
Abstract
Human immunodeficiency virus type 1 (HIV-1) transactivator of transcription (Tat) is a potent mediator involved in the development of HIV-1-associated neurocognitive disorders (HAND). Tat is expressed even in the presence of antiretroviral therapy (ART) and is able to enter the central nervous system (CNS) through a variety of ways, where Tat can interact with microglia, astrocytes, brain microvascular endothelial cells, and neurons. The presence of low concentrations of extracellular Tat alone has been shown to lead to dysregulated gene expression, chronic cell activation, inflammation, neurotoxicity, and structural damage in the brain. The reported effects of Tat are dependent in part on the specific HIV-1 subtype and amino acid length of Tat used. HIV-1 subtype B Tat is the most common subtype in North American and therefore, most studies have been focused on subtype B Tat; however, studies have shown many genetic, biologic, and pathologic differences between HIV subtype B and subtype C Tat. This review will focus primarily on subtype B Tat where the full-length protein is 101 amino acids, but will also consider variants of Tat, such as Tat 72 and Tat 86, that have been reported to exhibit a number of distinctive activities with respect to mediating CNS damage and neurotoxicity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
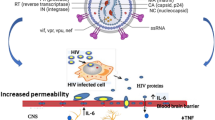
HIV-1 infection is known to involve the seeding of latent viral reservoirs at very early stages of primary infection. Subsequent treatment of infected individuals with life-long suppressive antiretroviral (ART) therapy has been shown to reduce viral burden in the peripheral blood and minimize risk of viral reactivation and possibly expansion of the latent reservoirs. Reactivation of latent reservoirs has been shown in notable cases such as the “Boston patients,” the “Mississippi baby,” and the VISCONTI Cohort in France, which reported viral rebounds after discontinuation of suppressive ART [1,2,3,4]. One of the major reservoirs for HIV-1 has been shown to involve the breach of the blood–brain barrier (BBB), which results in the establishment of a reservoir in the central nervous system (CNS) within hours to days after infection, in most cases before ART has been first administered [5,6,7,8]. Once HIV-1 has entered the CNS, it can result in a range of neurologic impairments identified as HIV-1-associated neurocognitive disorders (HAND), which is further categorized into levels of increasing impairment that interferes with daily living, and includes asymptomatic neurocognitive impairment (ANI), mild neurocognitive impairment (MND), and in its severest form, HIV-1-associated dementia (HAD) [9,10,11]. Patients with HAND have a wide range of clinical symptoms, such as cognitive, behavioral and motor deficits that can improve or worsen over the course of infection, making it difficult to incrementally track progression [5, 10]. Despite the success of ART in reducing viral loads, the prevalence of HAND has been on the increase and has been observed in 50–70% of HIV-1-infected individuals [5, 12]. The major mechanism proposed for how HIV-1 enters the CNS is the “Trojan Horse” model where HIV-1-infected immune cells, such as monocytes and CD4 + T cells cross the BBB, and where they can then infect cells of the CNS and propagate infection [13]. HIV-1-infected patients on ART who have been shown to be clinically avirmeric have also been shown to have detectable levels of the HIV-1 viral protein Tat, or the transactivator of transcription, in the CNS that contributes to CNS insult including inflammation and tissue damage and may lead to the development of HAND [5,10,12,14–26].
Tat is the first viral protein to be transcribed and translated from integrated HIV-1 provirus and the primary role of Tat is to recognize the 5′ TAR element in the HIV-1 RNA and recruit the host elongation factor p-TEFb, which increases the rate of viral transcription up to 100-fold, as compared to normal rates of transcription [11, 14, 15, 27,28,29,30,31]. Tat is a flexible and intrinsically disordered protein, allowing it to form high-affinity complexes with a number of cellular partners [27, 32]. Full-length Tat is 101 amino acids and is encoded by two separate exons, where exon one encodes the first 72 amino acids; while the second exon encodes the C-terminal domain [11, 27]. The functional domains of Tat often have overlapping functions, but include proline-rich, cysteine-rich, hydrophobic core, arginine-rich and glutamine-rich domains [11, 33, 34]. Together, the arginine-rich and glutamine-rich domains make up the basic region which is involved in cell entry and exit [33, 35]. The second exon encodes the C-terminus, including the RGD and ESKKKVE motifs, which are involved in immune cell activation and binding to cell-surface integrins (Fig. 1) [33, 36, 37]. The basic domain of Tat enables it to interact with heparin sulfate proteoglycans (HSPGs), located on many cells to promote Tat uptake into cells [36]. Additionally, the RGD motif in Tat allows it to interact with integrin receptors, creating another broad method for uptake into cells [36]. Extracellular Tat has been detected in the CSF at concentrations ranging from 5 to 35 ng/ml in patients on ART with undetectable HIV-1 viral load in the blood and CSF [39]. In a more recent paper, extracellular Tat was detected in the CSF at variable concentrations in one-third of patients who were well suppressed on ART and were clinically aviremic [38]. In this study, Tat was detected in the CSF of well-suppressed patients on ART at concentrations ranging from 200 pg/ml to 6.5 ng/ml; however, the concentration of detectable Tat was shown to fluctuate over time in the same patient [38]. Additionally, there may be patients who had extracellular Tat in the CSF; however, the level of Tat may be below the level of detection, because not all patients who had impairment had detectable Tat [38]. Together, these studies show that even with ART suppression of the viral load, Tat is still being made and is detectable in the CSF.

Motifs and associated functions of Tat 72, 86 and 101. Amino acids 1–72 are associated with the majority of Tat functions including binding to TAR, transcriptional activation, and cellular toxicity. The RGD motif, present in both Tat 86 and Tat 101, has been shown to interact with VEGF2 and integrin receptors. The ESKKKVE motif, only found in Tat 101, has been shown to be involved in Tat-mediated NF-κB activity
This review will describe the functional effects that each Tat length has been shown to have on different cells of the CNS. With regards to structure, all of the Tat variants (72, 86 and 101) have the major core region intact that is responsible for binding to the TAR region and transactivation of the LTR [27]. Additionally, amino acids 31–61 have been shown to primarily be related to toxicity-associated functions, and all three Tat length variants have been shown to cause toxicity in multiple cell types [40]. However, Tat 86 and Tat 101 both contain the RGD motif, that Tat 72 lacks and this motif interacts with VEGF2 and binds integrin receptors [36]. The RGD motif on Tat 86 and Tat 101 may help Tat bind integrin receptors on immune cells and BMECs at the BBB and improve Tat transmigration into the CNS [36]. Due to this effect, the presence of Tat 86 and Tat 101 in the periphery alone can lead to increased concentrations of Tat in the CNS, resulting in toxicity of cells in the CNS exposed to Tat and increased likelihood of the development of neurocognitive deficits. Finally, Tat 101 is the only variant to have an intact ESKKKVE motif which has been shown to be involved in NF-κB activity and can result in a cascade of further activation in these cells leading to increased production of pro-inflammatory cytokines in affected cells (Fig. 1) [37].
Tat has been shown to have multiple functions on cells throughout the CNS. The alterations observed in these cells have been dependent in part on the specific types of cells or tissues exposed, the input concentration, the length of exposure, the subtype, or amino acid length of Tat used, as well as whether the cells were exposed to intracellular or extracellular Tat. Tat subtypes have been derived from the HIV-1 classes and subtypes found throughout the world. Globally, HIV-1 has been divided into three classes: group M (major), group O (outlier) and group N (new, non-M, non-O); with group M shown to be responsible for more than 90% of HIV-1 infections worldwide. Group M has been divided into the 9 subtypes; A–D, F–H, J and K [41, 42]. In the United States, subtype B has been the predominant subtype; while subtypes A, C, and D are predominant in sub-Saharan Africa [41,42,43,44]. Although the 101 amino acid form of subtype B Tat is the predominant form found in HIV-1-infected patients, several less prevalent truncated forms also exist, including Tat 72, and Tat 86, which have been used extensively over the past three decades in both in vitro and in vivo animal model studies [10, 45].
In this review, we discuss the effect of extracellular and intracellular Tat on cells of the CNS. We define intracellular Tat as cells that have been transfected with Tat plasmid or cells that are expressing Tat. We also define extracellular Tat as recombinant or purified Tat added to media or injected into animal models. We have reported the method of Tat delivery as well as Tat length as listed in the original literature; however, these details have not been reported in all papers. Additionally, due to the ability of Tat to easily penetrate cells as well as its high rates of secretion from cells, it is difficult to isolate and identify the effects of intracellular versus extracellular Tat alone in all cases. Although the method of Tat exposure has been highlighted in this review, the physiological relevance of extracellular versus intracellular effects of Tat remain to be elucidated.
Oligodendrocytes
Oligodendrocytes are the myelin-forming cells in the CNS that sheath neuronal axons in the white matter of the brain [46]. Although oligodendrocytes do not express CD4 and are unlikely to be directly infected with HIV-1, oligodendrocytes have still been shown to be indirectly affected by HIV-1 and in post-mortem HIV-1 tissues oligodendrocytes have activated cell death signaling [47]. HIV-1 Tat has been shown to mediate many indirect effects on cells of the CNS including oligodendrocytes where Tat exposure on these cells has been shown to decrease immature oligodendrocyte viability [48]. As oligodendrocytes are the major myelin-forming cells in the CNS, it has been shown that Tat exposure to ex vivo rat brain slices resulted in myelin damage and decreased myelin basic protein expression in the corpus callosum and striatum [46]. Additionally, Tat exposure can also inhibit immature oligodendrocyte differentiation by reducing the formation of fine branching processes essential for mature functions [49]. In a study of immature oligodendrocytes exposed to 100 nM of Tat 86 for 18 h, there was observed decreased cell viability and impaired differentiation characterized by reduced processes and impaired branching (Fig. 2) [48, 49]. Furthermore, the genes Ugt8 and Cnp, which are essential for oligodendrocyte differentiation, have also been shown to be down regulated in immature oligodendrocytes exposed to Tat 86 [49]. A possible mechanism of Tat-impaired differentiation in oligodendrocytes is through autotaxin (ATX) inhibition [49]. ATX is an extracellular protein that has been shown to play a significant role in oligodendrocyte development, but in studies of Tat 86 exposure on oligodendrocytes, it was shown that Tat is capable of entering the cytoplasm of oligodendrocyte, bind to ATX, and inhibit its secretion [49]. Tat has also been shown to cause decreased viability of oligodendrocytes and in the doxycycline-inducible Tat transgenic mice, the expression of Tat for 7 days resulted in increased caspase-3 expression in oligodendrocytes leading to cell death [50]. A possible mechanism of the activation of cell death pathways is through the alteration of K + and Ca + 2 levels within the cells. Tat exposure on oligodendrocytes was shown to activate the voltage-gated K + channels in oligodendrocytes, leading to outward K + current, and cell death [46]. Another study showed that Tat exposure on primary rat oligodendrocyte cultures caused Ca + 2 influx into the cytoplasm resulting in glycogen synthase kinase 3b (GSK3b) and Ca + 2/Calmodulin-dependent kinase IIb (CAMKIIb) activation, where in immature oligodendrocytes, the increase in GSK3b is sufficient to the activate cell death signaling [47]. Overall, exposure of oligodendrocytes to Tat results in multiple mechanisms that reduce differentiation, enhance cell death, and contribute to HIV-1 neuropathogenesis.

Summary of effects of Tat length variants on the cells of the CNS. Divided into Tat 101, Tat 86 and Tat 72 and subdivided by the functional effects each Tat variant has been shown to cause on different cells of the CNS. Question marks indicate current gaps in the literature
Pericytes
Pericytes are important to blood–brain barrier integrity as they surround endothelial cells to help stabilize capillaries and regulate BBB permeability [51, 52]. Pericytes also have roles in the formation and maintenance of the BBB during brain development [51, 52]. However, due to their expression of CD4, CXCR4, and CCR5, pericytes have also been shown to support low infection rates of HIV-1 [51, 52]. In HIV-1-infected patients, pericytes have been shown to be dysfunctional and this may be relevant to the development of cognitive impairment as dysfunctional pericytes have been identified in other diseases such as multiple sclerosis, aging, brain tumors, and traumatic brain injuries [52]. When exposed to Tat 101, pericytes had activation of the expression of platelet-derived growth factor-b (PDGF-B) and this led to activation of the ERK and JNK pathways, which then led to downstream activation NF-κB, which is involved in production of pro-inflammatory cytokines (Fig. 2) [51]. Additionally, the activation of PDGF-B in pericytes that were exposed to Tat 101 resulted in other downstream effects such as pericyte migration and loss of micro-vessels in the brain where this may affect BBB permeability [51].
Monocytes, macrophages, and microglia
Although CD4 + T cells are the primary target of HIV-1 infection, they are likely not the primary cells capable of trafficking HIV-1 into the CNS. Monocytes and macrophages are susceptible and permissive to infection by HIV-1 and can cross the BBB to enter into the CNS [53, 54]. Cells of the monocyte–macrophage lineage can become infected with HIV-1 based on their surface accumulation of the necessary receptor CD4 and one or more coreceptor proteins including CXCR4 and CCR5 [55]. After these cells are infected in the periphery, they will cross the BBB to enter into the CNS, and interestingly, cross the BBB at a heightened rate compared to uninfected monocytes [56]. Once within the CNS, the infected cells may shed virions that infect tissue-resident cells of the monocyte–macrophage lineage including microglial cells [57]. These differentiated cells play a crucial role in the development of HAND and, once infected, act as viral reservoirs within the CNS [57].
