSF, p50 homodimer, CBF-1, CTIP2, and c-myc (Kilareski et al. 2009; Coiras et al. 2010). Based on current evidence the model that has emerged of the interaction between β-catenin/TCF-4 and HIV is depicted in Fig. 2.

Mechanism of β-catenin/TCF-4-mediated suppression of basel LTR activity: β-catenin/TCF-4/SMAR form a complex at −143 site of the HIV LTR. This multiprotein complex pulls the HIV DNA spanning this region into nuclear matrix and away from transcription machinery. Inhibition of β-catenin/TCF-4 disrupts this chromatin repression complex and allows for POLII docking and recruitment of transcription co-activators (TCoA) such as NFκB and C/EBP to drive basal LTR activity. Therefore, signals that inhibit β-catenin/TCF-4 signaling are likely to induce HIV transcription by disrupting this inhibitory complex. This image is a reprint (Henderson et al. 2012a), Copyright © American Society for Microbiology, [Journal of Virology, Vol. 86 no. 17 9495–9503, 2012, doi:10.1128/JVI.00486-12]
Tat-mediated transactivation of the HIV LTR is also negatively regulated by β-catenin/TCF-4 (Wortman et al. 2002; Henderson et al. 2012a). However, the mechanism by which Tat transactivation is affected is not dependent on tethering of TCF-4 or β-catenin on the −143 site of the HIV LTR (Henderson et al. 2012a). Rather, Tat overcomes the negative effect of β-catenin/TCF-4 on HIV transcription by inhibiting β-catenin signaling through binding to TCF-4 and sequestering it away from β-catenin (Henderson et al. 2012b). The intact core and cysteine-rich domains of Tat are required for Tat-mediated down regulation of β-catenin signaling (Henderson et al. 2012b). Consequently, a bi-directional negative interference occurs whereby docking of Tat at the TAR region of the HIV LTR is reduced leading to diminished RNA POL II processivity and TCF-4 is less likely to bind to β-catenin diminishing its net effect on HIV target genes. This model of bi-directional inhibition is illustrated in Fig. 3. The model, while based on convincing data, may not completely explain how Tat inhibits β-catenin signaling. Additional mechanisms may be at play. Tat, albeit in neurons, activates GSK3β, a negative regulator of β-catenin that is part of the β-catenin destruction complex, which phosphorylates and tags β-catenin for proteasomal degradation (Maggirwar et al. 1999; Sui et al. 2006). Further, Tat binds to low density lipoprotein (LRP) (Liu et al. 2000; Eugenin et al. 2007), a co-receptor for the Wnt/ β-catenin pathway, and this binding leads to LRP internalization which may sequester it away from Wnt ligands to initiate β-catenin signaling.

Mechanism of Tat/β-catenin interaction in astrocytes. β-catenin and TCF-4 repress basal and Tat transactivation of the HIV LTR through distinct mechanisms and are in turn antagonized by HIV Tat. a Under basal LTR activity (without significant Tat level), the TCF-4/β-catenin/SMAR1 complex represses LTR activity and transcription is low or silent. Low levels of Tat may be produced but are primarily retained in the cytoplasm by association with TCF-4. b When β-catenin signaling is disrupted by pro-inflammatory mediators (IFNγ) or any other signal that down regulates the β-catenin pathway, this complex is disrupted and LTR activity increases. Once the level of Tat reaches a certain threshold, Tat will a) transactivate the HIV LTR by inducing chromatin remodeling and recruiting positive transcription elongation factor b (pTEFb), allowing for efficient viral replication; and b) antagonize β-catenin signaling through mutual binding/inhibition with TCF-4 and enhanced degradation of β-catenin to maintain a permissive state for HIV replication. The broader biological consequences of productive infection in astrocytes include raising the CNS viral load and increasing production of neurotoxic HIV proteins (Tat, gp120, Vpr), potentially leading to neuronal injury and neuroinflammation. The image is a reprint (Henderson et al. 2012b), Copyright © Society of Neuroscience, [Journal of Neuroscience, in press, doi:10.1523/JNEUROSCI.3145-12.2012]
Based on the extensive studies demonstrating β-catenin/TCF-4 mediated repression of HIV replication at the transcription level, β-catenin/TCF-4 has joined a handful of host factors that restrict HIV such as tetherin/BST-2, which prevents virion release, and APOBEC3G, a member of the cytidine deaminases family that introduce mutations into the HIV genome (Sheehy et al. 2002; Neil et al. 2008). Further, the finding that Tat overcomes β-catenin-mediated repression of HIV provides a viral adaptation/escape mechanism for this suppressive effect. Many of the prominent host restriction factors of HIV also have viral products that evade their action. Vpu evades the action of tetherin, Vif evades the action of APOBEC(Malim and Bieniasz 2012), and now we can add Tat evading the action of β-catenin/TCF-4 to this list.
