
Abstract
Aging is associated with a significant deficiency in circulating insulin-like growth factor-1 (IGF-1), which has an important role in the pathogenesis of age-related vascular cognitive impairment (VCI). Impairment of moment-to-moment adjustment of regional cerebral blood flow via neurovascular coupling (NVC) importantly contributes to VCI. Previous studies established a causal link between circulating IGF-1 deficiency and neurovascular dysfunction. Release of vasodilator mediators from activated astrocytes plays a key role in NVC. To determine the impact of impaired IGF-1 signaling on astrocytic function, astrocyte-mediated NVC responses were studied in a novel mouse model of astrocyte-specific knockout of IGF1R (GFAP-CreERT2/Igf1rf/f) and accelerated neurovascular aging. We found that mice with disrupted astrocytic IGF1R signaling exhibit impaired NVC responses, decreased stimulated release of the vasodilator gliotransmitter epoxy-eicosatrienoic acids (EETs), and upregulation of soluble epoxy hydrolase (sEH), which metabolizes and inactivates EETs. Collectively, our findings provide additional evidence that IGF-1 promotes astrocyte health and maintains normal NVC, protecting cognitive health.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Vascular cognitive impairment (VCI) in the aging population has emerged as one of the major public health challenges in the Western world [1,2,3]. In addition to pathological alterations of the larger cerebral arteries (e.g., atherosclerosis), functional impairment of the cerebral microcirculation also contributes significantly to the pathogenesis of VCI [4]. In recent years, potentially reversible functional alterations of the aging neurovascular unit have garnered much attention as they have the potential to impair local regulation of cerebral blood flow (CBF) and have been causally linked to neuronal dysfunction [2, 4]. The neurovascular unit is a complex functional and anatomical structure, which consists of perivascular astrocytes (the most abundant glial cells in the brain, which outnumber neurons by fivefold), endothelial cells, vascular smooth muscle cells and pericytes, microglia, and neurons. One area of particular interest when it comes to understanding the contribution of the neurovascular unit to the pathogenesis of VCI is the role of impairment of neurovascular coupling (NVC), a critical homeostatic mechanism responsible for adjusting regional CBF to local neural activity [5]. Upon neuronal activation, the resulting functional hyperemia contributes to the maintenance of an optimal local microenvironment in the active brain region, ensuring adequate delivery of oxygen and glucose, and effective removal of potentially harmful metabolic by-products. Aging results in marked impairment of NVC, which likely contributes to cognitive dysfunction in elderly patients and aged laboratory animals [6,7,8,9,10,11,12].
The cellular mechanisms underlying NVC include astrocyte activation induced by neurotransmitters released from firing neurons and consequential astrocytic release of vasodilator mediators that induce prompt vasodilation in arterioles supplying the active brain region [13,14,15,16]. Gliotransmitters involved in mediation of NVC responses include vasodilator metabolites of arachidonic acid, including epoxyeicosatrienoic acids (EETs) produced by cytochrome p450 enzymes [17,18,19]. EETs released from astrocytes elicit dilation of resistance arterioles by activating K+ channels in the arteriolar smooth muscle cells [20]. Additional mechanisms that contribute to dilation of resistance arterioles induced by astrocyte activation include purinergic mechanisms, release of prostaglandins, and K+ channel activation [13, 16, 21,22,23,24,25,26,27,28,29,30]. Despite the critical role of astrocytes in mediation of functional hyperemia, the impact of shared molecular and cellular mechanisms of aging on astrocyte-mediated NVC responses is not yet fully understood.