Exposure to recombinant extracellular Tat 86 has been shown to cause an increase in the expression of CCR5 in primary human monocytes (Fig. 2) [58]. Increasing the expression of CCR5 may increase the susceptibility of uninfected bystander cells of the monocyte–macrophage lineage, which could directly affect the pathogenesis of the disease. Furthermore, interaction of CD40 ligand (CD40L) on the surface of T cells, and CD40, on the surface of monocytes and macrophages, has been shown to be crucial for antiviral immunity (Fig. 2) [59]. CD40L-deficient mice are severely immunocompromised and are unable to proliferate or induce isotype switching [60]. Primary human monocytes exposed to Tat 101 had increased expression of CD40 that resulted in increased expression of TNF-α in monocytes ex vivo [61]. Additionally, the conditioned media from monocytes exposed to both CD40L and Tat 101 elicited higher levels of neurotoxicity when compared to those exposed to either recombinant protein alone. However, when TNF-α was depleted using a neutralizing antibody from the conditioned media, neurotoxicity was ablated demonstrating the effect was dependent on this soluble inflammatory mediator [61].
In patients with HAND, increased monocyte chemoattractant protein-1 (CCL2/MCP-1), C–C motif chemokine ligand 5 (CCL5), and C-X-C motif chemokine ligand 10 (CXCL10) were detected in the brain and CSF [54]. The release of chemoattractants and pro-inflammatory cytokines helps recruit HIV-1-susceptible cells to the CNS and exacerbates disease progression [58, 62]. In a study of primary mouse microglial cells, exposure to extracellular Tat 101 was found to upregulate the novel miRNA-34a, which then downregulated the NLRC5 inflammasome to negatively regulate NF-κB p65 signaling, leading to an upregulation of the pro-inflammatory cytokines, Interleukin-1β (IL-1β) and IL-6 and microglial activation [63]. Additionally, injection of Tat 72 into the hippocampus of C57BL/6 mice resulted in increased monocyte transmigration across the BBB that was mediated by increased expression of MCP-1, a potent chemoattractant that induces monocytes to cross the BBB [64]. In cells of the monocyte–macrophage lineage, most pathways altered by extracellular Tat are pro-inflammatory and induce the expression of cytokines through signal cascades, such as TNF-α and IL-1β, which may result in neuronal death by processes such as increased reactive oxygen or nitric species [65].
Another cytokine altered by exposure to Tat has been TNF-related apoptosis-induced ligand (TRAIL), a member of the superfamily of tumor necrosis factor that has been shown to mediate apoptosis [62]. This process has been shown to involve TRAIL binding to death receptors DR4 (TRAIL-RI) and DR5 (TRAIL-RII), leading to the activation of caspases, though TRAIL has also been shown to bind to alternative receptors that lead to NF-κB activation, promoting inflammation rather than apoptosis [66]. Primary human macrophages exposed to HIV-1 Tat B were demonstrated to upregulate the expression of TRAIL [67]. This was initially reported as more macrophages producing TRAIL than there were cells infected with HIV-1, which may have been caused by interactions with extracellular Tat that was secreted by the infected cells, or perhaps due to secretion from bystander cells [67]. This mechanism was examined further in monocytes where exposure to Tat induced the expression of TRAIL, which directly caused the death of bystander CD4 + T cells [62]. This may be a mechanism for HIV-1 to alter tropism by removing specific immune cell populations or to evade an immune response by mediating the population of CD4 + T cells. Alterations in TRAIL expression have been observed in cell culture as well; U-937 cells, a monocytic cell line, were exposed to Tat 86, which resulted in increased expression of TRAIL [68]. Additionally, individual peptides representing distinct regions of Tat were exposed to the cells and it was determined that the cysteine-rich region (residues 16–35) were responsible for the induction of TRAIL [68].
Alternatively, Tat has been demonstrated to enhance the expression of anti-apoptotic factors, such as Bcl-2, which has prevented the activation of caspase-8 (Fig. 2) [58]. This mechanism has been relevant to the viral reservoir within the CNS, as monocytes and macrophages which are infected with HIV-1 were notably more resilient to virus-mediated apoptosis [62, 67]. Primary human monocytes exposed to Tat 86 demonstrated an increase in the expression of Bcl-2 [58]. Tat 86 reduced the apoptosis caused by TRAIL by 36%, from 51 to 15%, demonstrating that this mechanism may contribute to the virus-resistant phenotype observed in HIV-1-infected monocytes and macrophages (Fig. 2) [58].
The signaling cascade of TRAIL has been shown to involve the activation of caspase-8, which leads to the release of calcium (Ca+2) from the mitochondria, and expression of Bcl-2 directly inhibits this pathway [58]. Exposure to HIV-1 Tat has been shown to increase cytosolic Ca+2, leading to the activation, dysregulation, and/or apoptosis of cells of the monocytic lineage [69, 70]. Furthermore, this alteration in Ca+2 has been demonstrated to be correlated with TNF-α production [71]. Therefore, due to the expression of Bcl-2, virus-resistant long-lived cells of the monocyte–macrophage lineage can recruit additional susceptible cells to the CNS by producing of TNF-α [65]. Additionally, a study of mouse primary microglial cells exposed to extracellular Tat 101 had impaired mitochondrial membrane function, and upregulation of the mitophagy markers, PINK1, PRKN, and DNM1L, which indicated that Tat exposure to these cells induced mitophagy [72]. These Tat-induced alterations may lead to expansion of the viral reservoir and contribute to the development of HAND, and in a study of macrophages exposed to Tat (unknown subtype) for 72 h, there was increased levels of quinolinic acid [73]. This may indicate Tat was able to dysregulate the Kynureinine pathway, where tryptophan has been shown to be catabolized to quinolinic acid and increased quinolinic acid levels have been related to neuropathological damage in vitro, indicating a possible mechanism in the development of HAND [73].
While microglia have been shown to be capable of supporting HIV-1 replication in the CNS, the effects observed after microglial exposure to extracellular Tat have also suggested that infection was not required to elicit microglial activation and neurotoxic activities [74, 75]. Microglial activation was typically characterized by cytokine production and synthesis of ROS (Fig. 3) [76, 77]. Post-mortem brain tissue from HIV-1-infected patients showed high levels of pro-inflammatory IL-1β and IL-10 cytokine and superoxide dismutase (SOD) mRNA expression, which have been shown to be signs of microglial activation [78]. Similarly, treatment with recombinant Tat 86 on primary rat microglia cultures elicits TNFα and IL-1β production, as well as NO production [79]. Primary rat microglia stimulated with extracellular Tat 86 significantly upregulated iNOS activity and expression, resulting in a marked increase of NO, and moreover, pretreatment with IFNγ and extracellular Tat 86 resulted in a synergistic effect where NO accumulation in microglia was increased threefold, and this effect was blocked by addition of an anti-Tat antibody [79, 80]. While microglial cells represent the predominate immune presence in the CNS, and peripheral cells of the monocyte–macrophage lineage are the primary route of HIV-1 transmission into the CNS, they are not the only cells affected by extracellular Tat [81]. The comprehensive mechanisms of action operative during the development of all forms of HAND have yet to be defined; inflammatory chemokines and cytokines released by other cells, such as astrocytes, likely contribute integrally to the development of these forms of HIV-associated neurologic disease.

Effects of astrocytes, oligodendrocytes, microglial cells exposed to Tat on neurons. Astrocytes in orange, neurons in purple, microglia in green, and oligodendrocytes in blue. Tat in red. Arrows indicate the relationship the Tat has on and between each cell type leading to neurotoxicity
Astrocytes
Astrocytes represent an important cell type in the brain that exhibits a diverse array of functional properties including a contribution to the formation of the BBB, as well as signaling to nearby neuronal cells [15, 82]. Due to their proximity to neurons and endothelial cells, astrocytes have been shown to regulate BBB physiology through gap junctions and soluble factors [82]. In the CNS, HIV-1 has been shown to replicate primarily in microglia. In contrast, only a small percentage (5%) of astrocytes are infected, which occurs either by cell-to-cell interactions with T cells, where virological synapses formed by filopodia binding transfer the virion, or by virion interactions with CCR5 and CXCR4 on astrocytes, though it has not been elucidated which mechanism may be more operative in the intact brain [12, 14, 15]. Viral replication in astrocytes has been restricted due to impaired production of HIV structural proteins [12, 15, 82]. Although astrocytes are not productively infected by HIV-1, they have been shown to produce the non-structural proteins, Tat, the regulator of expression of viral proteins (Rev), and negative factor (Nef) [12, 83].
Tat can be secreted from astrocytes, and extracellular Tat is toxic to neurons, CD4 + T cells and astrocytes [12, 32, 84, 85]. Once intracellular, Tat has skewed astrocytes to a more aggressive, pro-inflammatory profile in both in vitro and in vivo models and has induced the expression of multiple cytokines. In primary astrocytes treated with extracellular Tat 86, there was an observed increase in TNFα; while in human SVG astrocytes exposed to Tat 101, there was an increase in CCL5/RANTES, CXCL10, IL-1β, and IL-6, and IL-8 [12, 85,86,87,88,89,90,91]. Tat 86 (plasmid) was shown to mediate ROS generation and activate MAPK-NF-κB/AP-1 signaling cascades leading to the upregulation of the chemokines CCL2, CXCL8, and CXCL10 and upregulation of HDAC6 expression in the human astroglial cell line, CRT-MG [92]. In this astrocyte cell line, treatment with Hindsiipropane B, a 1,3-diarylpropane (an anti-inflammatory drug), significantly suppressed the production of these pro-inflammatory cytokines, by inhibiting the activation of HDAC6 and MAPK-NF-κB/AP-1 signaling axes [92]. An additional study of primary human fetal astrocytes exposed to extracellular Tat 86 also exhibited enhanced secretion of IL-6, TNFα, and MCP-1 (Fig. 2) [15, 84, 85, 93,94,95].
As discussed previously, MCP-1 can induce the transmigration of inflammatory cells across the BBB. In a study of extracellular subtype B Tat 86, there was enhanced MCP-1 secretion in primary human astrocytes, an effect that was not observed with subtype C Tat [96]. Exposure of rat and human astrocytes to Tat in vitro resulted in induction of platelet-derived growth factor (PDGF) mRNA and protein levels, which led to increased expression of the pro-inflammatory cytokines MCP-1 and IL-1β [15, 83]. The upregulation of PDGF mRNA was due to activation of the ERK1/2 and JNK signal pathways, which have been shown to be downstream of the Erg-1 transcription factor, and both Erg-1 and PDGF were upregulated as early as 3 h after Tat exposure [15, 83, 97]. Additionally, Tat 72, Tat 86 and Tat 101, have all been shown to induce PDGF in primary astrocytes; while, heat-inactivated Tat had no effect on expression level [83]. In the nucleus, Tat interacted with and activated the transcription factor C/EBP, which stimulated MCP-1 transcription [98]. The release of MCP-1 can be mediated by the purinergic receptor P2X7R, which has been shown to be highly expressed on astrocytes [84]. When astrocytes were exposed to Tat in vitro, there was a significant increase in P2X7R expression on astrocytes; while inhibitors specific for P2X7R reduced MCP-1 release [84]. It has additionally been shown that P2X7R induced the ERK1/2 pathway, which also enhanced the expression of MCP-1 [15, 83, 84]. In addition, an increase in mRNA transcript expression of a number of apoptotic genes and pro-inflammatory cytokines, including DAXX (death-domain associated protein), Fas, FasL, TNFα, TNFα convertase, and Traf6 was also observed in Tat 86-transfected primary human astrocytes [87].
Astrocytes have been shown to utilize gap junctions for the transfer of ions and second messengers to neighboring cells, and during inflammatory conditions, gap junction communication was inhibited in an effort to control infection [16, 82]. In HIV-1 infection, however, astrocytes maintain gap junction communication, by upregulating Connexin 43 (Cx43), thus enhancing the spread of Tat to uninfected cells [16, 82]. Furthermore, in vitro and in vivo, both Tat 72 and Tat 101 mediated this effect by binding to the Cx43 promoter, which upregulated the expression of Cx43 [16]. Exposure of human CRT-MG astrocytes to recombinant Tat 86 stimulated the enriched expression of intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) mRNA and protein levels, mediated firm adhesion of THP-1 human monocytes to the CRT-MG astrocytes, and induced oxidative stress in exposed cells, in both time- and concentration-dependent manners [99]. Additionally, astrocytes exposed to Tat had activated NADPH oxidase, which resulted in the generation of ROS and activation NF-κB, and led to upregulated surface VCAM-1 and ICAM-1, as well as upregulated CXCL10 (IFN-γ inducible protein 10) (Fig. 2) [15, 27, 100]. Histone deacetylase 6 (HDAC6) has been located in the cytoplasm and shown to be a master regulator of the expression of pro-inflammatory mediators, and has regulated Nox2-based NADPH oxidase to mediate NADPH oxidation and ROS production, therefore Tat exposure resulted in increased HDAC6 and stimulated NADPH oxidase expression [100]. Additionally, in a study of U373 MG cells exposed to the pcDNA3 Tat 101-expressing plasmid, there was an induction in histone 3 tri-methylation at lysine 27 (H3K27me3), which has been shown to promote HIV latency [101].
Tat also repressed β-catenin, an adherens junction component, which has lead to neuro-inflammation (Fig. 2) [102]. Transfection of the human U87MG astrocyte cell line, as well as primary human progenitor-derived astrocytes with subtype B Tat 101 and Tat 72 plasmids resulted in suppression of β-catenin expression and signaling [103, 104]. This function of Tat was originally shown to be due to its core domain and cysteine-rich domain, located within the first exon, and more recently has been mapped to the lysine residues at positions 41 and 51, which are necessary for the suppression of β-catenin [102, 104]. Mechanistically, depletion of β-catenin by siRNA resulted in enhanced binding of Tat to the TAR element in U87MG astrocytes co-transfected with subtype B Tat and LTR constructs demonstrating a viral strategy for optimizing transactivation of the HIV-1 promotor in the CNS [104].
