
Abstract
Although the incidence of HIV-associated dementia (HAD) has declined, HIV-associated neurocognitive disorders (HAND) remain a significant health problem despite use of highly active antiretroviral therapy. In addition, the incidence and/or severity of HAND/HAD are increased with concomitant use of drugs of abuse, such as cocaine, marijuana, and methamphetamine. Furthermore, exposure to most drugs of abuse increases brain levels of dopamine, which has been implicated in the pathogenesis of HIV. This review evaluates the potential role of dopamine in the potentiation of HAND/HAD by drugs of abuse. In the brain, multiplication of HIV in infected macrophages/microglia could result in the release of HIV proteins such as gp120 and Tat, which can bind to and impair dopamine transporter (DAT) functions, leading to elevated levels of dopamine in the dopaminergic synapses in the early asymptomatic stage of HIV infection. Exposure of HIV-infected patients to drugs of abuse, especially cocaine and methamphetamine, can further increase synaptic levels of dopamine via binding to and subsequently impairing the function of DAT. This accumulated synaptic dopamine can diffuse out and activate adjacent microglia through binding to dopamine receptors. The activation of microglia may result in increased HIV replication as well as increased production of inflammatory mediators such as tumor necrosis factor (TNF)-alpha and chemokines. Increased HIV replication can lead to increased brain viral load and increased shedding of HIV proteins, gp120 and Tat. These proteins, as well as TNF-alpha, can induce cell death of adjacent dopaminergic neurons via apoptosis. Autoxidation and metabolism of accumulated synaptic dopamine can lead to generation of reactive oxygen species (hydrogen peroxide), quinones, and semiquinones, which can also induce apoptosis of neurons. Increased cell death of dopaminergic neurons can eventually lead to dopamine deficit that may exacerbate the severity and/or accelerate the progression of HAND/HAD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In the era of highly active antiretroviral therapy (HAART), although the incidence of HIV-associated dementia (HAD) has declined, HIV-associated neurocognitive disorders (HAND) remain a significant health problem. It is estimated that about 50% of the HIV-infected individuals are affected with HAND despite use of HAART [1]. This is probably due to the fact that HIV-infected individuals on HAART live longer [2] and the poor ability of most of the antiretroviral drugs, especially protease inhibitors, to penetrate blood–brain barrier (BBB) [3, 4]. In addition, incidence and/or severity of HAD/HAND are increased with concomitant use of drugs of abuse such as cocaine, marijuana, and methamphetamine [5–10]. Furthermore, exposure to most drugs of abuse increases brain levels of dopamine well beyond basal levels and levels associated with “natural rewards” (e.g., food, sex) [11–15]. Finally, dopamine has been implicated in HIV and simian immunodeficiency virus (SIV)-induced brain pathology [16, 17], and shown to promote HIV replication in macrophages [18]. The purpose of this review is to evaluate the potential role of dopamine in the development of HAND/HAD and in the exacerbation of these conditions by drugs of abuse.
HIV/AIDS and Brain Pathology in Dopamine-Rich Areas
Studies investigating relationship between HIV-I infection and brain pathology have revealed that this virus can cause selective damage to dopamine-rich areas of the brain. For example, using quantitative neuroimaging, selective basal ganglia atrophy was reported in HAD patients [19]. In addition, in autopsied patients who died of AIDS, significant degeneration of neurons was reported in the substantia nigra [20, 21]. Furthermore, atrophy of the caudate nucleus (a dopamine-rich part of basal ganglia) was significantly associated with cognitive impairment in HIV-infected patients [22–24]. Finally, severe neuronal degeneration was observed in dopamine-rich areas (substantia nigra and globus pallidus) of the brain in SIV-infected monkeys [25]. Neuronal degeneration in dopamine-rich areas appears to be associated with higher HIV load in these brain regions. Indeed, HIV-1 proviral DNA load was significantly higher in the medial temporal lobe (dopamine-rich area) than in the frontal lobe [26]. HIV-1 gp41-positive microglia were most numerous in globus pallidus obtained from the brain of AIDS encephalitis patients [27]. Relatively higher levels of HIV-1 RNA were found in dopamine-rich parts of human brain samples (substantia nigra, caudate nucleus, basal ganglia, and globus pallidus) obtained at autopsy [17, 28]. Overall, these data suggest that while HIV infection can generally cause brain atrophy, “dopamine-rich” areas can show proportionally more atrophy, suggesting a propensity for HIV in targeting these brain areas.
HIV/AIDS and Dopamine Levels
Several researchers have investigated a connection between HIV infection and dopamine levels measured in postmortem brain samples and cerebrospinal fluid (CSF). In a study, dopamine and its metabolite homovanillic acid (HVA) concentrations were both significantly reduced in caudate nucleus tissue (dopamine-rich area) taken at postmortem from patients with AIDS compared to tissues taken from age-matched controls [29]. This finding was associated with a loss of nigrostriatal dopaminergic neurons, suggesting a HIV-induced neuronal damage followed by dopamine depletion. In another postmortem sample study, Kumar et al. [17] found a significant decrease in dopamine levels in dopamine-rich brain regions, including caudate nucleus, putamen, globus pallidus, and substantia nigra obtained from HIV-1-positive subjects, who actually died of HIV/AIDS with no opportunistic infection at the time of death, compared to dopamine levels observed in HIV-negative cases. In contrast, in HIV-negative cases, the highest concentration of dopamine was found in dopamine-rich areas, including putamen, caudate, substantia nigra, and the basal ganglia. In this study, brain regions having the highest HIV-1 RNA had the maximum decrease in dopamine levels, indicating HIV-1-associated damage to dopaminergic neurons. There were no differences in dopamine and HVA content in the brain regions between HIV-positive cases who used HAART and HIV-positive cases who did not use HAART.