There is growing evidence in support of the concept that cell non-autonomous mechanisms of aging play a critical role in brain and neurovascular aging [31,32,33,34,35,36,37]. Insulin-like growth factor-1 (IGF-1) is an anabolic hormone produced by the liver and, locally, by diverse cells within the CNS, which exerts multifaceted neuroprotective, vasoprotective, and anti-geronic effects [5, 23, 31, 38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62]. Both circulating and brain concentrations of free IGF-1 decrease significantly with age in humans and in laboratory animals due to an age-related decline in GH production/release [31, 62,63,64,65]. Astrocytes abundantly express IGF1R, the receptor for IGF-1, and there is strong evidence that disruption of IGF1R signaling results in marked changes in astrocyte phenotype, which associate with cognitive impairment [50]. Previous studies also demonstrate that circulating IGF-1 deficiency also leads to impaired NVC responses [23]. Despite these advances, the specific role of IGF1R signaling in regulation of astrocytic mediation of NVC responses remains elusive.
The present study was designed to test the hypotheses that IGF1R signaling regulates astrocyte-mediated NVC responses and synthesis/release of vasodilator eicosanoid gliotransmitters in the brain and that disruption of IGF1R signaling in astrocytes impair gliovascular coupling, mimicking aspects of the aging phenotype. To test our hypotheses, we used a novel mouse model with adult-onset, astrocyte-specific disruption of IGF1R signaling using Cre-lox technology (GFAP-CreERT2/Igf1rf/f) [50]. NVC, synthesis of eicosanoid gliotransmitters, and expression of soluble epoxy hydrolase, which degrades EETs, were tested.
Methods
Animals
Igf1rf/f (B6;129-Igf1rtm2Arge/J; loxP sites flanking exon 3) and GFAP-Cre ERT2 (B6.Cg-TgGFAP-cre/ERT2/505Fmv/J) mice were obtained from Jackson laboratories. Mice were housed (3–4 per cage) in Allentown XJ cages with Anderson’s Enrich-o-cob bedding (Maumee, OH). Igf1rf/f mice were bred in house to generate experimental cohorts. Animals were housed under specific pathogen-free (including helicobacter and parvovirus free) barrier conditions in the Rodent Barrier Facility at University of Oklahoma Health Sciences Center. Mice were bred on a 14-h light/10-h dark cycle and weaned mice were maintained in a 12-h light/12-h dark cycle at 21 °C and were given access to standard irradiated bacteria-free rodent chow (5053 Pico Lab, Purina Mills, Richmond, IN) and reverse osmosis filtered water ad libitum. GFAP-CreERT2 (males) mice were bred with Igf1rf/f (females) to generate GFAP-CreERT2/Igf1r+/− males, which were bred with Igf1rf/f female mice to obtain the founder colony of Cre+/Igf1r homozygous floxed mice as previously described [50]. These mice were allowed to breed with Igf1rf/f mice to generate experimental cohorts of GFAP-CreERT2/Igf1rf/f and Cre-/Igf1rf/f control mice. Mice were injected intraperitoneally with tamoxifen (75 mg tamoxifen/kg body weight) dissolved in corn oil or sham (corn oil) only for 5 days at 3 months. Mice were allowed to recover for 2 months before initiation of experiments. All procedures were approved by the Institutional Animal Use and Care Committee of the University of Oklahoma Health Sciences Center.