β-catenin signaling is a check point for productive HIV replication in astrocytes
Historically, astrocytes were viewed as restricted to productive HIV replication. HIV enters astrocytes via the human mannose receptor and endocytosis (Liu et al. 2004; Vijaykumar et al. 2008) but robust and productive HIV replication is restricted. Studies that define the role of β-catenin/TCF-4 in restricting HIV replication in astrocytes and those demonstrating that signals that diminish/interrupt β-catenin/TCF-4 signaling lead to robust level of HIV replication suggest that astrocytes are “conditionally permissive” to productive HIV replication, whereby the environmental milieu dictates whether HIV infection is productive or not in astrocytes.
Multiple mechanisms driving restrictive HIV replication in astrocytes have been described. Most notable of which is the low level of Sam68 in astrocytes, which is required for efficient Rev function (Li et al. 2002). Astrocytes also have low expression of the TAR RNA binding protein (TRBP), which antagonizes the action of the interferon-induced double-stranded RNA-regulated protein kinase PKR, an inhibitor of HIV protein translation (Ong et al. 2005). The finding that β-catenin signaling limits HIV in astrocytes does not preclude the involvement of other factors in HIV restriction, especially because β-catenin signaling regulates hundred of genes which conceivably may also intersect with other pathways reported for HIV restriction in astrocytes. By defining the interaction between β-catenin and TCF-4 to restrict HIV transcription, β-catenin/TCF-4 could serve as a check point that dictates the degree of permissiveness to HIV replication in astrocytes. Inflammatory or other exogenous signals that down regulate β-catenin signaling are expected to promote HIV replication in astrocytes. Indeed, IFNγ down regulates β-catenin signaling through inducing an antagonist of the β-catenin pathway, DKK1, in a stat 3 dependent manner (Li et al. 2011). Methamphetamine, a stimulant of choice among HIV-infected individuals who abuse drugs, also inhibits β-catenin signaling (Sharma et al. 2011) and it leads to increased HIV replication in astrocytes (Al-Harthi, unpublished data). As such, negative regulators of the β-catenin pathway are likely to promote HIV replication in target cells.
Evidence for HIV infection of astrocytes in vivo exists. Post-mortem studies from HIV infected individuals demonstrate that astrocytes harbor HIV integrated DNA and that the degree of astrocyte infection, which can reach up to 19 %, is related to CNS disease severity and proximity to perivascular macrophages (Churchill et al. 2009). The association between the frequency of HIV DNA-positive astrocytes and proximity to perivascular macrophages is especially intriguing because under inflammatory conditions, macrophages secret IFNγ (a negative signal for β-catenin) (Munder et al. 1998; Schindler et al. 2001; Carvalho-Pinto et al. 2002). It is thus of interest to determine whether macrophages in the context of inflammatory responses secret IFNγ that drives down regulation of β-catenin signaling and hence higher level of HIV infection within astrocytes. Lastly, lack of significant number of HIV p24+ astrocytes within HIV infected brain is often used as an argument that these cells are not productively infected. Yet, even in the gut, an active source of HIV infection, p24 immunostaining is often negative either due to poor antibody immunoreactivity or perhaps due to much lower gene expression of p24 in comparison to other viral products. We compared HIV gag, env, and rev transcript levels from HIV infected PBMCs, a highly permissive population for HIV infection, and even in these cells HIV p24 mRNA is approximately 8-fold lower than HIV env and rev transcripts. Given the lower level of HIV infection in astrocytes, it is unlikely that p24 is expressed at a level that is above the detection limit in most assays. Measurement of HIV DNA or RNA env content may be a better tool to evaluate HIV infection of astrocytes than conventional immunostaining strategies for HIV p24 in tissue.