These postmortem studies show similarities to those studies using in vivo brain imaging technology such as magnetic resonance imaging and positron emission tomography, with HIV-1-positive patients. These reports show reduction in brain volume and metabolic activity in the basal ganglia, particularly the caudate nucleus [19, 30]. Interestingly, Rottenberg et al. [31] showed dynamic changes in the metabolic activity of the basal ganglia, over the course of HIV infection with hyper-metabolism in the early stages of infection followed by hypo-metabolism in late, more advanced, stages of the disease.
Researchers have also measured dopamine levels in CSF as a surrogate marker for dopamine levels in the brain. Both higher and lower levels of dopamine have been reported in the CSF of HIV-infected patients. In one study, CSF dopamine levels were significantly lower in the HIV-1-seropositive group than in the seronegative group [32]. Importantly, the decreased CSF dopamine levels were correlated with HIV status and neurological disease. CSF levels of dopamine in HIV-1-positive individuals without neurological disease were on average approximately 60% less than in seronegative controls, and those seropositive individuals with evidence of neurological disease on average had 80% less dopamine in CSF. In addition, decreased levels of HVA have also been reported in the CSF of HIV-infected patients [33, 34]. Importantly, Larsson et al. [33] observed the decrease only during the AIDS stage. Obermann at al [35] went further using transcranial sonographic (TCS) examination to assess the morphology of the substantia nigra, a region rich in dopaminergic cell bodies, and its relationship to dopamine levels in the CSF, and with neuropsychological performance in different stages of HIV infection. This group concluded that early nigrostriatal damage is mediated through HIV neurotoxicity, which can be detected using TCS and that this change is correlated with dopamine depletion in the CSF in therapy-naive asymptomatic HIV-infected patients. These data further suggest HIV infections can mediate dopaminergic hypofunction.
In contrast to the previous studies, in a recent study, dopamine levels were significantly increased by twofold, along with 3,4-dihydroxyphenylacetic acid (another metabolite of dopamine) in the CSF (collected from lumber puncture) of therapy-naive HIV-infected non-drug-abusing patients during early asymptomatic stage, when compared to uninfected control [36]. In this study, the increase in dopamine levels was accompanied by a decrease in dopamine turnover. In addition, the increase in dopamine levels was not accompanied by adaptive changes in dopamine transporter (DAT) or dopamine receptor 2 availability measured with receptor single-photon emission tomography imaging. These discrepant results may be accounted for if it is assumed that initially HIV infection leads to an increase in extracellular dopamine levels. These increased levels in turn may exert toxic effects on dopamine neurons through oxidative properties of catecholamines. This cytotoxicity may ultimately result in neurodegeneration of dopamine neurons and lower levels of brain dopamine.
Overall, these studies suggest a connection between HIV-1 infection, dopamine levels, and neuronal degeneration of dopamine-rich areas of the brain; however, they do not explain the underlying mechanisms of this correlation or directly test the interrelationship between changes in dopamine level and HIV infection and their role in neuronal degeneration or brain atrophy. To address this issue directly, researchers have used retroviral infection models in monkeys and rats.
Dopamine Studies in Simian Immunodeficient Virus -Infected Monkey Model
In the SIV model of HIV infection, affected Rhesus macaques show clinical symptoms and neurological complications that mimic those observed in human neuro-AIDS. Rhesus macaques inoculated with macrophage-tropic, neurovirulent SIV (SIVmac R71/17E) exhibit neuroAIDS-like neuro-pathological changes in brain, including microglial nodules, extensive perivascular cuffing, and the presence of multinucleated giant cells in dopamine-rich areas (globus pallidus and substantia nigra) of the brain [25]. Researchers have used this model to determine the interrelationship between dopamine and HIV-induced brain pathology [16, 37, 38].
Researcher [37–39] have shown that during early asymptomatic phase of 2–3 months post-SIV infection, dopamine levels were significantly decreased in dopamine-rich brain areas such as putamen, frontal cortex, hippocampus, and nuclear accumbens, and the levels were further decreased as the infection progressed to the stage of AIDS [38]. In addition, surface expression of major histocompatibility complex II (MHC II) (a marker for activated microglia) was increased on microglia isolated from animals sacrificed during the asymptomatic phase of SIV and the expression of this marker was further increased in animals with the development of AIDS [38].
Administration of selegiline, a monoamine oxidase-B (MAO-B) inhibitor, or L-3, 4-dihydroxyphenylalanine (l-DOPA) (a precursor of dopamine that can cross BBB) increased dopamine levels in the brain [37]. Both selegiline and l-DOPA increased SIV RNA expression in dopamine-rich forebrain/midbrain areas (basal ganglia, frontal cortex, substantia nigra) but not in dopamine-poor hind brain areas (nuclei of the reticular formation, the raphe nuclei, cerebellum) [16, 37]. Both substances aggravated SIV infection-related neuropathology and ultrastructural alterations of dendrites in dopamine-rich areas in asymptomatic animals [16, 37], and enhanced the expression of TNF-alpha, a marker of microglia activation [16].
Collectively, these findings suggest that (a) SIV infection-induced activation of microglia during asymptomatic phase may decrease brain dopamine levels due to degeneration of dopaminergic neurons. With the progression of disease, further activation of microglia may contribute to the advancement of neuronal degeneration, which may result in further decline in dopamine levels, and (b) selegiline or l-DOPA-induced elevated dopamine levels may be responsible for elevated brain SIV load and enhanced brain pathology in dopamine-rich areas. The enhanced pathology could have been mediated via activation of microglia as shown by increased expression of TNF-alpha and MHC II.