Measurement of neurovascular coupling responses
On the day of experimentation, mice in each group were anesthetized with isoflurane (4% induction and 1% maintenance), endotracheally intubated, and ventilated (MousVent G500; Kent Scientific Co, Torrington, CT). A thermostatic heating pad (Kent Scientific Co, Torrington, CT) was used to maintain rectal temperature at 37°C [12]. End-tidal CO2 was controlled between 3.2 and 3.7% to keep blood gas values within the physiological range, as described [23, 66]. The right femoral artery was canulated for arterial blood pressure measurement (Living Systems Instrumentations, Burlington, VT) [12]. The blood pressure was within the physiological range throughout the experiments (90–110 mmHg). Mice were immobilized and placed on a stereotaxic frame (Leica Microsystems, Buffalo Grove, IL), the scalp and periosteum were pulled aside, and the skull was gently thinned using a dental drill while cooled with dripping buffer. A laser speckle contrast imager (Perimed, Järfälla, Sweden) was placed 10 cm above the thinned skull, and to achieve the highest CBF response, the right whiskers were stimulated for 30 s at 10 Hz from side to side as described [67, 68]. Differential perfusion maps of the brain surface were captured. Changes in CBF were assessed above the left barrel cortex in six trials in each group, separated by 5–10-min intervals. To assess the role of EETs in mediation of NVC, CBF responses to whisker stimulation were repeated after administrating the epoxigenase inhibitor MSSPOH (N-(methylsulfonyl)-2-(2-propynyloxy)-benzenehexanamide; Cayman Chemicals; 20 mg/kg, dissolved in DMSO and diluted to final concentration with 45% cyclodextrin, Cayman Chemicals) which inhibits EET production [23, 69]. MS-PPOH is a selective inhibitor of the epoxygenation reactions catalyzed by specific CYP450 isozymes [70]. MS-PPOH inhibits the formation of arachidonate 11,12-epoxides by CYP4A2 and CYP4A3 enzymes with an IC50 value of 13 μM, but has no effect on the formation of 20-HETE, the ω-hydroxylation product of CYP4A1 [71]. Changes in CBF were averaged and expressed as percent (%) increase from the baseline value [72]. Experiments lasted <1 h/mouse, which permitted stable physiological parameters to be obtained. In each study, the experimenter was blinded to the treatment of the animals. At the end of the experiments, the animals (with the exception of those assigned to brain lipidomics studies) were transcardially perfused with ice-cold PBS and decapitated. Animals assigned to brain lipidomics studies were decapitated without perfusion to avoid wash-out of lipid mediators. The brains were immediately removed and samples were collected for subsequent studies. All reagents used in this study were purchased from Sigma-Aldrich (St Louis, MO) unless otherwise indicated.
Measurement of glutamate-induced release of EETs from acute hippocampal slices
To determine how disruption of IGF1R signaling affects synthesis of eicosanoid gliotransmitters, horizontal hippocampal slices of 325 μm thickness from mice in each cohort were prepared using a HM650V vibrating microtome (Thermo Scientific) in ice cold solution containing (in mmol/L) sucrose 110, NaCl 60, KCl 3, NaH2PO4 1.25, NaHCO3 28, sodium ascorbate acid 0.6, glucose 5, MgCl2 7, and CaCl2 0.5 as reported [73]. Slices were then transferred to a holding chamber containing oxygenated artificial cerebrospinal fluid (aCSF) of the following composition (in mM): NaCl 126, KCl 2.5, NaH2PO4 1.25, MgCl2 2, CaCl2 2, NaHCO3 26, glucose 10, pyruvic acid 2, ascorbic acid 0.4. Slices were left to recover for at least 60 min at room temperature prior to experimentation, then were transferred to a 24-well plate containing oxygenated aCSF, 2 slices per plate. Five minutes later, 500uL of aCSF was removed, mixed with 1mL of LC-MS grade methanol (ThermoFisher, A456-1), snap frozen, and used for control purposes. To activate astrocytes, glutamate (3×10−4 mol/L, for 5 min) was added to the chamber. Then, the aCSF was removed, mixed with 1mL of LC-MS grade methanol, and snap-frozen for analyses. Brain slices were snap frozen for protein concentration analyses to normalize the lipidomics data. Identification and quantification of EET gliotransmitters involved in NVC responses (5,6-EET, 8,9-EET, 11,12-EET, and 14,15-EET and their degradation products, 5,6-DiHET [5,6-dihydroxy-eicosatrienoic acid], 8,9-DiHET, 11,12-DiHET, and 14,15-DiHET), by LC-MS/MS was performed by the Schwartzman laboratory using a Shimadzu Triple Quadrupole Mass Spectrometer LCMS-8050 and multiple reaction monitoring mode [74]. Protein concentrations from frozen brain slices were used to normalize the data.