Astrocytes exposed to Tat that have altered functionality and a pro-inflammatory composition have been shown to lead to neuronal toxicity. Looking at primary rat astrocytes exposed to extracellular Tat 101, there was increased expression and secretion of miR-7, which when taken up by hippocampal neurons resulted in loss of both inhibitory and excitatory synapses [105]. Increased expression of glial fibrillary acidic protein (GFAP), a filament protein located in the cytosol of astrocytes, resulted in its aggregation, which induced the unfolded protein response (UPR), and leads to ER stress, astrocyte dysfunction, and neurotoxicity [27, 85, 97, 106]. Tat 86-transfected U373MG astrocytes and Tat 86 in Tat-transgenic mice have been shown to upregulate the mRNA and protein levels of GFAP, further demonstrating Tat’s role in the aberrant activation of astrocytes (Fig. 2) [97, 107, 108]. Tat influenced GFAP expression in astrocytes by activating the transcription factor STAT3, which initiated a transactivation cascade in the nucleus that included Erg-1 and p300, and resulted in upregulated expression of GFAP [97, 107, 108].
Astrocytes exposed to Tat also have metabolic changes and in primary murine astrocytes exposed to subtype B Tat 86, there was a reduced capacity for buffering glutamate, which was not observed with subtype C Tat 101 exposure, which has indicated exclusive subtype B Tat-mediated effects on glutamate uptake [107, 109]. Glutamate intake was also significantly impaired in subtype B Tat 86-transfected U373MG astrocytes, as well as in primary astrocytes from HIV-1HXB2 Tat 86 transgenic mice, demonstrating further negative consequences of intracellular Tat on astrocyte metabolism (Fig. 2) [107]. Overall, the many effects of Tat on astrocytes can result in dysfunction that negatively impairs the function of other major cells of the CNS.
In summary, a small percentage of astrocytes have been shown to be infected by HIV-1 and may be producers of Tat in the CNS. This is important because astrocytes support BMECs as well as neurons, and production of viral proteins and pro-inflammatory cytokines will contribute to CNS dysfunction. Astrocytes exposed to Tat can become dysfunctional and will have reduced processes leading to reduced BBB integrity. Additionally, astrocytes exposed to Tat can become activated leading to a pro-inflammatory phenotype that is directly damaging to neurons.
Neurons and neurotoxicity
It is generally accepted that HIV-1 does not infect neurons; therefore, injury and excitotoxicity on CNS neurons are believed to originate from HIV-1 infection of other CNS cell types, such as astrocytes and microglia, which can produce and secrete viral proteins, such as Tat, as well as pro-inflammatory cytokines that can affect neurons, even in the absence of actively replicating HIV-1 [89, 110,111,112,113,114]. Indirect consequences of Tat on neurons in the CNS have been shown by a study of primary rat neurons exposed to conditioned media from Tat 72-transfected astrocytes that resulted in augmented neurotoxicity [115]. Additionally, a co-culture model of Tat 86-transfected astrocytic cell lines with non-transfected primary human fetal neurons resulted in increased neurotoxicity, and manifested as hyperpolarization of the mitochondrial membrane, which led to increased release of calcium stores into the cytoplasm, MAP2 breakdown, diminished neurite length, and ultimately neuronal cell death (Fig. 3) [87, 116, 117]. When Tat 86-expressing C6 astrocytic cells were exposed to neuronal cultures in transwells, it resulted in significant neuronal death, showing that conditioned medium from Tat 86-expressing cells was sufficient to elicit neurotoxicity [87]. Furthermore, the addition of supernatant from Tat 72-treated microglial cell cultures to primary hippocampal neuron cultures resulted in observed neurotoxicity that could be blocked by the addition of NADPH oxidase inhibitors, where NADPH oxidase along with microglial activation has been found to cause hippocampal dysfunction in other neurodegenerative diseases and may account for some aspects of HAND [113, 118]. Additionally, primary microglia treated for 24 h with recombinant Tat 72 showed increased, dose-dependent glutamate release that could be abrogated by cysteine–glutamate antiporter inhibitors or NADPH oxidase inhibitors, indicating that glutamate release from primary mouse microglia due to Tat 72 exposure may induce neuronal excitotoxicity in an N-methyl-D-aspartate (NMDA) receptor manner, leading to neurotoxicity and cell death [119, 120].
A potential mechanism of Tat-induced toxic ROS production in neurons may originate from the induction of spermine oxidase (SMO) activity (Fig. 3). Spermine, a substrate for SMO, has been previously noted as a driving factor for NMDA receptor-dependent neuronal injury in a human neuroblastoma SH-SY5Y cell line (Fig. 2) [121]. One of the metabolic products of SMO has been shown to be H2O2, which may accumulate in neurons and drive apoptosis [121]. SH-SY5Y cells treated with either extracellular Tat 101 or supernatant from Tat 86-expressing U373MG astrocytic cells showed oxidative stress characterized by decreased intracellular glutathione, increased ROS in the form of H2O2, and decreased cell viability as well as increased SMO activity [121]. However, when SH-SY5Y cells were pretreated with the NMDA receptor antagonist MK801, the increased SMO activity could be completely prevented, indicating a mechanistic link between NMDA receptor activation and induction of ROS [121]. Additionally, H2O2 produced by SMO activity in response to extracellular Tat 101 treatment increased the expression of antioxidant related elements through translocation of the Nrf2 transcription factor to the nucleus, which indicated a potential role for cellular antioxidants to mitigate the downstream consequences of ROS toxicity triggered by Tat–NMDA receptor interactions [121, 122].
NMDA receptors have been shown to be a subclass of postsynaptic neuronal glutamate receptors that mediate cellular calcium entry, and are thought to be the main receptor that Tat interacts with to induce neuronal dysfunction, which can lead to excitotoxicity and cell death (Fig. 3) [123]. Subtype B Tat 72 has been shown to potentiate glutamate-dependent excitotoxicity via stimulation of NMDA receptors in vitro, which leads to increased apoptosis of neurons (Fig. 2) [124]. This effect has been shown to be dependent on pre-exposure to Tat 72 and has been shown to be caused by increased calcium flux from intracellular stores within neurons, and has been enhanced by the addition of glutamate [124]. In an in vitro model utilizing primary rat cortical neurons treated with extracellular Tat 72, the underlying cause of NMDA receptor stimulation was increased phosphorylation of NMDA receptor subunits, which potentiated excitotoxicity and was shown to be counteracted by the tyrosine kinase inhibitor genestein [124]. Tat 72 has been shown to bind directly to NMDA receptors via its arginine-rich domain and is immunoprecipitated with both the NR1 and NR2A subunits of the NMDA receptor as observed in a cell line model where HEK293 cells were transfected with and subsequently expressed with both subunits [115]. The addition of extracellular Tat 72 to the HEK-NMDAR cells resulted in cell death comparable to that caused by NMDA, which is a known excitotoxin and is capable of killing neuronal cells upon binding NMDA receptors [115, 125]. This binding was correlated with, and sufficient for, neurotoxicity in vitro, where HEK-NMDAR cell death induced by subtype B Tat 72 was more robust than subtype C Tat 72, and was abrogated by the deletion of the arginine-rich region of Tat [115]. The difference in neurotoxicity exhibited by HIV-1 Tat subtypes on in vitro models using HEK-NMDAR and primary fetal neuron models has been partially attributed to the di-cysteine motif normally present in subtype B Tat, but absent from subtype C Tat [115, 126]. Mutation of subtype C Tat 72 to include the di-cysteine motif resulted in increased neurotoxic potential of subtype C Tat 72 on primary fetal neurons in culture [126].
Neuronal cell death may be partly due to downstream caspase-3 activation in neurons exposed to extracellular Tat. Studies of striatal neuronal cultures from embryonic day 15 ICR mice showed activation of caspase-3 in neurons upon exposure to extracellular Tat protein [127,128,129]. In addition to caspase-3 activation and significant neuronal cell death, increased p38 and JNK-MAPK phosphorylation was observed with extracellular Tat 72 treatment of primary mouse neurons, suggesting that extracellular Tat interaction with neurons triggers apoptotic signaling pathways [128, 130]. Caspase-3 inhibitors and JNK inhibitors have been shown to prevent caspase-3-mediated apoptosis in primary rat and mouse neurons, respectively [128, 129]. Furthermore, treatment of primary mouse neurons with non-toxic deletion mutant Tat that lacks amino acids 31–61 targeting exon 1 of Tat, has not resulted in apoptosis, p38 or JNK-MAPK phosphorylation, or caspase-3 activation [128]. Amino acids 31–61 have been shown to be necessary for Tat functions associated with toxicity, which has implied that these residues are essential for extracellular Tat to trigger caspase-3-dependent apoptosis in neuronal cells (Fig. 2) [131]. In addition, a study of SH-SY5Y cells, a neuroblastoma cell line, exposed to the plasmid pcDNA3.1-Tat-flag, encoding Tat 101, for 48 h, indicated that there was activation of FOXO3, a regulator of apoptosis-related genes, through the JNK signaling pathway, resulting in increased neuronal apoptosis [132].
Dendritic spine size and morphology are major contributors to synapse integrity and detrimental changes to dendritic spine morphology and density are correlated with neurocognitive decline where dendritic loss is a predictive indicator of HAND [133,134,135,136,137]. Although extracellular Tat treatment of neurons can cause apoptosis, Tat at concentrations that are not sufficient to cause neuronal apoptosis may cause morphological changes to dendrites on affected neurons [138]. Morphological changes of dendrites upon exposure to extracellular Tat includes the development of abnormal, “spoke-like” dendritic processes, though dendritic spine shortening or loss has been more widely observed (Fig. 2) [87, 128, 139,140,141]. Exposure of primary neuronal cells to Tat 72 resulted in a significant decrease in the total length and number of dendrites per cell [142]. This was shown to be mediated by Tat inhibition of BDNF transcription, which has been shown to induce F-actin changes that are involved in dendrite and spine injury, leading to the development of neurotoxicity [142]. Extracellular full-length recombinant Tat 101 protein exposure also caused decreased presynaptic density, as measured by the amount of synaptophysin, a neurotransmitter release machinery protein, present in dendritic spines [143, 144].
Persistent stimulation of the NMDA receptor may also impact dendritic spine morphology and synaptic integrity, as NMDA receptor activation has been shown to potentially inhibit actin dynamics of dendritic spines, where a blockage in actin motility would cause spines to round resulting in increased spine stability [145]. The exact mechanisms have not been shown in vivo, though Tat-induced changes in dendrite physiology have been corroborated in premortem and postmortem imaging analyses of brains of HIV-1-infected patients both in the absence or presence of HIV encephalitis and have also been linked to learning and memory impairment in Tat 86-transgenic animals [146, 147]. The PSD-95 scaffold protein and associated signaling proteins present in postsynaptic dendritic spines help to determine the size and strength of dendritic spines [143]. LRP is required for normal motor function and intracellular signaling and binds to PSD-95, to connect the LRP signaling pathway with NMDA receptors via the NR2B subunit [148, 149]. Exposure of rat hippocampal neurons to extracellular Tat 86 caused a significant loss of PSD-95 in dendritic spines and the overall area of postsynaptic sites was decreased [141, 143]. This likely occurs through a mechanism mediated by LRP, where LRP antagonists that prevented Tat 86 association with LRP also blocked subsequent LRP association with PSD-95 and internalization of Tat 86 [141, 143]. The Tat core domain, which spans residues 37–48, has been shown to bind LRP to mediate rapid uptake of Tat into neurons grown in a human fetal neuron–astrocyte co-culture system [150]. The Tat 72–LRP interaction has been made more efficient by additional associations with heparan sulfate proteoglycans and leads to translocation of Tat 72 into the nucleus once internalized [150]. Extracellular Tat binding to LRP on the surface of neurons has been shown to trigger the formation of an LRP–PSD-95–NMDAR–nNOS macromolecular complex, which was shown to be inhibited by pretreatment with CCL2 chemokine, which was previously shown to protect neurons from Tat-mediated apoptosis [151]. The formation of this complex in response to Tat exposure ultimately has been shown to lead to apoptosis in neurons, though blocking LRP-mediated uptake, NMDA receptor activation, or neuronal nNOS activity significantly reduced apoptosis in neurons treated with extracellular Tat 72 [152]. Additionally, pretreatment with LRP antagonist, receptor-associated protein (RAP), has been shown to disrupt LRP–Tat binding and uptake of Tat 86 by its tight binding to surface LRP which directly inhibits LRP-Tat 86 interactions, and also reversed synapse loss that occurred post-Tat exposure, indicating that RAP was a potent modulator of synaptic stability [141, 144].
In summary, although neurons cannot be infected by HIV-1, they are subject to damage and excitotoxicity from exposure to HIV-1 viral proteins such as Tat as well as exposure to pro-inflammatory cytokines and other metabolites secreted from other cells of the CNS. Excitotoxicity mediated through Tat stimulation of NMDAR can lead to apoptosis as well-altered dendritic spine morphology which has been shown to be correlated with neurocognitive decline.
Brain microvascular endothelial cells (BMECs)
Tight junctions have been shown to be essential for maintaining the integrity of the BBB and are necessary to link brain microvascular endothelial cells (BMECs) together [14]. The major tight junction proteins (TJP) at the BBB are claudin-5, occludin, and zonula occludens-1 (ZO-1), where the impairment of any of these TJP resulted in increased BBB permeability [14]. When the endothelial cell line hCMEC/D3 was exposed to extracellular Tat, there was an activation of the RhoA/ROCK signaling pathway, which has been shown to be involved in regulating the cell cytoskeleton and the amount of actin filaments [14]. When the RhoA/ROCK pathway was activated in the presence of Tat, there was an observed decline in mRNA and protein levels of Occludin, ZO-1 and junctional adhesion molecule-A (JAM-A), resulting in increased BBB permeability [14, 15]. JAM-A was expressed at the lateral borders between adjacent endothelial cells and functions to help maintain barrier integrity by stabilizing cell junctions and reducing any paracellular permeability [5]. Exposure to subtype B Tat 86, but not subtype C Tat 86, also resulted in upregulated mRNA expression of JAM-2 in a time- and dose-dependent manner in primary human BMECs [153]. Extracellular Tat exposure of primary humans BMECs also increased the expression of E-selectin, a cell adhesion molecule involved in immune cell transmigration across the BBB [154]. When primary human BMECs were exposed to subtype B Tat 86, there was an overall downregulation of ZO-1 expression (Fig. 2) [153]. However, the nuclear localization of ZO-1 increased with subtype B Tat 72 [155]. Furthermore, rat BMEC exposure to subtype B Tat 72 led to a decline in ZO-2, claudin-1, claudin-5, and occludin mRNA and protein levels [156,157,158]. Mechanistically, the deregulated expression of tight junction complex components by Tat was found to be due to alterations in several pathways, including the previously discussed RhoA/ROCK pathway as well as the Ras/caveolin-1 pathway, responsible for changes in ZO-1, ZO-2, and occludin, and the VEGFR2/Ras/ERK1/2, PI3K/AKT pathway which modulates claudin-5 [155, 156, 158, 159]. Additionally, extracellular Tat 86 exposure to human cerebral microvascular endothelial cells resulted in downregulation of occludin and LRP-1 and upregulation of RhoA, which contributed to accumulation of amyloid beta in the brain [160]. The alteration of all these pathways highlights the multi-faceted downstream effects of Tat exposure on the BBB.