Role of Dopamine in a Retroviral Rat Model of Neurodegeneration Induced by Murine Leukemia Virus NT40
Murine leukemia virus NT40 is known to induce degeneration of the central nervous system in neonatal rats. In this model, administration of selegiline (1.0 mg/kg body weight at 17 days post-infection) significantly shortened the incubation period of neurological disease, increased viral load in CNS, increased number of cells expressing MHC II molecules, and exhibited inhibition of MAO-B activity in comparison to untreated but infected (control) animals [40]. These results suggest that selegiline-induced elevated levels of dopamine activated microglia that presumably resulted in viral replication and accelerated neurodegeneration.
Dopamine and HIV Replication
A few investigators have used in vitro studies investigating the role of dopamine in HIV replication using lymphocytes or macrophages [18, 41, 42]. In an early study, Rohr et al. [41] showed that dopamine (100 μM) stimulated HIV-1 transcription and replication (p24 antigen) in Jurkat T cells (T lymphocytes) and in peripheral blood mononuclear cells (PBMC). In this study, dopamine stimulated expression (transcription) of HIV-1 via nuclear factor-kappaB (NF-κB) activation. In another study, dopamine in 20–100 μM concentrations dose dependently increased HIV-1 replication (as measured by HIV-1 p24 antigen) in chronically HIV-infected T-lymphoblasts ACH-2 cells [42]. Treatment with antioxidants (glutathione and N-acetylcysteine) attenuated dopamine-induced-HIV activation indicating that changes in cellular redox states (oxidative stress) might have triggered HIV replication. Recently, dopamine (20 μM) was shown to increase HIV replication in human monocyte-derived macrophages (MDM) inoculated with R5 strain of HIV [18]. In this study, increased viral replication was associated with increased number of HIV-infected macrophages. In addition to dopamine, the dopamine receptor-2 agonist quinpirole (but not dopamine receptor-1 agonist, SKF82958) was also shown to increase viral replication similar to dopamine. Taken together, these results show in vitro that dopamine promotes HIV replication in lymphocytes and macrophages, and in human macrophages, this effect is mediated, at least in part through dopamine receptor-2 activation.
It is important to note that dopamine concentrations used in vitro studies (described above), to promote HIV replications were in micromolar range. Whether HIV infection and drugs of abuse combined can elevate dopamine levels in this range to increase HIV-infected macrophages in vivo is unknown. Recently, Scheller et al. [36] reported dopamine concentrations only in the range of 14–28 nM in the CSF (obtained from lumber puncture) collected from non-drug abusing HIV-infected patients during early asymptomatic stage of infection. It is likely that this concentration of dopamine represents only a fraction of what was actually available in the brain. However, dopamine concentrations in the lumber region are likely to be lower than those measured in dopamine-rich areas of the CNS. Furthermore, dopamine levels could have been higher if HIV-infected subjects were also exposed to drugs of abuse, such as cocaine or methamphetamine that act directly on dopaminergic neurons and increase brain extracellular dopamine. Alternatively, microglia in the dopamine-rich areas of brain could be more sensitive to dopamine activation than MDM used in vitro study referred above [18]. Thus, it is possible that dopamine concentrations, particularly in HIV-infected drug-abusing patients could reach a level that promotes HIV replication in microglia. The potential of dopamine to promote HIV replication is further corroborated in monkey studies wherein increasing endogenous dopamine levels by administering selegiline or l-DOPA promoted SIV replication [16, 37]. Similar results were also reported in a retroviral rat model of neurodegeneration where administration of seligiline increased murine leukemia viral replication in CNS [40].
Dopamine may promote HIV replication through various mechanisms: (1) Nair et al. [43] showed that in mature human dendritic cells, the dopamine agonist methamphetamine upregulated gene and protein expression of DC-SIGN, which is known to facilitate the attachment of HIV to the dendritic cells. This methamphetamine-induced increase in DC-SIGN was reversed by the dopamine receptor-1 antagonist (SCH23390) and by siRNA specific to dopamine receptor-1. Thus, dopamine may be involved in the attachment of HIV to dendritic cells via modulating the expression of DC-SIGN and thereby indirectly impact viral replication, although the underlying mechanism is not clear; (2) In human monocyte-derived dendritic cells, methamphetamine-induced HIV-1 infectivity was also associated with increased expression of CCR5, a HIV-1 co-receptor required for viral entry primarily into monocyte [44]. Methamphetamine-induced CCR5 upregulation was completely reversed by blocking the dopamine receptor-1 or dopamine receptor-2. Thus, dopamine may also facilitate HIV entry into monocytes/macrophages by modulating CCR5 expression and thereby indirectly modulating viral replication, although the underlying mechanism is not clear; (3) Methamphetamine increased reverse transcriptase activity (a marker of HIV replication and infectivity) in MDM infected with macrophage-tropic R5 HIV strain in a dose-dependent manner. Methamphetamine-induced reverse transcriptase activity was completely blocked by dopamine receptor-1 antagonists (SCH23390 and SKF83566) [45]. Thus, dopamine may regulate HIV replication by modulating HIV reverse transcriptase activity, although the underlying mechanism is not clear; (4) Dopamine has been shown to increase HIV transcription in Jurkat T lymphocytes, PBMC, and human brain cells [41, 46, 47]. This may be another mechanism by which dopamine may promote HIV replication. In summary, dopamine may promote HIV replication by increasing the expression of DC-SIGN and CCR5, increasing reverse transcriptase activity, and by increasing HIV transcription via NF-κB activation.
The data described thus far clearly show that dopamine can modulate HIV replication and the dopamine-mediated mechanisms involved in HIV replication may depend on the stages of HIV life cycle beginning with viral attachment to dendritic cells, monocytes, and lymphocytes through HIV reverse transcription to HIV transcription.
HIV and Dopamine Transporter
The DAT located on presynaptic axonal plasma membrane play an important role in regulating the concentration of dopamine in the synapse. Once dopamine is released from the pre- and post-synaptic receptors into the synapse, DAT transfer dopamine back into the cytoplasm of the pre-synaptic terminal [48–50].