Western blotting
Cortex samples were homogenized in RIPA buffer containing HALT protease and phosphatase inhibitors. The tissue homogenate was centrifuged at 12,000rpm for 10 min and the supernatant was isolated for protein estimation by Thermoscientific BCA assay. Equal amounts of protein (45μg) were resolved on NuPAGE 10% Bis-Tris Midi gel and then transferred on to a PVDF membrane using semi-dry transfer method (BioRad). The membranes were blocked using 5% BSA in Tris-buffered saline-Tween 20 (TBST) for 1h at RT followed by overnight incubation with the following primary antibodies at 4°C: IGFR1β (1:1000, Cell Signaling); sEH (1:200, Cayman Chemicals); and β-actin (1:5000, Abcam). The membranes were washed 3X with TBST and then incubated with the respective HRP-conjugated antibodies (Abcam, 1:10000) for 1 h at RT. The membranes were washed again 3X with TBST and developed using SuperSignal West Pico or SuperSignal West Femto chemiluminescent substrate solutions (Thermo Fisher Scientific). Digital images were obtained using myECL imaging system (Thermoscientific) and the densitometric analysis was performed using Fiji software.
Statistical analysis
Statistical analysis was carried out by unpaired t test or one-way ANOVA followed by Bonferroni multiple comparison test, as appropriate, using Prism 5.0 for Windows (Graphpad Software, La Jolla, CA). A p value less than 0.05 was considered statistically significant. Data are expressed as mean ± S.E.M.
Results
Astrocyte-specific disruption of IGF-1/IGF1R signaling impairs neurovascular coupling
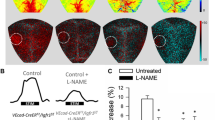
Changes in CBF in the whisker barrel cortex in response to contralateral whisker stimulation were significantly attenuated in GFAP-CreERT2/Igf1rf/f mice (Fig. 1a–c), indicating that astrocyte-specific disruption of IGF1R signaling leads to neurovascular uncoupling.

Astrocyte-specific disruption of IGF1R signaling impairs neurovascular coupling responses. a Representative pseudocolour laser speckle flowmetry maps of baseline CBF (upper row; shown for orientation purposes) and CBF changes in the whisker barrel field relative to baseline during contralateral whisker stimulation (bottom row, right oval, 30 s, 5 Hz) in control and GFAP-CreERT2/Igf1rf/f mice before and after administration of the P450 epoxygenase inhibitor MS-PPOH. Panel b shows the time-course of CBF changes after the start of contralateral whisker stimulation (horizontal bars). Summary data are shown in panel c. Data are mean ± S.E.M. (n=6–10 in each group), *P<0.05 vs. control; #P<0.05 vs. untreated. n.s. not significant
Upon activation by neuronal-derived glutamate, astrocytes were shown to convert arachidonic acid by P450 epoxygenase to vasodilator EETs [16, 23]. Consistent with this concept, we found that in control animals, administration of the P450 epoxygenase inhibitor MS-PPOH (Fig. 1b–c) significantly decreased CBF responses in the barrel cortex elicited by contralateral whisker stimulation. In GFAP-CreERT2/Igf1rf/f mice, the effects of MS-PPOH (Fig. 1b–c) were significantly decreased, suggesting that astrocyte-specific disruption of IGF1R signaling impairs mediation of NVC by EETs.
LC/MS/MS measurements demonstrated that astrocyte-specific disruption of IGF1R signaling resulted in a diminished cerebral production of EETs in response to glutamate stimulation of brain slices (Fig. 2).

Astrocyte-specific disruption of IGF1R signaling impairs glutamate-induced release of eicosanoid gliotransmitters. Shown is production of EETs in glutamate-activated brain slices from control and GFAP-CreERT2/Igf1rf/f mice, as measured by liquid chromatography/mass spectrometry (LC/MS). Data are mean ± S.E.M. (n=6–10 in each group), *P<0.05 vs. control (see the “Methods” section)
Astrocyte-specific disruption of IGF1R signaling results in upregulation of sEH
Western blot analysis showed that in whole brain samples derived from GFAP-CreERT2/Igf1rf/f, mice expression of IGF1R was significantly decreased, as compared to control mice, consistent with a successful knockdown of IGF1R in the astrocyte compartment but not in other cellular compartments (Fig. 3a, b). Protein expression of sEH was significantly increased in GFAP-CreERT2/Igf1rf/f mice as compared to control mice (Fig. 3c).