In addition to TJP, there are many other factors that can affect BMEC integrity and functionality. Matrix metalloproteinases (MMPs) are a class of enzymes that belong to the zinc metalloproteinases family that degrade components of the extracellular matrix and are commonly expressed during inflammatory conditions, where their activity is required for inflammatory cells to traverse the extracellular matrix [161]. Because Tat is known to produce a pro-inflammatory environment, studies have specifically examined changes in MMPs during Tat exposure to determine if elevated MMP levels contribute to breakdown of BMEC integrity. When primary human BMECs were exposed to Tat, there was increased expression of MMP-9, a major MMP, which cleaved occludin, to degrade it, resulting in reduced occludin levels and increased permeability of the BBB [14, 15, 54, 162]. Interestingly in another study, primary human BMECs exposed to subtype B Tat 86 exhibited transcriptional upregulation of MMP-3, MMP-10 and MMP-12, as well as upregulation of other proteases and peptidases involved in extracellular matrix processing and cleavage. However, these effects were not observed with Tat.AG, a Tat variant derived from HIV-1 subtypes A and G [43]. Importantly, this study did not find an increase in MMP-9, but this may have been due to variations in experimental approach, where this study used Tat at 100 ng/ml for 48 h, while the previous experiment that saw an MMP-9 increase used Tat at 200 ng/ml for 24 h [43].
We have observed that in multiple cell types, Tat exposure can increase the secretion of pro-inflammatory cytokines, and this effect has been repeated in BMECs. Exposure of primary human BMECs to subtype B Tat 86 enhanced the secretion of the pro-inflammatory cytokines TNFα, IL-6, and IL-8 [154, 163, 164]. In addition, conditioned media from Tat-exposed primary human BMECs stimulated the enhanced migration of PBMCs across a BBB model as a result of increased IL-8 [164]. BMECs exposed to Tat also increased MCP-1 secretion, which has previously been shown to induce the transmigration of immune cells across the BBB, and results in disrupted barrier integrity, and has been shown to contribute to neuro-inflammation [5, 165].
In multiple cell types, Tat exposure triggers cell stress and apoptosis; if the cell is damaged as a result of cell stress, it can trigger the activation of the unfolded protein response (UPR) and endoplasmic reticulum-associated degradation (ERAD), to mitigate ER stressors. However, if the stress continues, the cell will switch from a pro-survival to a pro-apoptotic phenotype via upregulation of C/EBP homologous protein (CHOP) [165]. Exposure of primary human BMECs to Tat 101 resulted in ER stress and induction of the ER stress sensors activating transcription factor (ATF) 6, and PKR-like ER kinase (p-PERK), as well as the downstream mediators; phosphorylated eukaryotic initiation factor 2 (p-eIF2a) and activating transcription factor (ATF) 4 (Fig. 2) [73, 165]. The ER stress induced by extracellular Tat resulted in a decline in BMEC viability and an increase in apoptosis, where Tat mediated the activation of PERK and ATF6 pathways, resulting in activation of the pro-apoptotic CHOP pathway [165]. ER stress in human BMECs may also play a role in amyloid beta pathology, as ER stress has been associated with inhibition of nuclear amyloid beta uptake [166]. Increased amyloid beta deposition has been observed in the brain of HIV-1-positive patients and has been shown to accumulate in the cytoplasm of HIV-1-infected hCMEC/D3 cells [166]. Furthermore, exposure to Tat increased the aggregation of amyloid beta fibrils in the brain, resulting in increased neurotoxicity both in vitro with Sprague Dawley rat hippocampal neuronal cell cultures free of mycoplasma and in six-month-old transgenic C57BL-6 mice expressing mutations in human amyloid precursor protein and human presenilin [167].
Although Tat can trigger cell stress leading to the UPR in BMECs, it can also trigger other stress-induced pathways and has been shown to increase the level of apoptosis in BMECs, resulting in increased BBB impairment [165]. Tat 101 exposure to primary human BMECS stimulated upregulation of the pro-apoptotic protein Bax, and decreased expression of the anti-apoptotic protein Bcl-2, resulting in the loss of mitochondrial membrane potential, and the release of cytochrome c, resulting in increased apoptosis [165].
Furthermore, Tat 86 elevated cytosolic Ca+2 levels in primary human BMECs, in vitro, and exposure of porcine BMECs to subtype B Tat 72 resulted in increased oxidative stress, coupled with a concomitant decrease in the glutathione concentration, in a concentration-dependent manner [168, 169]. Extracellular Tat exposure also mediated increases in both oxidative and nitrosative stress in primary human BMECs, which were linked to Tat-induced systematic apoptosis and increased permeability of exposed cells [170]. In primary human BMECs, subtype C Tat 86 upregulated the activity of NADPH oxidases (NOX2 and NOX4), which increased ROS and activated proline rich kinase 2 (PYK-2), which phosphorylated VE-cadherin and β-catenin, to result in disrupted adherens junctions and increased permeability [171].
Overall, these observations demonstrate that HIV-1 Tat was capable of modulating a variety of cellular genes, resulting in cascades of downstream effects that tend to be pro-inflammatory and pro-apoptotic in BMECs cells, leading to breaks in the BBB, which along with downregulated tight junction complexes, and upregulated adhesion molecules, enabled activated immune cells and various proteins, such as Tat, to more easily cross the BBB.
Blood–brain barrier
The BBB is composed primarily of BMECs and secondarily of astrocytes, pericytes and other cells, as well as other molecular components, such as tight junctions, adhesion molecules, and extracellular matrix components, which all combine to form a selective, semi-permeable barrier designed to protect the brain and maintain CNS homeostasis (Fig. 4) [5, 15, 82, 172]. This functionally selective barrier formed by astrocytes and BMECs still results in gaps about 20 nm wide, which would allow many solutes to easily pass through [173]. Therefore, tight junction complexes formed between the BMECs are essential to further restrict solute movement across the BBB [14]. The integrity of the BBB is essential to normal functioning in vivo, as immune surveillance in the brain occurs at a rate that is 100-fold less than in other organs (such as the spleen), exemplifying the importance of the BBB in regulating entry of immune cells and proteins into the CNS [5].

BBB under normal and Tat-impaired conditions. a BBB under normal homeostatic conditions: there is low entry of PBMCs into the brain, BMEC tight junctions and adherens junctions are intact, and astrocyte gap junctions and adherens junctions (1) are also intact, pericytes are also present, and provide additional support for BMECS. b BBB with Tat exposure: BMECs exposed to Tat have increased expression of cell adhesion molecules (2), and downregulation of tight junction and adherens junction complexes (3), which enhances PBMC migration across the BBB. Tat exposure also results in an increase in MMP-9 (4), which can cleave and inactivate occludin. Tat exposure enhances the secretion of MCP-1/CCL2 (5) from both BMECs and astrocytes, creating a chemokine gradient that enhances the migration of PBMCs across the BBB. In astrocytes, Tat exposure results in the upregulation of the gap junction component Connexin 43 (6), and cell adhesion molecules and while suppressing the expression of B-catenin (7), Additionally, pericytes become dysfunctional under Tat exposure where they do not provide BMEC support and instead support low-level HIV-1 infection, and contribute to BBB dysfunction
There are two main possibilities for how cells (such as monocytes), or proteins (such as Tat) cross the BBB. The first is paracellular diapedesis, which involves extravasation between tight junctions on adjacent cells, while the second possibility is transcellular diapedesis, which requires uptake into the BMECs and passage through the cytosol of the BMECs, with egress on the opposite side of the BBB [5, 14, 173]. BMECs express the HIV-1 co-receptors CCR5 and CXCR4, providing a potential mechanism for the virus to enter endothelial cells and gain access to the brain; however, productive infection of these cells by HIV-1 has not been reported [14]. Tat has been shown to be able to enter BMECs, likely by absorptive endocytosis, after interaction with surface receptors [14]. Notably, the predominant pathway of transmigration across the BBB and the effect that HIV-1 or Tat may have on the prevalence of each pathway remains to be elucidated [5].
Tat has been shown to impair BBB integrity by inhibiting essential tight junction protein expression, resulting in increased leukocyte transmigration across the BBB, which may contribute to the development of HAND (Fig. 4) [14, 165]. Throughout this review, we have discussed how Tat affects the cells of the CNS, but when combined Tat exposure to astrocytes and BMECs forming the BBB will alter tight junction proteins, increase apoptosis and increase secretion of pro-inflammatory cytokines. These effects together will result in increased transmigration of immune cells into the CNS, which creates a chronic feedback loop of inflammation and damage in the CNS, resulting in the development of neurocognitive decline.
Conclusion
This review has primarily focused on Tat and its effect on cells of the CNS; however, there are other HIV-1 proteins that have been shown to be involved in CNS dysfunction. Neuronal toxicity and activation-induced cell death has also been caused by the viral protein Nef which has shown to be released from HIV-1-infected cells via exosomes [12, 174]. Nef has also been shown to activate MMPs which can then contribute to BBB damage [175]. In a rat model recombinant Nef protein has also been shown to increase leukocyte recruitment to the CNS via increased levels of IL-6, TNF-alpha, and IFN-gamma [176]. Additionally, gp120, Vpr, and Nef can initiate apoptosis in BMECs at the BBB, indicating the ability of these proteins to alter BBB integrity [9]. At the BBB, gp120 has been shown to increase permeability by reducing the expression of ZO-1 in BMECs [177]. Gp120 binds to CCR5 and CXCR4 to mediate cellular entry, however, gp120 has been shown to be neurotoxic in endothelial cells and can contribute to decreased BBB integrity, resulting in increased permeability and transmigration of monocytes into the CNS [178]. At the BBB, gp120 has also been shown to reduce the ZO-1 and ZO-2 expression levels, as gp120 targets these proteins for degradation by the proteasome [177]. Finally, similar to Tat, gp120 has also been shown to activate astrocytes, and stimulates the production of pro-inflammatory cytokines, chemokines, and nitric oxide, and together these mediators can cause neuronal damage [179]. Extracellular Vpr has also been shown to increase ROS as well as the concentrations of IL-6, IL-8 and MCP-1 in astrocytes and this effect may be damaging to other cells of the CNS [180, 181]. Although other HIV-1 proteins have been shown to mediate effects similar to Tat, it is important to note that Tat remains the one protein still measurable in the CNS during ART.
It is strongly supported that Tat is a mediator of the development of HAND, and that Tat in the CNS can modulate gene regulation and skew the microenvironment of the brain to be more pro-inflammatory, thus enhancing HIV-1 pathogenesis. Despite ART, Tat is continually secreted from latently infected cells, indicating that current ART regimens are not adequate to inhibit the development of HAND, and more targeted therapies are required [5, 12, 15, 16, 31].
To fully understand the impact of Tat in the development of HAND, it will be necessary to compare all three Tat lengths in functional studies. Because Tat 86 and 101 can bind integrin receptors, it will be important to have studies incorporate BBB models to determine if Tat 86 and Tat 101 cross into the CNS at an accelerated rate as compared to Tat 72. These studies would also ideally determine if Tat 86 and Tat 101 are able to cross the BBB at an accelerated rate, if neurocognitive disorders are more likely to develop in animals exposed to these Tat lengths as compared to Tat 72. Additionally, because Tat 101 is involved in NF-kB activity via the ESKKKVE motif, it will be important to determine if there is increased NF-kB activity in the CNS as a result of Tat 101 exposure, and if this activity results in increased pro-inflammatory cytokines and increased transmigration of immune cells into the CNS [37]. An animal model of Tat is essential to study these effects further, where Tat 72, Tat 86 and Tat 101 can be separately introduced in the CNS at physiologically relevant concentrations and compared to determine if there is a difference in the ability of Tat 72, 86, and 101 to affect progression and severity of neurologic deficits. This study will be especially important because Tat 101 is the consensus length found in HIV-1-positive patients with subtype B [10, 34, 37]. Furthermore, Tat 101 has the combined effects of Tat 72 and Tat 86 plus the additional ability to interact with NF-kB [37]. A study such as this will definitively identify which Tat length is the most relevant for studying Tat in the CNS and determine the importance of Tat length in impacting the development of HAND.
A thorough understanding of the disparities in subtype-mediated effects of HIV-1 and Tat remains to be elucidated. At the present time, there are multiple crucial questions that remain to be answered. What are the areas of high conservation in Tat between subtypes? Does Tat genetic variation across the same subtype link to disease progression? Although ART does not inhibit the production of Tat, does it affect the toxicity of Tat? Are there differences in the amount of Tat in the CSF of well-suppressed patients versus patients with high viral burden? How can the establishment of viral reservoirs in the brain be inhibited? These questions highlight some of the current gaps of knowledge in the field and indicate that more research is necessary to fully understand the implications of Tat involvement in the CNS and its contribution to the development of HAND.
Author contributions statement
J.M.M. M.E.M., and M.R.N. conceived and designed the study. J.M.M., M.E.M., C.S, A.R.M., B.W. and M.R.N. prepared and designed the figures and drafted the manuscript. All authors have read and approved the final manuscript.