It is now well-recognized that within 2 weeks of primary systemic infection, HIV-1-infected monocytes can cross BBB and reach to the different brain regions [10]. In the brain, HIV-1 can further multiply in macrophages and microglia and release HIV proteins, gp120 and Tat, both of which can interact with neurons. Tat has been shown to impair the function of DAT in animal and in vitro studies [51–53]. Likewise, gp120 has also been shown to impair the function of DAT in cultured human dopaminergic neurons [54]. Clinical studies using positron emission tomography have found that HIV-1-infected patients with HAD display a significant reduction in DAT density in the putamen and ventral striatum [55, 56].
Like Tat and gp120, DAT is also a major target for drugs of abuse, especially cocaine and methamphetamine. Cocaine binds to DAT with high affinity [57–59], impairing its function of dopamine reuptake, which leads to an increase in extracellular levels of dopamine [60]. Methamphetamine impairs dopamine reuptake from synapse as well [61–63], and can also impair the function of intracellular vesicular dopamine transporter [64–66], thereby enhancing DAT-mediated transport of dopamine from the cytoplasm into the synapse [67]. These properties of methamphetamine explain, at least in part, why methamphetamine is more potent than cocaine and other drugs of abuse in increasing synaptic concentrations of dopamine [11].
By impairing the function of DAT, HIV proteins and drugs of abuse alone or together can increase dopamine levels in the synapse in an additive or synergistic manner. This dopamine could bind to the neighboring microglia through dopamine receptors in a paracrine manner and consequently promote HIV replication in HIV-infected microglia.
Dopamine, HIV, and Oxidative Stress
Metabolism of dopamine via MAO-B or amine autoxidation can give rise to radical species (hydrogen peroxide, semiquinones, and quinones), which, in turn, can damage cellular components such as lipids, proteins, and DNA [68]. Both l-DOPA and dopamine have been shown to induce neuronal (Neuro-2A cells) cell death, which was attenuated by antioxidants (ascorbic acid, glutathione, and N-acetylcysteine), suggesting a role for oxidative stress in cell toxicity [69]. Dopamine quinone was shown to cause a time-dependent activation of cultured microglia, as shown by increased expression of inflammatory genes, TNF-alpha, and chemokines [70]. These inflammatory mediators could induce injury to the neighboring dopaminergic neurons, leading to their degeneration.
HIV-dementia has been demonstrated to be associated with oxidative stress as shown by increased protein and lipid peroxidation in the brain and CSF, compared to HIV non-demented patients [71, 72]. The HIV protein, Tat, has been shown to induce oxidative stress in brain endothelial cells [73, 74] and synaptosomal membrane and neuronal cell cultures [75]. In addition, HIV protein gp120 has also been implicated in oxidative stress-induced damage to dopaminergic neurons. Cultured dopaminergic neurons were more sensitive than non-dopaminergic neurons (cholinergic and GABAergic) to HIV gp120-induced oxidative damage, as shown by reduced function (decreased dopamine uptake) and increased apoptosis [54, 76]. Increased sensitivity of dopaminergic neurons could be due to cumulative oxidative stress generated by HIV proteins (gp120 and Tat) as well as by dopamine metabolism. Increased levels of dopamine due to drugs of abuse may further exacerbate dopaminergic neuronal degeneration by potentiating oxidative stress via auto oxidation or releasing oxidative metabolites. Thus, it is plausible that cumulative oxidative stress generated by HIV proteins and dopamine metabolism may potentiate severity and accelerate progression of HAND/HAD. It is interesting to note that dopamine receptor-1 is involved in Tat-induced neurotoxicity [77] where oxidative stress may play a contributory role.
Summary
Summary of this report can be divided into two parts:
-
A.
Role of dopamine in the development of HAND/HAD in the absence of drugs of abuse exposure:
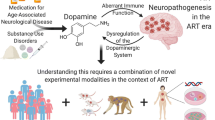
This part of the summary has been illustrated in Fig. 1. HIV-infected monocytes can cross BBB and reach different parts of brain within 2 weeks of infection. HIV can multiply in infected macrophages/microglia in the brain and release HIV proteins such as gp120 and Tat, which can bind to and impair DAT functions, leading to elevated levels of dopamine in the dopaminergic synapses in the early asymptomatic stage of HIV infection. Synaptic dopamine can diffuse out and bind to the adjacent microglia through dopamine receptors and promote HIV replication probably via increasing expression of DC-SIGN and CCR5, activation of reverse transcriptase, and/or increasing the transcription of HIV via generation of oxidative stress and activation of NF-κB. Increased HIV replication can lead to elevated brain viral load that can further increase shedding of gp120 and Tat. Both of these proteins can cause dopamine neuronal cell death via inducing oxidative stress. Autoxidation and metabolism of accumulated synaptic dopamine can also result in the generation of reactive oxygen species (ROS) (hydrogen peroxide), quinones, and semiquinones, which can also induce neuronal apoptosis. Synaptic dopamine can also activate microglia, leading to the production of inflammatory mediators such as cytokines (TNF-alpha) and chemokines. These inflammatory mediators can also cause damage to nearby dopaminergic neurons in a paracrine manner, ensuing apoptotic death of these cells. With the advancement of HIV infection from asymptomatic to symptomatic stage, increased multiplication of HIV, and development of AIDS, significant dopaminergic neurons are destroyed, which can lead to atrophy of dopamine-rich regions and dopamine deficiency observed in these areas in autopsy samples. These changes in dopamine system can lead to the development of HAND/HAD.
-
B.