Biologic impact of disrupted Wnt/β-catenin signaling in astrocytes
While considerable information exists on the role of Wnts in neuronal synaptic activity and plasticity, little is known about its role in astrocytes. Wnt/β-catenin signaling is robust in astrocytes (Caroll-Anzinger et al. 2007; Li et al. 2011), yet the functional consequence of this strong intact Wnt/β-catenin signal in astrocytes is not entirely clear. Nonetheless, hints at biologic events mediated by β-catenin signaling in astrocytes are beginning to emerge. Transcriptome studies indicate that β-catenin regulates the expression of approximately 150 genes in primary astrocytes. These genes fall under five broad categories: 1) inflammation/immunity; 2) uptake/transport; 3) vesicular transport/exocytosis; 4) apoptosis/cellular stress genes, and 5) cytoskeletal/trafficking (Narasipura et al. 2012). Most notably, knockdown of β-catenin down-regulates mRNA and protein expression of glutamine synthetase (GS), which catalyzes the conversion of the excitatory neurotransmitter glutamate to glutamine. These findings suggest that β-catenin may regulate the glutamate-glutamine cycle. Signals, whether inflammatory or viral mediated, that disrupt β-catenin signaling are likely to not only enhance HIV replication but also to drive inflammatory processes that compromise astrocyte function (e.g glutamate uptake), which will then contribute to a feedback loop of heightened inflammatory processes/dysregulation of astrocytes, as presented in Fig. 4.

Current model of effects and consequences of diminished β-catenin signaling in the CNS. Astrocytes express robust level of β-catenin signaling. Inflammatory signals that diminish β-catenin signaling in astrocytes, leads to enhanced HIV transcription and productive HIV replication in astrocytes. The consequence of these events include higher level of HIV in the CNS, release of cell permeable HIV neurotoxins, heightened overall inflammation, and dysregulation in astrocyte function, most notable of which is inhibition of EAAT2 and glutamate uptake by astrocytes. Dysregulated astrocytes will result in dysregulated cross talk with neurons that will in turn impact key events in neuronal health, including neurogenesis which is partly mediated by Wnt ligands events and synaptogenesis, which are partly mediated by Wnt ligands, culminating in neurodegenerative processes (e.g dendritic pruning and synaptic damage)
As indicated earlier, β-catenin/TCF-4 negatively regulates two key transcription factors involved in neuroinflammation (C/EBP and NFkB). C/EBPβ and C/EBPδ are linked to heightened neuroinflammation in HAND through increased production of inflammatory mediators by astrocytes such as IL-6, IL-1β and TNF-α (Poli 1998). NFκB drives the expression of several neuroinflammatory cytokines (e.g IL-1, IL-6) and in excess these cytokines are linked to neuropathology. C/EBPs and NFκB also synergize in mediating inflammatory processes. C/EBPs can form heterodimers with NFκB subunits to activate target genes including the promoter of HIV and the promoter of cytokines such as IL-6 and the chemokine IL-8 (Ruocco et al. 1996; Poli 1998). As such, signals that diminish β-catenin and/or TCF-4 mediated negative regulation of C/EBP and NFκB will likely drive robust production of pro-inflammatory cytokines and chemokines to recruit immune cells into the CNS, contributing to glial activation and neuronal injury.