References
Saez-Cirion A, Bacchus C, Hocqueloux L, Avettand-Fenoel V, Girault I, Lecuroux C, Potard V, Versmisse P, Melard A, Prazuck T, Descours B, Guergnon J, Viard JP, Boufassa F, Lambotte O, Goujard C, Meyer L, Costagliola D, Venet A, Pancino G, Autran B, Rouzioux C, Group AVS (2013) Post-treatment HIV-1 controllers with a long-term virological remission after the interruption of early initiated antiretroviral therapy ANRS VISCONTI Study. PLoS Pathog 9(3):e1003211. https://doi.org/10.1371/journal.ppat.1003211
Henrich TJ, Hu Z, Li JZ, Sciaranghella G, Busch MP, Keating SM, Gallien S, Lin NH, Giguel FF, Lavoie L, Ho VT, Armand P, Soiffer RJ, Sagar M, Lacasce AS, Kuritzkes DR (2013) Long-term reduction in peripheral blood HIV type 1 reservoirs following reduced-intensity conditioning allogeneic stem cell transplantation. J Infect Dis 207(11):1694–1702. https://doi.org/10.1093/infdis/jit086
Persaud D, Gay H, Ziemniak C, Chen YH, Piatak M Jr, Chun TW, Strain M, Richman D, Luzuriaga K (2013) Absence of detectable HIV-1 viremia after treatment cessation in an infant. N Engl J Med 369(19):1828–1835. https://doi.org/10.1056/NEJMoa1302976
Luzuriaga K, Gay H, Ziemniak C, Sanborn KB, Somasundaran M, Rainwater-Lovett K, Mellors JW, Rosenbloom D, Persaud D (2015) Viremic relapse after HIV-1 remission in a perinatally infected child. N Engl J Med 372(8):786–788. https://doi.org/10.1056/NEJMc1413931
Williams DW, Eugenin EA, Calderon TM, Berman JW (2012) Monocyte maturation, HIV susceptibility, and transmigration across the blood brain barrier are critical in HIV neuropathogenesis. J Leukoc Biol 91(3):401–415. https://doi.org/10.1189/jlb.0811394
Gray LR, Roche M, Flynn JK, Wesselingh SL, Gorry PR, Churchill MJ (2014) Is the central nervous system a reservoir of HIV-1? Curr Opin HIV AIDS 9(6):552–558. https://doi.org/10.1097/COH.0000000000000108
Fois AF, Brew BJ (2015) The Potential of the CNS as a Reservoir for HIV-1 Infection: Implications for HIV Eradication. Curr HIV/AIDS Rep 12(2):299–303. https://doi.org/10.1007/s11904-015-0257-9
Joseph SB, Arrildt KT, Sturdevant CB, Swanstrom R (2015) HIV-1 target cells in the CNS. J Neurovirol 21(3):276–289. https://doi.org/10.1007/s13365-014-0287-x
Toborek M, Lee YW, Flora G, Pu H, Andras IE, Wylegala E, Hennig B, Nath A (2005) Mechanisms of the blood-brain barrier disruption in HIV-1 infection. Cell Mol Neurobiol 25(1):181–199
Link RW, Mele AR, Antell GC, Pirrone V, Zhong W, Kercher K, Passic S, Szep Z, Malone K, Jacobson JM, Dampier W, Wigdahl B, Nonnemacher MR (2019) Investigating the distribution of HIV-1 Tat lengths present in the Drexel Medicine CARES cohort. Virus Res. https://doi.org/10.1016/j.virusres.2019.197727
Spector C, Mele AR, Wigdahl B, Nonnemacher MR (2019) Genetic variation and function of the HIV-1 Tat protein. Med Microbiol Immunol 208(2):131–169. https://doi.org/10.1007/s00430-019-00583-z
Dickens AM, Yoo SW, Chin AC, Xu J, Johnson TP, Trout AL, Hauser KF, Haughey NJ (2017) Chronic low-level expression of HIV-1 Tat promotes a neurodegenerative phenotype with aging. Sci Rep 7(1):7748. https://doi.org/10.1038/s41598-017-07570-5
Fischer-Smith T, Rappaport J (2005) Evolving paradigms in the pathogenesis of HIV-1-associated dementia. Expert Rev Mol Med 7(27):1–26. https://doi.org/10.1017/S1462399405010239
Atluri VS, Hidalgo M, Samikkannu T, Kurapati KR, Jayant RD, Sagar V, Nair MP (2015) Effect of human immunodeficiency virus on blood-brain barrier integrity and function: an update. Front Cell Neurosci 9:212. https://doi.org/10.3389/fncel.2015.00212
Bagashev Asen SB (2013) Roles and functions of HIV-1 Tat protein in the CNS: an overview. Virol J 10:358
Berman JW, Carvallo L, Buckner CM, Luers A, Prevedel L, Bennett MV, Eugenin EA (2016) HIV-tat alters Connexin43 expression and trafficking in human astrocytes: role in NeuroAIDS. J Neuroinflammation 13(1):54. https://doi.org/10.1186/s12974-016-0510-1
Mediouni S, Darque A, Baillat G, Ravaux I, Dhiver C, Tissot-Dupont H, Mokhtari M, Moreau H, Tamalet C, Brunet C, Paul P, Dignat-George F, Stein A, Brouqui P, Spector SA, Campbell GR, Loret EP (2012) Antiretroviral therapy does not block the secretion of the human immunodeficiency virus tat protein. Infect Disord Drug Targets 12(1):81–86
Falkensammer B, Freissmuth D, Hubner L, Speth C, Dierich MP, Stoiber H (2007) Changes in HIV-specific antibody responses and neutralization titers in patients under ART. Front Biosci 12:2148–2158
Cysique LA, Maruff P, Brew BJ (2004) Prevalence and pattern of neuropsychological impairment in human immunodeficiency virus-infected/acquired immunodeficiency syndrome (HIV/AIDS) patients across pre- and post-highly active antiretroviral therapy eras: a combined study of two cohorts. J Neurovirol 10(6):350–357. https://doi.org/10.1080/13550280490521078
Robertson KR, Smurzynski M, Parsons TD, Wu K, Bosch RJ, Wu J, McArthur JC, Collier AC, Evans SR, Ellis RJ (2007) The prevalence and incidence of neurocognitive impairment in the HAART era. AIDS 21(14):1915–1921. https://doi.org/10.1097/QAD.0b013e32828e4e27
Tozzi V, Balestra P, Bellagamba R, Corpolongo A, Salvatori MF, Visco-Comandini U, Vlassi C, Giulianelli M, Galgani S, Antinori A, Narciso P (2007) Persistence of neuropsychologic deficits despite long-term highly active antiretroviral therapy in patients with HIV-related neurocognitive impairment: prevalence and risk factors. J Acquir Immune Defic Syndr 45(2):174–182. https://doi.org/10.1097/QAI.0b013e318042e1ee
Heaton RK, Clifford DB, Franklin DR Jr, Woods SP, Ake C, Vaida F, Ellis RJ, Letendre SL, Marcotte TD, Atkinson JH, Rivera-Mindt M, Vigil OR, Taylor MJ, Collier AC, Marra CM, Gelman BB, McArthur JC, Morgello S, Simpson DM, McCutchan JA, Abramson I, Gamst A, Fennema-Notestine C, Jernigan TL, Wong J, Grant I, Group C (2010) HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy: CHARTER Study. Neurology 75(23):2087–2096. https://doi.org/10.1212/WNL.0b013e318200d727
Heaton RK, Franklin DR, Ellis RJ, McCutchan JA, Letendre SL, Leblanc S, Corkran SH, Duarte NA, Clifford DB, Woods SP, Collier AC, Marra CM, Morgello S, Mindt MR, Taylor MJ, Marcotte TD, Atkinson JH, Wolfson T, Gelman BB, McArthur JC, Simpson DM, Abramson I, Gamst A, Fennema-Notestine C, Jernigan TL, Wong J, Grant I, Group C, Group H (2011) HIV-associated neurocognitive disorders before and during the era of combination antiretroviral therapy: differences in rates, nature, and predictors. J Neurovirol 17(1):3–16. https://doi.org/10.1007/s13365-010-0006-1
Simioni S, Cavassini M, Annoni JM, Rimbault Abraham A, Bourquin I, Schiffer V, Calmy A, Chave JP, Giacobini E, Hirschel B, Du Pasquier RA (2010) Cognitive dysfunction in HIV patients despite long-standing suppression of viremia. AIDS 24(9):1243–1250. https://doi.org/10.1097/QAD.0b013e3283354a7b
Cysique LA, Brew BJ (2011) Prevalence of non-confounded HIV-associated neurocognitive impairment in the context of plasma HIV RNA suppression. J Neurovirol 17(2):176–183. https://doi.org/10.1007/s13365-011-0021-x
Wang T, Jiang Z, Hou W, Li Z, Cheng S, Green LA, Wang Y, Wen X, Cai L, Clauss M, Wang Z (2014) HIV Tat protein affects circadian rhythmicity by interfering with the circadian system. HIV Med 15(9):565–570. https://doi.org/10.1111/hiv.12154
Clark E, Nava B, Caputi M (2017) Tat is a multifunctional viral protein that modulates cellular gene expression and functions. Oncotarget 8(16):27569–27581. https://doi.org/10.18632/oncotarget.15174
Frankel AD, Young JA (1998) HIV-1: fifteen proteins and an RNA. Annu Rev Biochem 67:1–25. https://doi.org/10.1146/annurev.biochem.67.1.1
Friedrich BM, Dziuba N, Li G, Endsley MA, Murray JL, Ferguson MR (2011) Host factors mediating HIV-1 replication. Virus Res 161(2):101–114. https://doi.org/10.1016/j.virusres.2011.08.001
Ramakrishnan R, Chiang K, Liu H, Budhiraja S, Donahue H, Rice AP (2012) Making a Short Story Long: Regulation of P-TEFb and HIV-1 Transcriptional Elongation in CD4+ T Lymphocytes and Macrophages. Biology (Basel) 1(1):94–115. https://doi.org/10.3390/biology1010094
Carvallo L, Lopez L, Fajardo JE, Jaureguiberry-Bravo M, Fiser A, Berman JW (2017) HIV-Tat regulates macrophage gene expression in the context of neuroAIDS. PLoS ONE 12(6):e0179882. https://doi.org/10.1371/journal.pone.0179882
Debaisieux S, Rayne F, Yezid H, Beaumelle B (2012) The ins and outs of HIV-1 Tat. Traffic 13(3):355–363. https://doi.org/10.1111/j.1600-0854.2011.01286.x
Li L, Dahiya S, Kortagere S, Aiamkitsumrit B, Cunningham D, Pirrone V, Nonnemacher MR, Wigdahl B (2012) Impact of tat genetic variation on HIV-1 disease. Adv Virol 2012:123605. https://doi.org/10.1155/2012/123605
Jeang KT, Xiao H, Rich EA (1999) Multifaceted activities of the HIV-1 transactivator of transcription. Tat J Biol Chem 274(41):28837–28840
Mediouni S, Chinthalapudi K, Ekka MK, Usui I, Jablonski JA, Clementz MA, Mousseau G, Nowak J, Macherla VR, Beverage JN, Esquenazi E, Baran P, de Vera IMS, Kojetin D, Loret EP, Nettles K, Maiti S, Izard T, Valente ST (2019) Didehydro-Cortistatin A Inhibits HIV-1 by Specifically Binding to the Unstructured Basic Region of Tat. mBio 10.1128/mBio.02662–18
Mediouni S, Jablonski J, Paris JJ, Clementz MA, Thenin-Houssier S, McLaughlin JP, Valente ST (2015) Didehydro-cortistatin A inhibits HIV-1 Tat mediated neuroinflammation and prevents potentiation of cocaine reward in Tat transgenic mice. Curr HIV Res 13(1):64–79
Lopez-Huertas MR, Mateos E, Sanchez Del Cojo M, Gomez-Esquer F, Diaz-Gil G, Rodriguez-Mora S, Lopez JA, Calvo E, Lopez-Campos G, Alcami J, Coiras M (2013) The presence of HIV-1 Tat protein second exon delays fas protein-mediated apoptosis in CD4+ T lymphocytes: a potential mechanism for persistent viral production. J Biol Chem 288(11):7626–7644. https://doi.org/10.1074/jbc.M112.408294
Henderson LJ, Johnson TP, Smith BR, Reoma LB, Santamaria UA, Bachani M, Demarino C, Barclay RA, Snow J, Sacktor N, McArthur J, Letendre S, Steiner J, Kashanchi F, Nath A (2019) Presence of Tat and transactivation response element in spinal fluid despite antiretroviral therapy. AIDS 33(Suppl 2):S145–S157. https://doi.org/10.1097/QAD.0000000000002268
Johnson TN, Avindra (2013) Induction of IL-17 and nonclassical T-cell activation by HIV-Tat protein. PNAS
Buscemi L, Ramonet D, Geiger JD (2007) Human immunodeficiency virus type-1 protein Tat induces tumor necrosis factor-alpha-mediated neurotoxicity. Neurobiol Dis 26(3):661–670. https://doi.org/10.1016/j.nbd.2007.03.004
Sacktor N, Nakasujja N, Skolasky RL, Rezapour M, Robertson K, Musisi S, Katabira E, Ronald A, Clifford DB, Laeyendecker O, Quinn TC (2009) HIV subtype D is associated with dementia, compared with subtype A, in immunosuppressed individuals at risk of cognitive impairment in Kampala. Uganda Clin Infect Dis 49(5):780–786. https://doi.org/10.1086/605284
Liner KJ 2nd, Hall CD, Robertson KR (2007) Impact of human immunodeficiency virus (HIV) subtypes on HIV-associated neurological disease. J Neurovirol 13(4):291–304. https://doi.org/10.1080/13550280701422383
Woollard SM, Bhargavan B, Yu F, Kanmogne GD (2014) Differential effects of Tat proteins derived from HIV-1 subtypes B and recombinant CRF02_AG on human brain microvascular endothelial cells: implications for blood-brain barrier dysfunction. J Cereb Blood Flow Metab 34(6):1047–1059. https://doi.org/10.1038/jcbfm.2014.54