Role of dopamine in drugs of abuse-mediated exacerbation of HAND/HAD
This part of the summary has been illustrated in Fig. 2. Exposure of HIV-infected patients to drugs of abuse, especially cocaine and methamphetamine, can further increase synaptic levels of dopamine via binding to and subsequently impairing the function of DAT. This accumulated synaptic dopamine can diffuse out and activate adjacent microglia through binding to dopamine receptors. The activation of microglia may result in increased HIV replication as well as increased production of inflammatory mediators such as TNF-alpha and chemokines. Increased HIV replication can lead to increased brain viral load and increased shedding of HIV proteins, gp120 and Tat. These proteins, as well as TNF-alpha, can induce cell death of adjacent dopaminergic neurons via apoptosis. Autoxidation and metabolism of accumulated synaptic dopamine can lead to generation of ROS (hydrogen peroxide), quinones, and semiquinones, which can also induce apoptosis of neurons. Increased cell death of dopaminergic neurons can eventually lead to dopamine deficit that may exacerbate the severity and/or accelerate the progression of HAND/HAD.

Role of dopamine (DA) in the development of HAND/HAD. HIV-infected monocytes can cross blood–brain barrier (BBB) and reach different parts of brain where they are transformed into macrophages, which can further infect microglia. HIV can multiply in infected macrophages/microglia in the brain, which results in increased viral load in the brain and shedding of HIV proteins, gp120 and Tat. These proteins can bind to and impair functions of DAT, leading to elevated levels of DA in the dopaminergic synapses. Synaptic dopamine can diffuse out and bind to the adjacent microglia through dopamine receptors and activates these cells to promote HIV replication and stimulate production of inflammatory mediators such as cytokines (TNF-alpha) and chemokines. Increased HIV replication can lead to elevated brain viral load that can further increase shedding of gp120 and Tat. Both of these proteins can cause dopamine neuronal cell death. Inflammatory mediators can also induce apoptosis of dopaminergic neurons. Increased cell death of these neurons can lead to significant dopamine deficit and subsequently development of HAND/HAD

Role of dopamine (DA) in drugs of abuse-mediated exacerbation of HAND/HAD. Exposure of HIV-infected patients to drugs of abuse, especially cocaine and methamphetamine, can further increase synaptic levels of dopamine via binding to and subsequently impairing the function of DAT. This accumulated synaptic dopamine can diffuse out and activate adjacent microglia through binding to dopamine receptors. The activation of microglia may result in increased HIV replication as well as increased production of inflammatory mediators such as TNF-alpha and chemokines. Increased HIV replication can lead to increased brain viral load and increased shedding of HIV proteins, gp120 and Tat. These proteins, as well as TNF-alpha, can induce cell death of adjacent dopaminergic neurons via apoptosis. Autoxidation and metabolism of accumulated synaptic dopamine can lead to generation of ROS (hydrogen peroxide), quinones, and semiquinones, which can also induce apoptosis of neurons. Increased cell death of dopaminergic neurons can eventually lead to dopamine deficit that may exacerbate the severity and/or accelerate the progression of HAND/HAD
Conclusions
It is hypothesized that dopamine accumulated in the synaptic areas of the brain of HIV-infected (asymptomatic early stage) drug-abusing individuals has potential to activate adjacent microglia and subsequently promote HIV replication in these cells. Inflammatory mediators released from the activated microglia and ROS generated from dopamine metabolism have potential to cause damage to dopaminergic neurons, which may exacerbate the severity and/or accelerate the progression of HAND/HAD.
Future Directions
-
Determine dopamine levels in the brain and CSF at different stages of HIV infection, especially in the early asymptomatic stage in the absence of microglial activation.
-
Determine brain dopamine levels in HIV-infected individuals exposed to drugs of abuse.
-
Investigate the ability of dopamine to stimulate HIV replication in a cell line more close to human microglia located in dopamine-rich areas of the brain.
-
Evaluate the effect of dopamine and its metabolites in the generation of ROS in the dopamine synaptic areas.
-
Evaluate the role of dopamine and its metabolites in inducing cell death of dopaminergic neurons via inducing oxidative stress.
-
Determine the combined effect of HIV proteins (Tat and gp120) and drugs of abuse on the function of DAT.
-
Determine the effect of dopamine and its metabolites on BBB permeability for transmigration of HIV-infected monocytes.
-
Develop an in vitro model of dopaminergic neurons and microglia to understand the interactive effect of HIV proteins and dopamine on inducing neuronal cell death.