Remaining questions
Significant progress has been made to define the relationship between β-catenin signaling and HIV at the molecular level and to begin to understand the effect of signals that diminish β-catenin signaling in astrocytes on HIV-mediated neuropathogenesis. Several questions, which are categorized under four themes, still remain. These highlighted themes include deciphering the role of Wnt/β-catenin in: 1) HIV-mediated neuropathogenic processes (e.g inflammation, interplay between astrocytes and neurons); 2) HIV and aging processes, 3) HIV evolution and compartmentalization in the CNS, and 4) HIV and drug abuse. Under these four categories, pertinent questions include: 1) Does the integrity of β-catenin signaling correlate with various degrees of HIV associated neurocognitive disorders (HAND)? Perturbed β-catenin signaling is associated with a number of neurodegenerative and psychiatric diseases such as Alzheimer’s, bipolar disorder, schizophrenia, and even emerging data in autism. HAND exhibits several key pathologic features of neurodegenerative diseases such as glial activation and neuronal pruning/apoptosis. In vitro, as highlighted here, some inflammatory signals diminish β-catenin signaling in astrocytes (Caroll-Anzinger et al. 2007; Li et al. 2011). Given that β-catenin is a cellular protein, post-mortem studies can probe the relationship between β-catenin signaling and HAND. However, it is the Wnt ligands, which are secreted glycoproteins, rather than intracellular β-catenin, that may potentially be a viable biomarker for CNS integrity. To this end, much remains to be revealed. Specifically, 2) Which of the 19 Wnt ligands are secreted by astrocytes, how do they signal in astrocytes, does HIV perturb their production and/or function, and how does their expression profile relate to overall inflammatory process in the CNS in context of HIV? 3) Can β-catenin signaling be harnessed in HIV therapy? Although current drugs used in antiretroviral therapy (ART) successfully target components of the viral life cycle, such as fusion, reverse transcription, and viral protein processing, viral mutations continue to diminish the effectiveness of these drugs. Thus, new therapeutic approaches are needed. Inducing β-catenin signaling in HIV target cells can represent a novel pathway to treat drug-resistant HIV. Further, because drug intensification alone has not been able to alter the size of the latent HIV reservoir pool (Dinoso et al. 2009), strategies to purge the latent reservoir are needed. Suppressing β-catenin in latently infected cells can reactivate HIV to become susceptible to cART. Nonetheless, these reactivation strategies should be viewed with caution. Virus reactivation in the CNS, even if transient, may have long-term negative effects in the CNS by establishing inflammatory responses that are neurotoxic. Theoretically, one can envision that suppressing β-catenin in HIV infected cells can be used in HIV purging strategies, while activating β-catenin can be used to treat patients harboring drug-resistant HIV. If so, 4) What are the ideal tools to manipulate β-catenin signaling (either an up or a downward signal) in the CNS? A number of small molecules have been described that modulate β-catenin signaling (Miyabayashi et al. 2007; Chen et al. 2009). However, these small molecules may have a different effect in the CNS, as β-catenin signaling is context dependent. In fact, some small molecules defined as repressor of β-catenin/TCF-4 interaction in colon cancer cells have the opposite effect in astrocytes (Al-Harthi unpublished data). This highlights the challenge in harnessing β-catenin signaling for HIV therapy as the aim is to target it to specific cells. 5) How does β-catenin-mediated perturbation of astrocyte protein signature impact key functions of astrocytes (e.g release of neurotrophic factors, glutamate uptake, etc) and how does HIV in turn impact astrocyte/neuron interaction through Wnt ligands? 6) What is the role of β-catenin/TCF-4 in driving HIV evolution/compartmentalization in the CNS? There is clear evidence for genetic evolution of HIV compartmentalization in the brain that is different from that of lymphoid tissue (Holman et al. 2010) (Ellis et al. 2000) (Harrington et al. 2009; Schnell et al. 2010; Schnell et al. 2011). The factors that drive this evolution are not clear. TCF-4 sites are found in 1/3 of HIV isolates evaluated (Henderson et al. 2012a). Whether these isolates would be preferentially compartmentalized within astrocytes in particular and in the CNS in general is also not clear. 7) What is the effect of the aging HIV-infected brain on Wnt signaling? Wnt activation is linked to either delayed or accelerated aging (DeCarolis et al. 2008). Much of this uncertainty about the effect of Wnt on aging probably stems from reports using different cell types and model systems and the realization that Wnt effects are context dependent. The integrity of the Wnt pathway in the CNS as a function of age is not clear and neither are the effects of HIV on this process. In other words, is Wnt activity diminished as the brain ages? How does HIV affect Wnt in a younger vs. older brain? 8) What is the effect of drug abuse and HIV on Wnt signaling? Substance abuse is a major debilitating co-morbidity in the HIV/AIDS population. HIV neuropathogenesis is more severe in those who abuse drugs than those who do not (Ferris et al. 2008; Nath 2010; Purohit et al. 2011; Hauser et al. 2012). Methamphetamine (Meth), in particular, is a frequently abused psychostimulant that is neurotoxic to dopaminergic regions in the brain, which are also negatively impacted by HIV. Meth inhibits β-catenin signaling (Sharma et al. 2011). Whether β-catenin signaling is a mechanism whereby Meth and HIV interface leading to enhanced pathogenesis in the CNS is not clear. Addressing some of these questions will propel the field forward and further highlight a significant role for β-catenin signaling in HIV disease and homeostasis of the CNS.