Kanki PJ, Hamel DJ, Sankale JL, Hsieh C, Thior I, Barin F, Woodcock SA, Gueye-Ndiaye A, Zhang E, Montano M, Siby T, Marlink R, ND I, Essex ME, MB S (1999) Human immunodeficiency virus type 1 subtypes differ in disease progression. J Infect Dis 179(1):68–73. https://doi.org/10.1086/314557
Mele AR, Marino J, Dampier W, Wigdahl B, Nonnemacher MR (2020) HIV-1 Tat Length: Comparative and Functional Considerations. Front Microbiol 11:444. https://doi.org/10.3389/fmicb.2020.00444
Liu H, Liu J, Xu E, Tu G, Guo M, Liang S, Xiong H (2017) Human immunodeficiency virus protein Tat induces oligodendrocyte injury by enhancing outward K(+) current conducted by KV1.3 Neurobiol Dis 97 (Pt A):1–10. 10.1016/j.nbd.2016.10.007
Zou S, Balinang JM, Paris JJ, Hauser KF, Fuss B, Knapp PE (2019) Effects of HIV-1 Tat on oligodendrocyte viability are mediated by CaMKIIbeta-GSK3beta interactions. J Neurochem 149(1):98–110. https://doi.org/10.1111/jnc.14668
Zou S, Fuss B, Fitting S, Hahn YK, Hauser KF, Knapp PE (2015) Oligodendrocytes Are Targets of HIV-1 Tat: NMDA and AMPA Receptor-Mediated Effects on Survival and Development. J Neurosci 35(32):11384–11398. https://doi.org/10.1523/JNEUROSCI.4740-14.2015
Wheeler NA, Fuss B, Knapp PE, Zou S (2016) HIV-1 Tat Inhibits Autotaxin Lysophospholipase D Activity and Modulates Oligodendrocyte Differentiation. ASN Neuro. https://doi.org/10.1177/1759091416669618
Hauser KF, Hahn YK, Adjan VV, Zou S, Buch SK, Nath A, Bruce-Keller AJ, Knapp PE (2009) HIV-1 Tat and morphine have interactive effects on oligodendrocyte survival and morphology. Glia 57(2):194–206. https://doi.org/10.1002/glia.20746
Niu F, Yao H, Liao K, Buch S (2015) HIV Tat 101-mediated loss of pericytes at the blood-brain barrier involves PDGF-BB. Ther Targets Neurol Dis 2(1) 10.14800/ttnd.471
Nakagawa S, Castro V, Toborek M (2012) Infection of human pericytes by HIV-1 disrupts the integrity of the blood-brain barrier. J Cell Mol Med 16(12):2950–2957. https://doi.org/10.1111/j.1582-4934.2012.01622.x
Nonnemacher MR QS, Allen AG, Mele AR, Pirrone V, Wigdahl B (2017) Myelomonocytic Cell Lines in Modeling HIV-1 Infection of the Bone Marrow. In: A G (ed) Biology of Myelomonocytic Cells. InTech, pp 129–162
Eugenin EA, Osiecki K, Lopez L, Goldstein H, Calderon TM, Berman JW (2006) CCL2/monocyte chemoattractant protein-1 mediates enhanced transmigration of human immunodeficiency virus (HIV)-infected leukocytes across the blood-brain barrier: a potential mechanism of HIV-CNS invasion and NeuroAIDS. J Neurosci 26(4):1098–1106. https://doi.org/10.1523/JNEUROSCI.3863-05.2006
Alkhatib G (2009) The biology of CCR5 and CXCR4. Curr Opin HIV AIDS 4(2):96–103. https://doi.org/10.1097/COH.0b013e328324bbec
Persidsky Y, Ghorpade A, Rasmussen J, Limoges J, Liu XJ, Stins M, Fiala M, Way D, Kim KS, Witte MH, Weinand M, Carhart L, Gendelman HE (1999) Microglial and astrocyte chemokines regulate monocyte migration through the blood-brain barrier in human immunodeficiency virus-1 encephalitis. Am J Pathol 155(5):1599–1611. https://doi.org/10.1016/S0002-9440(10)65476-4
Cenker JJ, Stultz RD, McDonald D (2017) Brain Microglial Cells Are Highly Susceptible to HIV-1 Infection and Spread. AIDS Res Hum Retroviruses 33(11):1155–1165. https://doi.org/10.1089/AID.2017.0004
Zheng L, Yang Y, Guocai L, Pauza CD, Salvato MS (2007) HIV Tat protein increases Bcl-2 expression in monocytes which inhibits monocyte apoptosis induced by tumor necrosis factor-alpha-related apoptosis-induced ligand. Intervirology 50(3):224–228. https://doi.org/10.1159/000100565
Alderson MR, Armitage RJ, Tough TW, Strockbine L, Fanslow WC, Spriggs MK (1993) CD40 expression by human monocytes: regulation by cytokines and activation of monocytes by the ligand for CD40. J Exp Med 178(2):669–674
Borrow P, Tishon A, Lee S, Xu J, Grewal IS, Oldstone MB, Flavell RA (1996) CD40L-deficient mice show deficits in antiviral immunity and have an impaired memory CD8+ CTL response. J Exp Med 183(5):2129–2142
Sui Z, Sniderhan LF, Schifitto G, Phipps RP, Gelbard HA, Dewhurst S, Maggirwar SB (2007) Functional synergy between CD40 ligand and HIV-1 Tat contributes to inflammation: implications in HIV type 1 dementia. J Immunol 178(5):3226–3236
Yang Y, Tikhonov I, Ruckwardt TJ, Djavani M, Zapata JC, Pauza CD, Salvato MS (2003) Monocytes treated with human immunodeficiency virus Tat kill uninfected CD4(+) cells by a tumor necrosis factor-related apoptosis-induced ligand-mediated mechanism. J Virol 77(12):6700–6708
Periyasamy P, Thangaraj A, Bendi VS, Buch S (2019) HIV-1 Tat-mediated microglial inflammation involves a novel miRNA-34a-NLRC5-NFκB signaling axis. Brain Behav Immun 80:227–237. https://doi.org/10.1016/j.bbi.2019.03.011
Pu H, Tian J, Flora G, Lee YW, Nath A, Hennig B, Toborek M (2003) HIV-1 Tat protein upregulates inflammatory mediators and induces monocyte invasion into the brain. Mol Cell Neurosci 24(1):224–237
Brabers NA, Nottet HS (2006) Role of the pro-inflammatory cytokines TNF-alpha and IL-1beta in HIV-associated dementia. Eur J Clin Invest 36(7):447–458. https://doi.org/10.1111/j.1365-2362.2006.01657.x
Song JJ, Lee YJ (2008) Differential cleavage of Mst1 by caspase-7/-3 is responsible for TRAIL-induced activation of the MAPK superfamily. Cell Signal 20(5):892–906. https://doi.org/10.1016/j.cellsig.2008.01.001
Zhang M, Li X, Pang X, Ding L, Wood O, Clouse K, Hewlett I, Dayton AI (2001) Identification of a potential HIV-induced source of bystander-mediated apoptosis in T cells: upregulation of trail in primary human macrophages by HIV-1 tat. J Biomed Sci 8(3):290–296
Chen P, Mayne M, Power C, Nath A (1997) The Tat protein of HIV-1 induces tumor necrosis factor-alpha production. Implications for HIV-1-associated neurological diseases. J Biol Chem 272 (36):22385–22388
Sheng WS, Hu S, Hegg CC, Thayer SA, Peterson PK (2000) Activation of human microglial cells by HIV-1 gp41 and Tat proteins. Clin Immunol 96(3):243–251. https://doi.org/10.1006/clim.2000.4905
Yim HC, Li JC, Lau JS, Lau AS (2009) HIV-1 Tat dysregulation of lipopolysaccharide-induced cytokine responses: microbial interactions in HIV infection. AIDS 23(12):1473–1484. https://doi.org/10.1097/QAD.0b013e32832d7abe
Contreras X, Bennasser Y, Chazal N, Moreau M, Leclerc C, Tkaczuk J, Bahraoui E (2005) Human immunodeficiency virus type 1 Tat protein induces an intracellular calcium increase in human monocytes that requires DHP receptors: involvement in TNF-alpha production. Virology 332(1):316–328. https://doi.org/10.1016/j.virol.2004.11.032
Thangaraj A, Periyasamy P, Liao K, Bendi VS, Callen S, Pendyala G, Buch S (2018) HIV-1 TAT-mediated microglial activation: role of mitochondrial dysfunction and defective mitophagy. Autophagy 14(9):1596–1619. https://doi.org/10.1080/15548627.2018.1476810
Williams ME, Zulu SS, Stein DJ, Joska JA, Naudé PJW (2020) Signatures of HIV-1 subtype B and C Tat proteins and their effects in the neuropathogenesis of HIV-associated neurocognitive impairments. Neurobiol Dis. https://doi.org/10.1016/j.nbd.2019.104701
Koenig S, Gendelman HE, Orenstein JM, Dal Canto MC, Pezeshkpour GH, Yungbluth M, Janotta F, Aksamit A, Martin MA, Fauci AS (1986) Detection of AIDS virus in macrophages in brain tissue from AIDS patients with encephalopathy. Science 233(4768):1089–1093
Wahl SM, Allen JB, McCartney-Francis N, Morganti-Kossmann MC, Kossmann T, Ellingsworth L, Mai UE, Mergenhagen SE, Orenstein JM (1991) Macrophage- and astrocyte-derived transforming growth factor beta as a mediator of central nervous system dysfunction in acquired immune deficiency syndrome. J Exp Med 173(4):981–991
Dheen ST, Kaur C, Ling EA (2007) Microglial activation and its implications in the brain diseases. Curr Med Chem 14(11):1189–1197
Lull ME, Block ML (2010) Microglial activation and chronic neurodegeneration. Neurotherapeutics 7(4):354–365. https://doi.org/10.1016/j.nurt.2010.05.014
Boven LA, Gomes L, Hery C, Gray F, Verhoef J, Portegies P, Tardieu M, Nottet HS (1999) Increased peroxynitrite activity in AIDS dementia complex: implications for the neuropathogenesis of HIV-1 infection. J Immunol 162(7):4319–4327
Nicolini A, Ajmone-Cat MA, Bernardo A, Levi G, Minghetti L (2001) Human immunodeficiency virus type-1 Tat protein induces nuclear factor (NF)-kappaB activation and oxidative stress in microglial cultures by independent mechanisms. J Neurochem 79(3):713–716
Polazzi E, Levi G, Minghetti L (1999) Human immunodeficiency virus type 1 Tat protein stimulates inducible nitric oxide synthase expression and nitric oxide production in microglial cultures. J Neuropathol Exp Neurol 58(8):825–831
Filiano AJ, Gadani SP, Kipnis J (2015) Interactions of innate and adaptive immunity in brain development and function. Brain Res 1617:18–27. https://doi.org/10.1016/j.brainres.2014.07.050
Eugenin EA, Clements JE, Zink MC, Berman JW (2011) Human immunodeficiency virus infection of human astrocytes disrupts blood-brain barrier integrity by a gap junction-dependent mechanism. J Neurosci 31(26):9456–9465. https://doi.org/10.1523/JNEUROSCI.1460-11.2011
Bethel-Brown C, Yao H, Callen S, Lee YH, Dash PK, Kumar A, Buch S (2011) HIV-1 Tat-mediated induction of platelet-derived growth factor in astrocytes: role of early growth response gene 1. J Immunol 186(7):4119–4129. https://doi.org/10.4049/jimmunol.1002235
Tewari M, Monika VRK, Menon M, Seth P (2015) Astrocytes mediate HIV-1 Tat-induced neuronal damage via ligand-gated ion channel P2X7R. J Neurochem 132(4):464–476. https://doi.org/10.1111/jnc.12953
Fan Y, He JJ (2016) HIV-1 Tat Promotes Lysosomal Exocytosis in Astrocytes and Contributes to Astrocyte-mediated Tat Neurotoxicity. J Biol Chem 291(43):22830–22840. https://doi.org/10.1074/jbc.M116.731836
Kim BO, Liu Y, Zhou BY, He JJ (2004) Induction of C chemokine XCL1 (lymphotactin/single C motif-1 alpha/activation-induced, T cell-derived and chemokine-related cytokine) expression by HIV-1 Tat protein. J Immunol 172(3):1888–1895
Chauhan A, Turchan J, Pocernich C, Bruce-Keller A, Roth S, Butterfield DA, Major EO, Nath A (2003) Intracellular human immunodeficiency virus Tat expression in astrocytes promotes astrocyte survival but induces potent neurotoxicity at distant sites via axonal transport. J Biol Chem 278(15):13512–13519. https://doi.org/10.1074/jbc.M209381200
Nookala AR, Shah A, Noel RJ, Kumar A (2013) HIV-1 Tat-mediated induction of CCL5 in astrocytes involves NF-kappaB, AP-1, C/EBPalpha and C/EBPgamma transcription factors and JAK, PI3K/Akt and p38 MAPK signaling pathways. PLoS ONE 8(11):e78855. https://doi.org/10.1371/journal.pone.0078855
Nookala AR, Kumar A (2014) Molecular mechanisms involved in HIV-1 Tat-mediated induction of IL-6 and IL-8 in astrocytes. J Neuroinflammation 11:214. https://doi.org/10.1186/s12974-014-0214-3
El-Hage N, Gurwell JA, Singh IN, Knapp PE, Nath A, Hauser KF (2005) Synergistic increases in intracellular Ca2+, and the release of MCP-1, RANTES, and IL-6 by astrocytes treated with opiates and HIV-1 Tat. Glia 50(2):91–106. https://doi.org/10.1002/glia.20148
El-Hage N, Bruce-Keller AJ, Knapp PE, Hauser KF (2008) CCL5/RANTES gene deletion attenuates opioid-induced increases in glial CCL2/MCP-1 immunoreactivity and activation in HIV-1 Tat-exposed mice. J Neuroimmune Pharmacol 3(4):275–285. https://doi.org/10.1007/s11481-008-9127-1
Jo H, Jang HY, Youn GS, Kim D, Lee CY, Jang JH, Choi SY, Jun JG, Park J (2018) Hindsiipropane B alleviates HIV-1 Tat-induced inflammatory responses by suppressing HDAC6-NADPH oxidase-ROS axis in astrocytes. BMB Rep 51(8):394–399. https://doi.org/10.5483/bmbrep.2018.51.8.061