Upregulation of sEH in mice with astrocyte-specific disruption of IGF1R signaling. a Representative Western blot showing upregulation of soluble epoxide hydrolase (sEH), a key enzyme in the metabolism of vasodilatory epoxyeicosatrienoic acids, and decreased expression of IGF1Rβ in cortical samples derived from tamoxifen-treated GFAP-CreERT2/Igf1rf/f mice, as compared to control mice. b–c Bar graphs are summary densitometric data showing expression of IGF1Rβ (a) and sEH (b), normalized to β-actin expression. Data are normalized to control mean values and are expressed as fold changes. Data are mean ± S.E.M. (n=6–10 in each group), *P<0.05 vs. control
Discussion
In astrocytes, upon neuronal activation and glutamate release from the synapses, a calcium wave is initiated, leading to activation of the synthesis/release of vasodilator metabolites of arachidonic acid, including EETs that have an important role in mediation of NVC responses [16, 24, 75]. The present study provides critical evidence that cell-specific disruption of IGF1R signaling in astrocytes alters their phenotype, impairing EET-mediated gliovascular coupling responses. These findings are consistent with the results of previous studies showing that circulating IGF-1 deficiency, which decreases cerebral IGF-1 levels, also impairs the astrocyte-mediated NVC [23]. The mechanisms by which disruption of astrocytic IGF-1/IGF1R signaling impairs EET-mediated gliovascular coupling may include upregulation of sEH and altered expression of cytochrome P-450 enzymes [23]. Further studies are warranted to investigate transcriptional changes in detail in the astrocytic cellular compartment in GFAP-CreERT2/Igf1rf/f mice. Future studies should also determine the effects of pharmacological inhibitors of sEH on NVC responses and EET release in GFAP-CreERT2/Igf1rf/f mice.
On the basis of the available evidence [66, 76], we propose that neurovascular dysfunction associated with disruption of IGF-1/IGF1R signaling in astrocytes may contribute to the cognitive defects observed in GFAP-CreERT2/Igf1rf/f mice [50]. Previous studies showed that in addition to regulating gliovascular coupling mechanisms, IGF-1/IGF1R signaling also regulates many other important aspects of astrocyte function, including mitochondrial energy metabolism [50, 77], ROS metabolism [50, 78, 79], glucose uptake [80,81,82], and glutamate transport [49, 83]. Perivascular astrocytic endfeet contain a large number of mitochondria [84], and based on previous research [50], disruption of energy metabolism and increased oxidative stress from IGF-1/IGF1R signaling deficiency in astrocytes could impair calcium buffering necessary for NVC. Thus, disruption of IGF-1/IGF1R signaling may also impact these aspects of astrocyte function, which are also likely to contribute to cognitive impairment in GFAP-CreERT2/Igf1rf/f mice. IGF-1 can enter the brain from the circulation by transcytosis across the blood brain barrier [85]. On the basis of the aforementioned observations, one may expect that interventions that increase circulating IGF-1 will also increase IGF-1 levels in the aged brain, which would positively impact astrocyte function and may improve NVC and cognition. In humans, IGF-1 deficiency leads to cognitive dysfunction that can be ameliorated by interventions increasing circulating IGF-1 levels [31]. Aged rodents exhibit a similar decline in circulating IGF-1 levels and treatment of aged rats with IGF-1 was shown to partially rescue cognitive impairment [31]. Future studies are warranted to determine how astrocytic NVC responses are impacted in the aforementioned experimental settings. The effects of strategies aimed at selectively increasing central IGF-1 action on neurovascular and cognitive outcomes have been discordant. Specifically, studies on an inducible, brain-specific (TRE-IGF-1 × Camk2a-tTA) IGF-1 overexpression mouse model showed that increases in central IGF-1 in aging can improve various domains of cognition without significantly affecting gliovascular coupling responses [39]. This further adds to the complexity of IGF-1 actions that may be cell-type and region specific within the brain. IGF-1 has been shown to have pleiotropic, sex-, and tissue-specific effects [86, 87]. In that regard, it should be noted that the effects of age-related circulating IGF-1 deficiency on the cellular mechanisms involved in NVC are likely multifaceted. Cerebromicrovascular endothelial cells are directly exposed to circulating IGF-1 and are known to abundantly express IGF-1 receptors [32]. Ample evidence supports the view that endothelial cells also play a key role in mediation of functional hyperemia [22] and that aging critically impairs endothelium-mediated NVC responses [11, 12, 67, 68]. Because previous studies demonstrate that mouse models of circulating IGF-1 deficiency also exhibit cerebromicrovascular endothelial dysfunction and impaired endothelium-mediated NVC responses [23], in future studies, the effects of endothelium-specific disruption of IGFR-1 signaling on functional hyperemia should also be determined.