References
Caroll-Anzinger D, Kumar A, Adarichev V, Kashanchi F, Al-Harthi L (2007) HIV restricted replication in astrocytes and the ability of IFNg to modulate this restriction is regulated by a downstream effector of the Wnt signaling pathway. J Virol 81:5864–5871
Carvalho-Pinto CE, Garcia MI, Mellado M, Rodriguez-Frade JM, Martin-Caballero J, Flores J, Martinez AC, Balomenos D (2002) Autocrine production of IFN-gamma by macrophages controls their recruitment to kidney and the development of glomerulonephritis in MRL/lpr mice. J Immunol 169:1058–1067
Chen B, Dodge ME, Tang W, Lu J, Ma Z, Fan CW, Wei S, Hao W, Kilgore J, Williams NS, Roth MG, Amatruda JF, Chen C, Lum L (2009) Small molecule-mediated disruption of Wnt-dependent signaling in tissue regeneration and cancer. Nat Chem Biol 5:100–107
Churchill MJ, Wesselingh SL, Cowley D, Pardo CA, McArthur JC, Brew BJ, Gorry PR (2009) Extensive astrocyte infection is prominent in human immunodeficiency virus-associated dementia. Ann Neurol 66:253–258
Coiras M, Lopez-Huertas MR, Sanchez del Cojo M, Mateos E, Alcami J (2010) Dual role of host cell factors in HIV-1 replication: restriction and enhancement of the viral cycle. AIDS Rev 12:103–112
DeCarolis NA, Wharton KA Jr, Eisch AJ (2008) Which way does the Wnt blow? Exploring the duality of canonical Wnt signaling on cellular aging. Bioessays 30:102–106
Deng J, Miller SA, Wang HY, Xia W, Wen Y, Zhou BP, Li Y, Lin SY, Hung MC (2002) Beta-catenin interacts with and inhibits NF-kappa B in human colon and breast cancer. Cancer Cell 2:323–334
Dinoso JB, Kim SY, Wiegand AM, Palmer SE, Gange SJ, Cranmer L, O’Shea A, Callender M, Spivak A, Brennan T, Kearney MF, Proschan MA, Mican JM, Rehm CA, Coffin JM, Mellors JW, Siliciano RF, Maldarelli F (2009) Treatment intensification does not reduce residual HIV-1 viremia in patients on highly active antiretroviral therapy. Proc Natl Acad Sci U S A 106:9403–9408
Ellis RJ, Gamst AC, Capparelli E, Spector SA, Hsia K, Wolfson T, Abramson I, Grant I, McCutchan JA (2000) Cerebrospinal fluid HIV RNA originates from both local CNS and systemic sources. Neurology 54:927–936
Eugenin EA, King JE, Nath A, Calderon TM, Zukin RS, Bennett MV, Berman JW (2007) HIV-tat induces formation of an LRP-PSD-95- NMDAR-nNOS complex that promotes apoptosis in neurons and astrocytes. Proc Natl Acad Sci U S A 104:3438–3443
Ferris MJ, Mactutus CF, Booze RM (2008) Neurotoxic profiles of HIV, psychostimulant drugs of abuse, and their concerted effect on the brain: current status of dopamine system vulnerability in NeuroAIDS. Neurosci Biobehav Rev 32:883–909
Harrington PR, Schnell G, Letendre SL, Ritola K, Robertson K, Hall C, Burch CL, Jabara CB, Moore DT, Ellis RJ, Price RW, Swanstrom R (2009) Cross-sectional characterization of HIV-1 env compartmentalization in cerebrospinal fluid over the full disease course. AIDS 23:907–915
Hauser KF, Fitting S, Dever SM, Podhaizer EM, Knapp PE (2012) Opiate drug use and the pathophysiology of NeuroAIDS. Curr HIV Res 10:435–452
Heaton RK, Franklin DR, Ellis RJ, McCutchan JA, Letendre SL, Leblanc S, Corkran SH, Duarte NA, Clifford DB, Woods SP, Collier AC, Marra CM, Morgello S, Mindt MR, Taylor MJ, Marcotte TD, Atkinson JH, Wolfson T, Gelman BB, McArthur JC, Simpson DM, Abramson I, Gamst A, Fennema-Notestine C, Jernigan TL, Wong J, Grant I (2011) HIV-associated neurocognitive disorders before and during the era of combination antiretroviral therapy: differences in rates, nature, and predictors. J Neurovirol 17:3–16