References
Clifford DB (2008) HIV-associated neurocognitive disease continues in the antiretroviral era. Top HIV Med 16:94–98
Anthony IC, Bell JE (2008) The neuropathology of HIV/AIDS. Int Rev Psychiatr 20:15–24
Dore GJ, Correll PK, Li Y, Kaldor JM, Cooper DA, Brew BJ (1999) Changes to AIDS dementia complex in the era of highly active antiretroviral therapy. AIDS 13:1249–1253
Thomas SA (2004) Anti-HIV drug distribution to the central nervous system. Curr Pharm Des 10:1313–1324
Davies J, Everall IP, Weich S, McLaughlin J, Scaravilli F, Lantos PL (1997) HIV-associated brain pathology in the United Kingdom: an epidemiological study. AIDS 11:1145–1150
Nath A, Maragos WF, Avison MJ, Schmitt FA, Berger JR (2001) Acceleration of HIV dementia with methamphetamine and cocaine. J Neurovirol 7:66–71
Cristiani SA, Pukay-Martin ND, Bornstein RA (2004) Marijuana use and cognitive function in HIV-infected people. J Neuropsychiatry Clin Neurosci 16:330–335
Rippeth JD, Heaton RK, Carey CL, Marcotte TD, Moore DJ, Gonzalez R, Wolfson T, Grant I, HNRC Group (2004) Methamphetamine dependence increases risk of neuropsychological impairment in HIV infected persons. J Int Neuropsychol Soc 10:1–14
Burdo TH, Katner SN, Taffe MA, Fox HS (2006) Neuroimmunity, drugs of abuse, and neuroAIDS. J Neuroimmune Pharmacol 1:41–49
Ferris MJ, Mactutus CF, Booze RM (2008) Neurotoxic profiles of HIV, psychostimulant drugs of abuse, and their concerted effect on the brain: current status of dopamine system vulnerability in NeuroAIDS. Neurosci Biobehav Rev 32:883–909
Di Chiara G, Imperato A (1988) Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci U S A 85:5274–5278
Pontieri FE, Tanda G, Di Chiara G (1995) Intravenous cocaine, morphine, and amphetamine preferentially increase extracellular dopamine in the “shell” as compared with the “core” of the rat nucleus accumbens. Proc Natl Acad Sci U S A 92:12304–12308
Bradberry CW (2000) Acute and chronic dopamine dynamics in a nonhuman primate model of recreational cocaine use. J Neurosci 20:7109–7115
Walker QD, Kuhn CM (2008) Cocaine increases stimulated dopamine release more in periadolescent than adult rats. Neurotoxicol Teratol 30:412–418
Brody AL, London ED, Olmstead RE, Allen-Martinez Z, Shulenberger S, Costello MR, Abrams AL, Scheibal D, Farahi J, Shoptaw S, Mandelkern MA (2010) Smoking-induced change in intrasynaptic dopamine concentration: effect of treatment for tobacco dependence. Psychiatr Res 183:218–224
Czub S, Czub M, Koutsilieri E, Sopper S, Villinger F, Müller JG, Stahl-Hennig C, Riederer P, Ter Meulen V, Gosztonyi G (2004) Modulation of simian immunodeficiency virus neuropathology by dopaminergic drugs. Acta Neuropathol 107:216–226
Kumar AM, Fernandez JB, Singer EJ, Commins D, Waldrop-Valverde D, Ownby RL, Kumar M (2009) Human immunodeficiency virus type 1 in the central nervous system leads to decreased dopamine in different regions of postmortem human brains. J Neurovirol 15:257–274
Gaskill PJ, Calderon TM, Luers AJ, Eugenin EA, Javitch JA, Berman JW (2009) Human immunodeficiency virus (HIV) infection of human macrophages is increased by dopamine: a bridge between HIV-associated neurologic disorders and drug abuse. Am J Pathol 175:1148–1159
Aylward EH, Henderer JD, McArthur JC, Brettschneider PD, Harris GJ, Barta PE, Pearlson GD (1993) Reduced basal ganglia volume in HIV-1-associated dementia: results from quantitative neuroimaging. Neurology 43:2099–2104
Reyes MG, Faraldi F, Senseng CS, Flowers C, Fariello R (1991) Nigral degeneration in acquired immune deficiency syndrome (AIDS). Acta Neuropathol 82:39–44
Itoh K, Mehraein P, Weis S (2000) Neuronal damage of the substantia nigra in HIV-1 infected brains. Acta Neuropathol 99:376–384
Dal Pan GJ, McArthur JH, Aylward E, Selnes OA, Nance-Sproson TE, Kumar AJ, Mellits ED, McArthur JC (1992) Patterns of cerebral atrophy in HIV-1-infected individuals: results of a quantitative MRI analysis. Neurology 42:2125–2130
Hestad K, McArthur JH, Dal Pan GJ, Selnes OA, Nance-Sproson TE, Aylward E, Mathews VP, McArthur JC (1993) Regional brain atrophy in HIV-1 infection: association with specific neuropsychological test performance. Acta Neurol Scand 88:112–118
Paul R, Cohen R, Navia B, Tashima K (2002) Relationships between cognition and structural neuroimaging findings in adults with human immunodeficiency virus type-1. Neurosci Biobehav Rev 26:353–359
Marcario JK, Manaye KF, SantaCruz KS, Mouton PR, Berman NE, Cheney PD (2004) Severe subcortical degeneration in macaques infected with neurovirulent simian immunodeficiency virus. J Neurovirol 10:387–399
Fujimura RK, Goodkin K, Petito CK, Douyon R, Feaster DJ, Concha M, Shapshak P (1997) HIV-1 proviral DNA load across neuroanatomic regions of individuals with evidence for HIV-1-associated dementia. J Acquir Immune Defic Syndr Hum Retrovirol 16:146–152
Kure K, Weidenheim KM, Lyman WD, Dickson DW (1990) Morphology and distribution of HIV-1 gp41-positive microglia in subacute AIDS encephalitis. Pattern of involvement resembling a multisystem degeneration. Acta Neuropathol 80:393–400