McManus CM, Weidenheim K, Woodman SE, Nunez J, Hesselgesser J, Nath A, Berman JW (2000) Chemokine and chemokine-receptor expression in human glial elements: induction by the HIV protein, Tat, and chemokine autoregulation. Am J Pathol 156(4):1441–1453. https://doi.org/10.1016/S0002-9440(10)65013-4
Nath A, Conant K, Chen P, Scott C, Major EO (1999) Transient exposure to HIV-1 Tat protein results in cytokine production in macrophages and astrocytes. A hit and run phenomenon. J Biol Chem 274(24):17098–17102
Mayne M, Bratanich AC, Chen P, Rana F, Nath A, Power C (1998) HIV-1 tat molecular diversity and induction of TNF-alpha: implications for HIV-induced neurological disease. NeuroImmunoModulation 5(3–4):184–192
Rao VR, Sas AR, Eugenin EA, Siddappa NB, Bimonte-Nelson H, Berman JW, Ranga U, Tyor WR, Prasad VR (2008) HIV-1 clade-specific differences in the induction of neuropathogenesis. J Neurosci 28(40):10010–10016. https://doi.org/10.1523/JNEUROSCI.2955-08.2008
Fan Y, Zou W, Green LA, Kim BO, He JJ (2011) Activation of Egr-1 expression in astrocytes by HIV-1 Tat: new insights into astrocyte-mediated Tat neurotoxicity. J Neuroimmune Pharmacol 6(1):121–129. https://doi.org/10.1007/s11481-010-9217-8
Abraham S, Sweet T, Sawaya BE, Rappaport J, Khalili K, Amini S (2005) Cooperative interaction of C/EBP beta and Tat modulates MCP-1 gene transcription in astrocytes. J Neuroimmunol 160(1–2):219–227. https://doi.org/10.1016/j.jneuroim.2004.11.009
Song HY, Ryu J, Ju SM, Park LJ, Lee JA, Choi SY, Park J (2007) Extracellular HIV-1 Tat enhances monocyte adhesion by up-regulation of ICAM-1 and VCAM-1 gene expression via ROS-dependent NF-kappaB activation in astrocytes. Exp Mol Med 39(1):27–37. https://doi.org/10.1038/emm.2007.4
Youn GS, Cho H, Kim D, Choi SY, Park J (2017) Crosstalk between HDAC6 and Nox2-based NADPH oxidase mediates HIV-1 Tat-induced pro-inflammatory responses in astrocytes. Redox Biol 12:978–986. https://doi.org/10.1016/j.redox.2017.05.001
Liu Y, Niu Y, Li L, Timani KA, He VL, Sanburns C, Xie J, He JJ (2019) Tat expression led to increased histone 3 tri-methylation at lysine 27 and contributed to HIV latency in astrocytes through regulation of MeCP2 and Ezh2 expression. J NeuroVirol 25(4):508–519. https://doi.org/10.1007/s13365-019-00751-0
Sardo L, Vakil PR, Elbezanti W, El-Sayed A, Klase Z (2016) The inhibition of microRNAs by HIV-1 Tat suppresses beta catenin activity in astrocytes. Retrovirology 13:25. https://doi.org/10.1186/s12977-016-0256-y
Sharma A, Hu XT, Napier TC, Al-Harthi L (2011) Methamphetamine and HIV-1 Tat down regulate beta-catenin signaling: implications for methampetamine abuse and HIV-1 co-morbidity. J Neuroimmune Pharmacol 6(4):597–607. https://doi.org/10.1007/s11481-011-9295-2
Henderson LJ, Sharma A, Monaco MC, Major EO, Al-Harthi L (2012) Human immunodeficiency virus type 1 (HIV-1) transactivator of transcription through its intact core and cysteine-rich domains inhibits Wnt/beta-catenin signaling in astrocytes: relevance to HIV neuropathogenesis. J Neurosci 32(46):16306–16313. https://doi.org/10.1523/JNEUROSCI.3145-12.2012
Hu G, Niu F, Liao K, Periyasamy P, Sil S, Liu J, Dravid SM, Buch S (2019) HIV-1 Tat-Induced Astrocytic Extracellular Vesicle miR-7 Impairs Synaptic Architecture. J Neuroimmune Pharmacol. https://doi.org/10.1007/s11481-019-09869-8
Fan Y, He JJ (2016) HIV-1 Tat Induces unfolded protein response and endoplasmic reticulum stress in astrocytes and causes neurotoxicity through glial fibrillary acidic protein (GFAP) activation and aggregation. J Biol Chem 291(43):22819–22829. https://doi.org/10.1074/jbc.M116.731828
Zhou BY, Liu Y, Kim B, Xiao Y, He JJ (2004) Astrocyte activation and dysfunction and neuron death by HIV-1 Tat expression in astrocytes. Mol Cell Neurosci 27(3):296–305. https://doi.org/10.1016/j.mcn.2004.07.003
Fan Y, Timani KA, He JJ (2015) STAT3 and its phosphorylation are involved in HIV-1 Tat-induced transactivation of glial fibrillary acidic protein. Curr HIV Res 13(1):55–63
Zou S, Fitting S, Hahn YK, Welch SP, El-Hage N, Hauser KF, Knapp PE (2011) Morphine potentiates neurodegenerative effects of HIV-1 Tat through actions at mu-opioid receptor-expressing glia. Brain 134(Pt 12):3616–3631. https://doi.org/10.1093/brain/awr281
Nath A, Hartloper V, Furer M, Fowke KR (1995) Infection of human fetal astrocytes with HIV-1: viral tropism and the role of cell to cell contact in viral transmission. J Neuropathol Exp Neurol 54(3):320–330
Castellano P, Prevedel L, Eugenin EA (2017) HIV-infected macrophages and microglia that survive acute infection become viral reservoirs by a mechanism involving Bim. Sci Rep 7(1):12866. https://doi.org/10.1038/s41598-017-12758-w
El-Hage N, Podhaizer EM, Sturgill J, Hauser KF (2011) Toll-like receptor expression and activation in astroglia: differential regulation by HIV-1 Tat, gp120, and morphine. Immunol Invest 40(5):498–522. https://doi.org/10.3109/08820139.2011.561904
Turchan-Cholewo J, Dimayuga VM, Gupta S, Gorospe RM, Keller JN, Bruce-Keller AJ (2009) NADPH oxidase drives cytokine and neurotoxin release from microglia and macrophages in response to HIV-Tat. Antioxid Redox Signal 11(2):193–204. https://doi.org/10.1089/ARS.2008.2097
Tatro ET, Soontornniyomkij B, Letendre SL, Achim CL (2014) Cytokine secretion from brain macrophages infected with human immunodeficiency virus in vitro and treated with raltegravir. BMC Infect Dis 14:386. https://doi.org/10.1186/1471-2334-14-386
Li W, Huang Y, Reid R, Steiner J, Malpica-Llanos T, Darden TA, Shankar SK, Mahadevan A, Satishchandra P, Nath A (2008) NMDA receptor activation by HIV-Tat protein is clade dependent. J Neurosci 28(47):12190–12198. https://doi.org/10.1523/JNEUROSCI.3019-08.2008
Norman JP, Perry SW, Reynolds HM, Kiebala M, De Mesy Bentley KL, Trejo M, Volsky DJ, Maggirwar SB, Dewhurst S, Masliah E, Gelbard HA (2008) HIV-1 Tat activates neuronal ryanodine receptors with rapid induction of the unfolded protein response and mitochondrial hyperpolarization. PLoS ONE 3(11):e3731. https://doi.org/10.1371/journal.pone.0003731
Aprea S, Del Valle L, Mameli G, Sawaya BE, Khalili K, Peruzzi F (2006) Tubulin-mediated binding of human immunodeficiency virus-1 Tat to the cytoskeleton causes proteasomal-dependent degradation of microtubule-associated protein 2 and neuronal damage. J Neurosci 26(15):4054–4062. https://doi.org/10.1523/JNEUROSCI.0603-06.2006
Di Filippo M, de Iure A, Giampa C, Chiasserini D, Tozzi A, Orvietani PL, Ghiglieri V, Tantucci M, Durante V, Quiroga-Varela A, Mancini A, Costa C, Sarchielli P, Fusco FR, Calabresi P (2016) Persistent activation of microglia and NADPH oxidase [corrected] drive hippocampal dysfunction in experimental multiple sclerosis. Sci Rep 6:20926. https://doi.org/10.1038/srep20926
Gupta S, Knight AG, Gupta S, Knapp PE, Hauser KF, Keller JN, Bruce-Keller AJ (2010) HIV-Tat elicits microglial glutamate release: role of NAPDH oxidase and the cystine-glutamate antiporter. Neurosci Lett 485(3):233–236. https://doi.org/10.1016/j.neulet.2010.09.019
Piani D, Frei K, Do KQ, Cuenod M, Fontana A (1991) Murine brain macrophages induced NMDA receptor mediated neurotoxicity in vitro by secreting glutamate. Neurosci Lett 133(2):159–162
Capone C, Cervelli M, Angelucci E, Colasanti M, Macone A, Mariottini P, Persichini T (2013) A role for spermine oxidase as a mediator of reactive oxygen species production in HIV-Tat-induced neuronal toxicity. Free Radic Biol Med 63:99–107. https://doi.org/10.1016/j.freeradbiomed.2013.05.007
Mastrantonio R, Cervelli M, Pietropaoli S, Mariottini P, Colasanti M, Persichini T (2016) HIV-Tat Induces the Nrf2/ARE Pathway through NMDA Receptor-Elicited Spermine Oxidase Activation in Human Neuroblastoma Cells. PLoS ONE 11(2):e0149802. https://doi.org/10.1371/journal.pone.0149802
Zhou Q, Sheng M (2013) NMDA receptors in nervous system diseases. Neuropharmacology 74:69–75. https://doi.org/10.1016/j.neuropharm.2013.03.030
Haughey NJ, Nath A, Mattson MP, Slevin JT, Geiger JD (2001) HIV-1 Tat through phosphorylation of NMDA receptors potentiates glutamate excitotoxicity. J Neurochem 78(3):457–467
Gupta K, Hardingham GE, Chandran S (2013) NMDA receptor-dependent glutamate excitotoxicity in human embryonic stem cell-derived neurons. Neurosci Lett 543:95–100. https://doi.org/10.1016/j.neulet.2013.03.010
Mishra M, Vetrivel S, Siddappa NB, Ranga U, Seth P (2008) Clade-specific differences in neurotoxicity of human immunodeficiency virus-1 B and C Tat of human neurons: significance of dicysteine C30C31 motif. Ann Neurol 63(3):366–376. https://doi.org/10.1002/ana.21292
Singh IN, Goody RJ, Dean C, Ahmad NM, Lutz SE, Knapp PE, Nath A, Hauser KF (2004) Apoptotic death of striatal neurons induced by human immunodeficiency virus-1 Tat and gp120: Differential involvement of caspase-3 and endonuclease G. J Neurovirol 10(3):141–151. https://doi.org/10.1080/13550280490441103
Singh IN, El-Hage N, Campbell ME, Lutz SE, Knapp PE, Nath A, Hauser KF (2005) Differential involvement of p38 and JNK MAP kinases in HIV-1 Tat and gp120-induced apoptosis and neurite degeneration in striatal neurons. Neuroscience 135(3):781–790. https://doi.org/10.1016/j.neuroscience.2005.05.028
Kruman II, Nath A, Mattson MP (1998) HIV-1 protein Tat induces apoptosis of hippocampal neurons by a mechanism involving caspase activation, calcium overload, and oxidative stress. Exp Neurol 154(2):276–288. https://doi.org/10.1006/exnr.1998.6958
Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME (1995) Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 270(5240):1326–1331
Kuppuswamy M, Subramanian T, Srinivasan A, Chinnadurai G (1989) Multiple functional domains of Tat, the trans-activator of HIV-1, defined by mutational analysis. Nucleic Acids Res 17(9):3551–3561
Dong H, Ye X, Zhong L, Xu J, Qiu J, Wang J, Shao Y, Xing H (2019) Role of FOXO3 activated by HIV-1 Tat in HIV-associated neurocognitive disorder neuronal apoptosis. Front Neurosci. https://doi.org/10.3389/fnins.2019.00044
Masliah E, Heaton RK, Marcotte TD, Ellis RJ, Wiley CA, Mallory M, Achim CL, McCutchan JA, Nelson JA, Atkinson JH, Grant I (1997) Dendritic injury is a pathological substrate for human immunodeficiency virus-related cognitive disorders. HNRC Group. The HIV Neurobehavioral Research Center. Ann Neurol 42(6):963–972. https://doi.org/10.1002/ana.410420618
Kasai H, Matsuzaki M, Noguchi J, Yasumatsu N, Nakahara H (2003) Structure-stability-function relationships of dendritic spines. Trends Neurosci 26(7):360–368. https://doi.org/10.1016/S0166-2236(03)00162-0
Sa MJ, Madeira MD, Ruela C, Volk B, Mota-Miranda A, Paula-Barbosa MM (2004) Dendritic changes in the hippocampal formation of AIDS patients: a quantitative Golgi study. Acta Neuropathol 107(2):97–110. https://doi.org/10.1007/s00401-003-0781-3
Fiala JC, Spacek J, Harris KM (2002) Dendritic spine pathology: cause or consequence of neurological disorders? Brain Res Brain Res Rev 39(1):29–54
Swann JW, Al-Noori S, Jiang M, Lee CL (2000) Spine loss and other dendritic abnormalities in epilepsy. Hippocampus 10(5):617–625. https://doi.org/10.1002/1098-1063(2000)10:5<617:AID-HIPO13>3.0.CO;2-R
Kolson DL, Buchhalter J, Collman R, Hellmig B, Farrell CF, Debouck C, Gonzalez-Scarano F (1993) HIV-1 Tat alters normal organization of neurons and astrocytes in primary rodent brain cell cultures: RGD sequence dependence. AIDS Res Hum Retroviruses 9(7):677–685. https://doi.org/10.1089/aid.1993.9.677