Hedgepeth CM, Conrad LJ, Zhang J, Huang HC, Lee VM, Klein PS (1997) Activation of the Wnt signaling pathway: a molecular mechanism for lithium action. Dev Biol 185:82–91
Henderson LJ, Al-Harthi L (2011) Role of beta-catenin/TCF-4 signaling in HIV replication and pathogenesis: insights to informing novel anti-HIV molecular therapeutics. J Neuroimmune Pharmacol 6:247–259
Henderson LJ, Narasipura SD, Adarichev V, Kashanchi F, Al-Harthi L (2012a) Identification of novel T cell factor 4 (TCF-4) binding sites on the HIV long terminal repeat which associate with TCF-4, beta-catenin, and SMAR1 to repress HIV transcription. J Virol 86:9495–9503
Henderson LJ, Sharma A, Monaco-Kushner MC, Major EO, Al-Harthi L (2012b) HIV Tat through its intact core and cysteine-rich domains inhibits Wnt/beta-catenin signaling in astrocytes: Relevance to HIV neuropathogenesis. J Neurosci in press
Holman AG, Mefford ME, O’Connor N, Gabuzda D (2010) HIVBrainSeqDB: a database of annotated HIV envelope sequences from brain and other anatomical sites. AIDS Res Ther 7:43
Hoverter NP, Waterman ML (2008) A Wnt-fall for gene regulation: repression. Sci Signal 1:pe43
Kilareski EM, Shah S, Nonnemacher MR, Wigdahl B (2009) Regulation of HIV-1 transcription in cells of the monocyte-macrophage lineage. Retrovirology 6:118
Kumar A, Zloza A, Moon RT, Watts J, Tenorio AR, Al-Harthi L (2008) Active {beta}-catenin signaling is an inhibitory pathway of HIV replication in peripheral blood mononuclear cells. J Virol 82:2813–2820
Li J, Liu Y, Park IW, He JJ (2002) Expression of exogenous Sam68, the 68-kilodalton SRC-associated protein in mitosis, is able to alleviate impaired Rev function in astrocytes. J Virol 76:4526–4535
Li W, Henderson LJ, Major EO, Al-Harthi L (2011) IFN-{gamma} mediates enhancement of HIV replication in astrocytes by inducing an antagonist of the {beta}-catenin pathway (DKK1) in a STAT 3-dependent manner. J Immunol 186:6771–6778
Liu Y, Jones M, Hingtgen CM, Bu G, Laribee N, Tanzi RE, Moir RD, Nath A, He JJ (2000) Uptake of HIV-1 tat protein mediated by low-density lipoprotein receptor-related protein disrupts the neuronal metabolic balance of the receptor ligands. Nat Med 6:1380–1387
Liu Y, Liu H, Kim BO, Gattone VH, Li J, Nath A, Blum J, He JJ (2004) CD4-independent infection of astrocytes by human immunodeficiency virus type 1: requirement for the human mannose receptor. J Virol 78:4120–4133
Maggirwar SB, Tong N, Ramirez S, Gelbard HA, Dewhurst S (1999) HIV-1 Tat-mediated activation of glycogen synthase kinase-3beta contributes to Tat-mediated neurotoxicity. J Neurochem 73:578–586
Malim MH, Bieniasz PD (2012) HIV restriction factors and mechanisms of evasion. Cold Spring Harb Perspect Med 2:a006940
Miyabayashi T, Teo JL, Yamamoto M, McMillan M, Nguyen C, Kahn M (2007) Wnt/beta-catenin/CBP signaling maintains long-term murine embryonic stem cell pluripotency. Proc Natl Acad Sci U S A 104:5668–5673
Munder M, Mallo M, Eichmann K, Modolell M (1998) Murine macrophages secrete interferon gamma upon combined stimulation with interleukin (IL)-12 and IL-18: A novel pathway of autocrine macrophage activation. J Exp Med 187:2103–2108
Narasipura SD, Henderson LJ, Fu SW, Chen L, Kashanchi F, Al-Harthi L (2012) Role of beta-catenin and TCF/LEF family members in transcriptional activity of HIV in astrocytes. J Virol 86:1911–1921
Nath A (2010) Human immunodeficiency virus-associated neurocognitive disorder: pathophysiology in relation to drug addiction. Ann N Y Acad Sci 1187:122–128
Neil SJ, Zang T, Bieniasz PD (2008) Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 451:425–430