Kumar AM, Borodowsky I, Fernandez B, Gonzalez L, Kumar M (2007) Human immunodeficiency virus type 1 RNA Levels in different regions of human brain: quantification using real-time reverse transcriptase-polymerase chain reaction. J Neurovirol 13:210–224
Sardar AM, Czudek C, Reynolds GP (1996) Dopamine deficits in the brain: the neurochemical basis of parkinsonian symptoms in AIDS. NeuroReport 7:910
Kieburtz K, Ketonen L, Cox C, Grossman H, Holloway R, Booth H, Hickey C, Feigin A, Caine ED (1996) Cognitive performance and regional brain volume in human immunodeficiency virus type 1 infection. Arch Neurol 53:155–158
Rottenberg DA, Sidtis JJ, Strother SC, Schaper KA, Anderson JR, Nelson MJ, Price RW (1996) Abnormal cerebral glucose metabolism in HIV-1 seropositive subjects with and without dementia. J Nucl Med 37:1133–1141
Berger JR, Kumar M, Kumar A, Fernandez JB, Levin B (1994) Cerebrospinal fluid dopamine in HIV-1 infection. AIDS 8:67–71
Larsson M, Hagberg L, Forsman A, Norkrans G (1991) Cerebrospinal fluid catecholamine metabolites in HIV-infected patients. J Neurosci Res 28:406–409
di Rocco A, Bottiglieri T, Dorfman D, Werner P, Morrison C, Simpson D (2000) Decreased homovanilic acid in cerebrospinal fluid correlates with impaired neuropsychologic function in HIV-1-infected patients. Clin Neuropharmacol 23:190–194
Obermann M, Küper M, Kastrup O, Yaldizli O, Esser S, Thiermann J, Koutsilieri E, Arendt G, Diener HC, Maschke M, German Competence Network HIV/AIDS (2009) Substantia nigra hyperechogenicity and CSF dopamine depletion in HIV. J Neurol 256:948–953
Scheller C, Arendt G, Nolting T, Antke C, Sopper S, Maschke M, Obermann M, Angerer A, Husstedt IW, Meisner F, Neuen-Jacob E, Müller HW, Carey P, Ter Meulen V, Riederer P, Koutsilieri E (2010) Increased dopaminergic neurotransmission in therapy-naïve asymptomatic HIV patients is not associated with adaptive changes at the dopaminergic synapses. J Neural Transm 117:699–705
Czub S, Koutsilieri E, Sopper S, Czub M, Stahl-Hennig C, Müller JG, Pedersen V, Gsell W, Heeney JL, Gerlach M, Gosztonyi G, Riederer P, ter Meulen V (2001) Enhancement of central nervous system pathology in early simian immunodeficiency virus infection by dopaminergic drugs. Acta Neuropathol 101:85–91
Scheller C, Sopper S, Jenuwein M, Neuen-Jacob E, Tatschner T, Grünblatt E, ter Meulen V, Riederer P, Koutsilieri E (2005) Early impairment in dopaminergic neurotransmission in brains of SIV-infected rhesus monkeys due to microglia activation. J Neurochem 95:377–387
Jenuwein M, Scheller C, Neuen-Jacob E, Sopper S, Tatschner T, ter Meulen V, Riederer P, Koutsilieri E (2004) Dopamine deficits and regulation of the cAMP second messenger system in brains of simian immunodeficiency virus-infected rhesus monkeys. J Neurovirol 10:163–170
Czub M, Czub S, Gosztonyi G, Koutsilieri E, Sopper S, Müller JG, Gerlach M, Riederer P, ter Meulen V (1999) Effects of selegiline in a retroviral rat model for neurodegenerative disease. J Neurovirol 5:458–464
Rohr O, Sawaya BE, Lecestre D, Aunis D, Schaeffer E (1999) Dopamine stimulates expression of the human immunodeficiency virus type 1 via NF-kappaB in cells of the immune system. Nucleic Acids Res 27:3291–3299
Scheller C, Sopper S, Jassoy C, ter Meulen V, Riederer P, Koutsilieri E (2000) Dopamine activates HIV in chronically infected T lymphoblasts. J Neural Transm 107:1483–1489
Nair MP, Mahajan S, Sykes D, Bapardekar MV, Reynolds JL (2006) Methamphetamine modulates DC-SIGN expression by mature dendritic cells. J Neuroimmune Pharmacol 1:296–304
Nair MP, Saiyed ZM, Nair N, Gandhi NH, Rodriguez JW, Boukli N, Provencio-Vasquez E, Malow RM, Miguez-Burbano MJ (2009) Methamphetamine enhances HIV-1 infectivity in monocyte derived dendritic cells. J Neuroimmune Pharmacol 4:129–139
Liang H, Wang X, Chen H, Song L, Ye L, Wang SH, Wang YJ, Zhou L, Ho WZ (2008) Methamphetamine enhances HIV infection of macrophages. Am J Pathol 172:1617–1624
Sawaya BE, Rohr O, Aunis D, Schaeffer E (1996) Chicken ovalbumin upstream promoter transcription factor, a transcriptional activator of HIV-1 gene expression in human brain cells. J Biol Chem 271:23572–23576
Rohr O, Schwartz C, Aunis D, Schaeffer E (1999) CREB and COUP-TF mediate transcriptional activation of the human immunodeficiency virus type 1 genome in Jurkat T cells in response to cyclic AMP and dopamine. J Cell Biochem 75:404–413
Cami J, Farre D (2003) Drug addiction. N Engl J Med 349:975–986
Volz TJ, Hanson GR, Fleckenstein AE (2007) The role of the plasmalemmal dopamine and vesicular monoamine transporters in methamphetamine-induced dopaminergic deficits. J Neurochem 101:883–888
Seger D (2010) Cocaine, metamfetamine, and MDMA abuse: the role and clinical importance of neuroadaptation. Clin Toxicol (Phila) 48:695–708
Ferris MJ, Frederick-Duus D, Fadel J, Mactutus CF, Booze RM (2009) In vivo microdialysis in awake, freely moving rats demonstrates HIV-1 Tat-induced alterations in dopamine transmission. Synapse 63:181–185
Ferris MJ, Frederick-Duus D, Fadel J, Mactutus CF, Booze RM (2009) The human immunodeficiency virus-1-associated protein, Tat1-86, impairs dopamine transporters and interacts with cocaine to reduce nerve terminal function: a no-net-flux microdialysis study. Neuroscience 159:1292–1299