Taken together, our findings add to the growing evidence that deficient IGF-1 input to astrocytes compromises their function, impairing gliovascular coupling responses and likely multiple other aspects of brain health. The findings that disruption of IGF-1/IGF1R signaling results in functional and phenotypic alterations in astrocytes have important clinical relevance for cognitive impairment associated both with advanced age and genetic IGF-1 deficiency (e.g., growth hormone releasing hormone-receptor [GHRH-R] defect; isolated GH deficiency; GH receptor gene defects [Laron syndrome]). Furthermore, expression of IGF1R and downstream signaling transcripts are decreased in astrocytes in human brain with progression of Alzheimer’s neuropathology [88]. Of note, multiple IGF1R mutations have been diagnosed in children born small for gestational age (SGA) [89, 90], who have lower IQ scores than age-matched control subjects [91]. Future studies determining how IGF1R mutations in humans affect gliovascular coupling and regulation of CBF should be quite revealing. Our present findings, taken together with the results of previous studies [31], point to potential multifaceted benefits of interventions improving IGF-1 input to the brain in aging.
References
Gorelick PB, Counts SE, Nyenhuis D. Vascular cognitive impairment and dementia. Biochim Biophys Acta. 1862;2016:860–8.
Iadecola C, Duering M, Hachinski V, Joutel A, Pendlebury ST, Schneider JA, et al. Vascular cognitive impairment and dementia: JACC Scientific Expert Panel. J Am Coll Cardiol. 2019;73:3326–44.
Zlokovic BV, Gottesman RF, Bernstein KE, Seshadri S, McKee A, Snyder H, Greenberg SM, Yaffe K, Schaffer CB, Yuan C, Hughes TM, Daemen MJ, Williamson JD, Gonzalez HM, Schneider J, Wellington CL, Katusic ZS, Stoeckel L, Koenig JI, Corriveau RA, Fine L, Galis ZS, Reis J, Wright JD and Chen J. Vascular contributions to cognitive impairment and dementia (VCID): a report from the 2018 National Heart, Lung, and Blood Institute and National Institute of Neurological Disorders and Stroke Workshop. Alzheimers Dement. 2020.
Toth P, Tarantini S, Csiszar A, Ungvari Z. Functional vascular contributions to cognitive impairment and dementia: mechanisms and consequences of cerebral autoregulatory dysfunction, endothelial impairment, and neurovascular uncoupling in aging. Am J Physiol Heart Circ Physiol. 2017;312:H1–H20.
Tarantini S, Tran CHT, Gordon GR, Ungvari Z, Csiszar A. Impaired neurovascular coupling in aging and Alzheimer’s disease: contribution of astrocyte dysfunction and endothelial impairment to cognitive decline. Exp Gerontol. 2017;94:52–8.
Zaletel M, Strucl M, Pretnar-Oblak J, Zvan B. Age-related changes in the relationship between visual evoked potentials and visually evoked cerebral blood flow velocity response. Funct Neurol. 2005;20:115–20.