Ong CL, Thorpe JC, Gorry PR, Bannwarth S, Jaworowski A, Howard JL, Chung S, Campbell S, Christensen HS, Clerzius G, Mouland AJ, Gatignol A, Purcell DF (2005) Low TRBP levels support an innate human immunodeficiency virus type 1 resistance in astrocytes by enhancing the PKR antiviral response. J Virol 79:12763–12772
Poli V (1998) The role of C/EBP isoforms in the control of inflammatory and native immunity functions. J Biol Chem 273:29279–29282
Purohit V, Rapaka R, Shurtleff D (2011) Drugs of abuse, dopamine, and HIV-associated neurocognitive disorders/HIV-associated dementia. Mol Neurobiol 44:102–110
Ruocco MR, Chen X, Ambrosino C, Dragonetti E, Liu W, Mallardo M, De Falco G, Palmieri C, Franzoso G, Quinto I, Venuta S, Scala G (1996) Regulation of HIV-1 long terminal repeats by interaction of C/EBP(NF-IL6) and NF-kappaB/Rel transcription factors. J Biol Chem 271:22479–22486
Schindler H, Lutz MB, Rollinghoff M, Bogdan C (2001) The production of IFN-gamma by IL-12/IL-18-activated macrophages requires STAT4 signaling and is inhibited by IL-4. J Immunol 166:3075–3082
Schnell G, Price RW, Swanstrom R, Spudich S (2010) Compartmentalization and clonal amplification of HIV-1 variants in the cerebrospinal fluid during primary infection. J Virol 84:2395–2407
Schnell G, Joseph S, Spudich S, Price RW, Swanstrom R (2011) HIV-1 replication in the central nervous system occurs in two distinct cell types. PLoS Pathog 7:e1002286
Sharma A, Hu XT, Napier TC, Al-Harthi L (2011) Methamphetamine and HIV-1 Tat down regulate beta-catenin signaling: implications for methampetamine abuse and HIV-1 co-morbidity. J Neuroimmune Pharmacol 6:597–607
Sheehy AM, Gaddis NC, Choi JD, Malim MH (2002) Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 418:646–650
Sreenath K, Pavithra L, Singh S, Sinha S, Dash PK, Siddappa NB, Ranga U, Mitra D, Chattopadhyay S (2010) Nuclear matrix protein SMAR1 represses HIV-1 LTR mediated transcription through chromatin remodeling. Virology 400:76–85
Sui Z, Sniderhan LF, Fan S, Kazmierczak K, Reisinger E, Kovacs AD, Potash MJ, Dewhurst S, Gelbard HA, Maggirwar SB (2006) Human immunodeficiency virus-encoded Tat activates glycogen synthase kinase-3beta to antagonize nuclear factor-kappaB survival pathway in neurons. Eur J Neurosci 23:2623–2634
Vijaykumar TS, Nath A, Chauhan A (2008) Chloroquine mediated molecular tuning of astrocytes for enhanced permissiveness to HIV infection. Virology 381:1–5
Williams RS, Harwood AJ (2000) Lithium therapy and signal transduction. Trends Pharmacol Sci 21:61–64
Wortman B, Darbinian N, Sawaya BE, Khalili K, Amini S (2002) Evidence for regulation of long terminal repeat transcription by Wnt transcription factor TCF-4 in human astrocytic cells. J Virol 76:11159–11165
Acknowledgment
I thank colleagues in the Wnt signaling field that have paved the way and discovered key tools to benefit the greater Wnt field community. I also thank present and past members of the Al-Harthi lab who propelled these studies forward (Lisa J. Henderson, Srinivasa Narasipura, Maureen Richards, Deborah Carrol-Anzinger; Anvita Kumar, and Wei Li). This work was supported by the following grants from the National Institutes of Health: R01 NS060632, R03 DA 026723, R01 DA 033966, and PO1A1082971.
Conflict of interest
The author declares no conflict of interest
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Al-Harthi, L. Interplay Between Wnt/β-Catenin Signaling and HIV: Virologic and Biologic Consequences in the CNS. J Neuroimmune Pharmacol 7, 731–739 (2012). https://doi.org/10.1007/s11481-012-9411-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11481-012-9411-y