Zhu J, Mactutus CF, Wallace DR, Booze RM (2009) HIV-1 Tat protein-induced rapid and reversible decrease in [3H]dopamine uptake: dissociation of [3H]dopamine uptake and [3H]2beta-carbomethoxy-3-beta-(4-fluorophenyl)tropane (WIN 35,428) binding in rat striatal synaptosomes. J Pharmacol Exp Ther 329:1071–1083
Hu S, Sheng WS, Lokensgard JR, Peterson PK, Rock RB (2009) Preferential sensitivity of human dopaminergic neurons to gp120-induced oxidative damage. J Neurovirol 15:401–410
Wang GJ, Chang L, Volkow ND, Telang F, Logan J, Ernst T, Fowler JS (2004) Decreased brain dopaminergic transporters in HIV-associated dementia patients. Brain 127:2452–2458
Chang L, Wang GJ, Volkow ND, Ernst T, Telang F, Logan J, Fowler JS (2008) Decreased brain dopamine transporters are related to cognitive deficits in HIV patients with or without cocaine abuse. NeuroImage 42:869–878
Ritz MC, Lamb RJ, Goldberg SR, Kuhar MJ (1987) Cocaine receptors on dopamine transporters are related to self-administration of cocaine. Science 237:1219–1223
Amara SG, Kuhar MJ (1993) Neurotransmitter transporters: recent progress. Annu Rev Neurosci 16:73–93
Han DD, Gu HH (2006) Comparison of the monoamine transporters from human and mouse in their sensitivities to psychostimulant drugs. BMC Pharmacol 6:6
Reith ME, Li MY, Yan QS (1997) Extracellular dopamine, norepinephrine, and serotonin in the ventral tegmental area and nucleus accumbens of freely moving rats during intracerebral dialysis following systemic administration of cocaine and other uptake blockers. Psychopharmacology (Berl) 134:309–317
Fleckenstein AE, Metzger RR, Wilkins DG, Gibb JW, Hanson GR (1997) Rapid and reversible effects of methamphetamine on dopamine transporters. J Pharmacol Exp Ther 282:834–838
Kokoshka JM, Vaughan RA, Hanson GR, Fleckenstein AE (1998) Nature of methamphetamine-induced rapid and reversible changes in dopamine transporters. Eur J Pharmacol 361:269–275
Metzger RR, Hanson GR, Gibb JW, Fleckenstein AE (1998) 3-4-Methylenedioxymethamphetamine-induced acute changes in dopamine transporter function. Eur J Pharmacol 349:205–210
Brown JM, Hanson GR, Fleckenstein AE (2000) Methamphetamine rapidly decreases vesicular dopamine uptake. J Neurochem 74:2221–2223
Hogan KA, Staal RG, Sonsalla PK (2000) Analysis of VMAT2 binding after methamphetamine or MPTP treatment: disparity between homogenates and vesicle preparations. J Neurochem 74:2217–2220
Brown JM, Hanson GR, Fleckenstein AE (2001) Regulation of the vesicular monoamine transporter-2: a novel mechanism for cocaine and other psychostimulants. J Pharmacol Exp Ther 296:762–767
Khoshbouei H, Wang H, Lechleiter JD, Javitch JA, Galli A (2003) Amphetamine-induced dopamine efflux. A voltage-sensitive and intracellular Na+-dependent mechanism. J Biol Chem 278:12070–12077
Graham DG (1978) Oxidative pathways for catecholamines in the genesis of neuromelanin and cytotoxic quinones. Mol Pharmacol 14:633–643
Pedrosa R, Soares-da-Silva P (2002) Oxidative and non-oxidative mechanisms of neuronal cell death and apoptosis by L-3,4-dihydroxyphenylalanine (l-DOPA) and dopamine. Br J Pharmacol 137:1305–1313
Kuhn DM, Francescutti-Verbeem DM, Thomas DM (2006) Dopamine quinones activate microglia and induce a neurotoxic gene expression profile: relationship to methamphetamine-induced nerve ending damage. Ann N Y Acad Sci 1074:31–41
Aksenov MY, Hasselrot U, Bansal AK, Wu G, Nath A, Anderson C, Mactutus CF, Booze RM (2001) Oxidative damage induced by the injection of HIV-1 Tat protein in the rat striatum. Neurosci Lett 305:5–8
Pocernich CB, Sultana R, Mohmmad-Abdul H, Nath A, Butterfield DA (2005) HIV-dementia, Tat-induced oxidative stress, and antioxidant therapeutic considerations. Brain Res Brain Res Rev 50:14–26
Toborek M, Lee YW, Pu H, Malecki A, Flora G, Garrido R, Hennig B, Bauer HC, Nath A (2003) HIV-Tat protein induces oxidative and inflammatory pathways in brain endothelium. Neurochem 84:169–179
Price TO, Ercal N, Nakaoke R, Banks WA (2005) HIV-1 viral proteins gp120 and Tat induce oxidative stress in brain endothelial cells. Brain Res 1045:57–63
Pocernich CB, Sultana R, Hone E, Turchan J, Martins RN, Calabrese V, Nath A, Butterfield DA (2004) Effects of apolipoprotein E on the human immunodeficiency virus protein Tat in neuronal cultures and synaptosomes. J Neurosci Res 77:532–539
Agrawal L, Louboutin JP, Marusich E, Reyes BA, Van Bockstaele EJ, Strayer DS (2010) Dopaminergic neurotoxicity of HIV-1 gp120: reactive oxygen species as signaling intermediates. Brain Res 1306:116–130
Silvers JM, Aksenova MV, Aksenov MY, Mactutus CF, Booze RM (2007) Neurotoxicity of HIV-1 Tat protein: involvement of D1 dopamine receptor. NeuroToxicology 28:1184–1190
Acknowledgments
We are thankful to Dr. Gayathri Dowling for the critical review of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Purohit, V., Rapaka, R. & Shurtleff, D. Drugs of Abuse, Dopamine, and HIV-Associated Neurocognitive Disorders/HIV-Associated Dementia. Mol Neurobiol 44, 102–110 (2011). https://doi.org/10.1007/s12035-011-8195-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-011-8195-z