Haughey NJ, Holden CP, Nath A, Geiger JD (1999) Involvement of inositol 1,4,5-trisphosphate-regulated stores of intracellular calcium in calcium dysregulation and neuron cell death caused by HIV-1 protein tat. J Neurochem 73(4):1363–1374
Perry SW, Norman JP, Litzburg A, Zhang D, Dewhurst S, Gelbard HA (2005) HIV-1 transactivator of transcription protein induces mitochondrial hyperpolarization and synaptic stress leading to apoptosis. J Immunol 174(7):4333–4344
Kim HJ, Martemyanov KA, Thayer SA (2008) Human immunodeficiency virus protein Tat induces synapse loss via a reversible process that is distinct from cell death. J Neurosci 28(48):12604–12613. https://doi.org/10.1523/JNEUROSCI.2958-08.2008
Liu Y, Zhou D, Feng J, Liu Z, Hu Y, Liu C, Kong X (2018) HIV-1 protein Tat1–72 impairs neuronal dendrites via activation of PP1 and regulation of the CREB/BDNF pathway. Virologica Sinica 33(3):261–269. https://doi.org/10.1007/s12250-018-0031-4
Kim E, Sheng M (2004) PDZ domain proteins of synapses. Nat Rev Neurosci 5(10):771–781. https://doi.org/10.1038/nrn1517
Shin AH, Thayer SA (2013) Human immunodeficiency virus-1 protein Tat induces excitotoxic loss of presynaptic terminals in hippocampal cultures. Mol Cell Neurosci 54:22–29. https://doi.org/10.1016/j.mcn.2012.12.005
Fischer M, Kaech S, Wagner U, Brinkhaus H, Matus A (2000) Glutamate receptors regulate actin-based plasticity in dendritic spines. Nat Neurosci 3(9):887–894. https://doi.org/10.1038/78791
Fitting S, Ignatowska-Jankowska BM, Bull C, Skoff RP, Lichtman AH, Wise LE, Fox MA, Su J, Medina AE, Krahe TE, Knapp PE, Guido W, Hauser KF (2013) Synaptic dysfunction in the hippocampus accompanies learning and memory deficits in human immunodeficiency virus type-1 Tat transgenic mice. Biol Psychiatry 73(5):443–453. https://doi.org/10.1016/j.biopsych.2012.09.026
Archibald SL, Masliah E, Fennema-Notestine C, Marcotte TD, Ellis RJ, McCutchan JA, Heaton RK, Grant I, Mallory M, Miller A, Jernigan TL (2004) Correlation of in vivo neuroimaging abnormalities with postmortem human immunodeficiency virus encephalitis and dendritic loss. Arch Neurol 61(3):369–376. https://doi.org/10.1001/archneur.61.3.369
Sheng M, Sala C (2001) PDZ domains and the organization of supramolecular complexes. Annu Rev Neurosci 24:1–29. https://doi.org/10.1146/annurev.neuro.24.1.1
May P, Rohlmann A, Bock HH, Zurhove K, Marth JD, Schomburg ED, Noebels JL, Beffert U, Sweatt JD, Weeber EJ, Herz J (2004) Neuronal LRP1 functionally associates with postsynaptic proteins and is required for normal motor function in mice. Mol Cell Biol 24(20):8872–8883. https://doi.org/10.1128/MCB.24.20.8872-8883.2004
Liu Y, Jones M, Hingtgen CM, Bu G, Laribee N, Tanzi RE, Moir RD, Nath A, He JJ (2000) Uptake of HIV-1 tat protein mediated by low-density lipoprotein receptor-related protein disrupts the neuronal metabolic balance of the receptor ligands. Nat Med 6(12):1380–1387. https://doi.org/10.1038/82199
Eugenin EA, D'Aversa TG, Lopez L, Calderon TM, Berman JW (2003) MCP-1 (CCL2) protects human neurons and astrocytes from NMDA or HIV-tat-induced apoptosis. J Neurochem 85(5):1299–1311
Eugenin EA, King JE, Nath A, Calderon TM, Zukin RS, Bennett MV, Berman JW (2007) HIV-tat induces formation of an LRP-PSD-95- NMDAR-nNOS complex that promotes apoptosis in neurons and astrocytes. Proc Natl Acad Sci U S A 104(9):3438–3443. https://doi.org/10.1073/pnas.0611699104
Gandhi N, Saiyed ZM, Napuri J, Samikkannu T, Reddy PV, Agudelo M, Khatavkar P, Saxena SK, Nair MP (2010) Interactive role of human immunodeficiency virus type 1 (HIV-1) clade-specific Tat protein and cocaine in blood-brain barrier dysfunction: implications for HIV-1-associated neurocognitive disorder. J Neurovirol 16(4):294–305. https://doi.org/10.3109/13550284.2010.499891
Hofman FM, Dohadwala MM, Wright AD, Hinton DR, Walker SM (1994) Exogenous tat protein activates central nervous system-derived endothelial cells. J Neuroimmunol 54(1–2):19–28
Zhong Y, Zhang B, Eum SY, Toborek M (2012) HIV-1 Tat triggers nuclear localization of ZO-1 via Rho signaling and cAMP response element-binding protein activation. J Neurosci 32(1):143–150. https://doi.org/10.1523/JNEUROSCI.4266-11.2012
Zhong Y, Smart EJ, Weksler B, Couraud PO, Hennig B, Toborek M (2008) Caveolin-1 regulates human immunodeficiency virus-1 Tat-induced alterations of tight junction protein expression via modulation of the Ras signaling. J Neurosci 28(31):7788–7796. https://doi.org/10.1523/JNEUROSCI.0061-08.2008
Andras IE, Pu H, Deli MA, Nath A, Hennig B, Toborek M (2003) HIV-1 Tat protein alters tight junction protein expression and distribution in cultured brain endothelial cells. J Neurosci Res 74(2):255–265. https://doi.org/10.1002/jnr.10762
Xu R, Feng X, Xie X, Zhang J, Wu D, Xu L (2012) HIV-1 Tat protein increases the permeability of brain endothelial cells by both inhibiting occludin expression and cleaving occludin via matrix metalloproteinase-9. Brain Res 1436:13–19. https://doi.org/10.1016/j.brainres.2011.11.052
Andras IE, Pu H, Tian J, Deli MA, Nath A, Hennig B, Toborek M (2005) Signaling mechanisms of HIV-1 Tat-induced alterations of claudin-5 expression in brain endothelial cells. J Cereb Blood Flow Metab 25(9):1159–1170. https://doi.org/10.1038/sj.jcbfm.9600115
Zou M, Huang W, Jiang W, Wu Y, Chen Q (2019) Role of Cav-1 in HIV-1 Tat-Induced Dysfunction of Tight Junctions and Aβ-Transferring Proteins. Oxidat Med Cell Long 2019:1–8. https://doi.org/10.1155/2019/3403206
Rao VR, Ruiz AP, Prasad VR (2014) Viral and cellular factors underlying neuropathogenesis in HIV associated neurocognitive disorders (HAND). AIDS Res Ther 11:13. https://doi.org/10.1186/1742-6405-11-13
Johnston JB, Zhang K, Silva C, Shalinsky DR, Conant K, Ni W, Corbett D, Yong VW, Power C (2001) HIV-1 Tat neurotoxicity is prevented by matrix metalloproteinase inhibitors. Ann Neurol 49(2):230–241
Acheampong E, Mukhtar M, Parveen Z, Ngoubilly N, Ahmad N, Patel C, Pomerantz RJ (2002) Ethanol strongly potentiates apoptosis induced by HIV-1 proteins in primary human brain microvascular endothelial cells. Virology 304(2):222–234
Hofman FM, Chen P, Incardona F, Zidovetzki R, Hinton DR (1999) HIV-1 tat protein induces the production of interleukin-8 by human brain-derived endothelial cells. J Neuroimmunol 94(1–2):28–39
Ma R, Yang L, Niu F, Buch S (2016) HIV Tat-mediated induction of human brain microvascular endothelial cell apoptosis involves endoplasmic reticulum stress and mitochondrial dysfunction. Mol Neurobiol 53(1):132–142. https://doi.org/10.1007/s12035-014-8991-3
Andras IE, Rampersaud E, Eum SY, Toborek M (2014) Transcriptional profile of HIV-induced nuclear translocation of amyloid beta in brain endothelial cells. Arch Med Res 45(8):744–752. https://doi.org/10.1016/j.arcmed.2014.11.003
Hategan A, Bianchet MA, Steiner J, Karnaukhova E, Masliah E, Fields A, Lee MH, Dickens AM, Haughey N, Dimitriadis EK, Nath A (2017) HIV Tat protein and amyloid-beta peptide form multifibrillar structures that cause neurotoxicity. Nat Struct Mol Biol 24(4):379–386. https://doi.org/10.1038/nsmb.3379
Mahajan SD, Aalinkeel R, Sykes DE, Reynolds JL, Bindukumar B, Fernandez SF, Chawda R, Shanahan TC, Schwartz SA (2008) Tight junction regulation by morphine and HIV-1 tat modulates blood-brain barrier permeability. J Clin Immunol 28(5):528–541. https://doi.org/10.1007/s10875-008-9208-1
Toborek M, Lee YW, Pu H, Malecki A, Flora G, Garrido R, Hennig B, Bauer HC, Nath A (2003) HIV-Tat protein induces oxidative and inflammatory pathways in brain endothelium. J Neurochem 84(1):169–179
Kim TA, Avraham HK, Koh YH, Jiang S, Park IW, Avraham S (2003) HIV-1 Tat-mediated apoptosis in human brain microvascular endothelial cells. J Immunol 170(5):2629–2637
Mishra R, Singh SK (2014) HIV-1 Tat C phosphorylates VE-cadherin complex and increases human brain microvascular endothelial cell permeability. BMC Neurosci 15:80. https://doi.org/10.1186/1471-2202-15-80
Weiss JM, Nath A, Major EO, Berman JW (1999) HIV-1 Tat induces monocyte chemoattractant protein-1-mediated monocyte transmigration across a model of the human blood-brain barrier and up-regulates CCR5 expression on human monocytes. J Immunol 163(5):2953–2959
Ivey NS, MacLean AG, Lackner AA (2009) Acquired immunodeficiency syndrome and the blood-brain barrier. J Neurovirol 15(2):111–122. https://doi.org/10.1080/13550280902769764
Hu G, Yang L, Cai Y, Niu F, Mezzacappa F, Callen S, Fox HS, Buch S (2016) Emerging roles of extracellular vesicles in neurodegenerative disorders: focus on HIV-associated neurological complications. Cell Death Dis 7(11):e2481. https://doi.org/10.1038/cddis.2016.336
Bergonzini V, Calistri A, Salata C, Del Vecchio C, Sartori E, Parolin C, Palu G (2009) Nef and cell signaling transduction: a possible involvement in the pathogenesis of human immunodeficiency virus-associated dementia. J Neurovirol 15(3):238–248. https://doi.org/10.1080/13550280902939748
Koedel U, Kohleisen B, Sporer B, Lahrtz F, Ovod V, Fontana A, Erfle V, Pfister HW (1999) HIV type 1 Nef protein is a viral factor for leukocyte recruitment into the central nervous system. J Immunol 163(3):1237–1245
Nakamuta S, Endo H, Higashi Y, Kousaka A, Yamada H, Yano M, Kido H (2008) Human immunodeficiency virus type 1 gp120-mediated disruption of tight junction proteins by induction of proteasome-mediated degradation of zonula occludens-1 and -2 in human brain microvascular endothelial cells. J Neurovirol 14(3):186–195. https://doi.org/10.1080/13550280801993630
Kanmogne Georgette SK, Persidsky Yuri (2007) HIV-1 gp120 compromises blood-brain barrier integrity and enhance monocyte migration across blood-brain barrier: implication for viral neuropathogenesis. Cereb Blood Flow Metab
Cotto B, Natarajaseenivasan K, Langford D (2019) Astrocyte activation and altered metabolism in normal aging, age-related CNS diseases, and HAND. J Neurovirol 25(5):722–733. https://doi.org/10.1007/s13365-019-00721-6
Ferrucci A, Nonnemacher MR, Cohen EA, Wigdahl B (2012) Extracellular human immunodeficiency virus type 1 viral protein R causes reductions in astrocytic ATP and glutathione levels compromising the antioxidant reservoir. Virus Res 167(2):358–369. https://doi.org/10.1016/j.virusres.2012.06.002
Ferrucci A, Nonnemacher MR, Wigdahl B (2013) Extracellular HIV-1 viral protein R affects astrocytic glyceraldehyde 3-phosphate dehydrogenase activity and neuronal survival. J Neurovirol 19(3):239–253. https://doi.org/10.1007/s13365-013-0170-1
Funding
The authors were funded in part by the Public Health Service, National Institutes of Health, through grants from the National Institute of Neurological Disorders and Stroke (NINDS) R01 NS089435 (PI, Michael R. Nonnemacher), the NIMH Comprehensive NeuroAIDS Center (CNAC) P30 MH092177 (Kamel Khalili, PI; Brian Wigdahl, PI of the Drexel subcontract involving the Clinical and Translational Research Support Core) and under the Ruth L. Kirschstein National Research Service Award T32 MH079785 (PI, Kamel Khalili and Tricia Burdo; with Brian Wigdahl serving as the PI of the Drexel University College of Medicine component and Olimpia Meucci as Co-Director). The contents of the paper are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Marino, J., Maubert, M.E., Mele, A.R. et al. Functional impact of HIV-1 Tat on cells of the CNS and its role in HAND. Cell. Mol. Life Sci. 77, 5079–5099 (2020). https://doi.org/10.1007/s00018-020-03561-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-020-03561-4