Topcuoglu MA, Aydin H, Saka E. Occipital cortex activation studied with simultaneous recordings of functional transcranial Doppler ultrasound (fTCD) and visual evoked potential (VEP) in cognitively normal human subjects: effect of healthy aging. Neurosci Lett. 2009;452:17–22.
Stefanova I, Stephan T, Becker-Bense S, Dera T, Brandt T, Dieterich M. Age-related changes of blood-oxygen-level-dependent signal dynamics during optokinetic stimulation. Neurobiol Aging. 2013;34:2277–86.
Fabiani M, Gordon BA, Maclin EL, Pearson MA, Brumback-Peltz CR, Low KA, et al. Neurovascular coupling in normal aging: a combined optical. Neuroimage: ERP and fMRI study; 2013.
Sorond FA, Hurwitz S, Salat DH, Greve DN, Fisher ND. Neurovascular coupling, cerebral white matter integrity, and response to cocoa in older people. Neurology. 2013;81:904–9.
Park L, Anrather J, Girouard H, Zhou P, Iadecola C. Nox2-derived reactive oxygen species mediate neurovascular dysregulation in the aging mouse brain. J Cereb Blood Flow Metab. 2007;27:1908–18.
Toth P, Tarantini S, Tucsek Z, Ashpole NM, Sosnowska D, Gautam T, et al. Resveratrol treatment rescues neurovascular coupling in aged mice: role of improved cerebromicrovascular endothelial function and down-regulation of NADPH oxidas. Am J Physiol Heart Circ Physiol. 2014;306:H299–308.
Attwell D, Buchan AM, Charpak S, Lauritzen M, Macvicar BA, Newman EA. Glial and neuronal control of brain blood flow. Nature. 2010;468:232–43.
Lind BL, Brazhe AR, Jessen SB, Tan FC, Lauritzen MJ. Rapid stimulus-evoked astrocyte Ca2+ elevations and hemodynamic responses in mouse somatosensory cortex in vivo. Proc Natl Acad Sci U S A. 2013;110:E4678–87.
Otsu Y, Couchman K, Lyons DG, Collot M, Agarwal A, Mallet JM, et al. Calcium dynamics in astrocyte processes during neurovascular coupling. Nat Neurosci. 2015;18:210–8.
Petzold GC, Murthy VN. Role of astrocytes in neurovascular coupling. Neuron. 2011;71:782–97.
Peng X, Carhuapoma JR, Bhardwaj A, Alkayed NJ, Falck JR, Harder DR, et al. Suppression of cortical functional hyperemia to vibrissal stimulation in the rat by epoxygenase inhibitors. Am J Physiol Heart Circ Physiol. 2002;283:H2029–37.
Takano T, Tian GF, Peng W, Lou N, Libionka W, Han X, et al. Astrocyte-mediated control of cerebral blood flow. Nat Neurosci. 2006;9:260–7.
Zonta M, Angulo MC, Gobbo S, Rosengarten B, Hossmann KA, Pozzan T, et al. Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation. Nat Neurosci. 2003;6:43–50.
Gebremedhin D, Ma YH, Falck JR, Roman RJ, VanRollins M, Harder DR. Mechanism of action of cerebral epoxyeicosatrienoic acids on cerebral arterial smooth muscle. Am J Phys. 1992;263:H519–25.
Chen BR, Kozberg MG, Bouchard MB, Shaik MA, Hillman EM. A critical role for the vascular endothelium in functional neurovascular coupling in the brain. J Am Heart Assoc. 2014;3:e000787.
Toth P, Tarantini S, Davila A, Valcarcel-Ares MN, Tucsek Z, Varamini B, et al. Purinergic glio-endothelial coupling during neuronal activity: role of P2Y1 receptors and eNOS in functional hyperemia in the mouse somatosensory cortex. Am J Physiol Heart Circ Physiol. 2015;309:H1837–45.
Toth P, Tarantini S, Ashpole NM, Tucsek Z, Milne GL, Valcarcel-Ares NM, et al. IGF-1 deficiency impairs neurovascular coupling in mice: implications for cerebromicrovascular aging. Aging Cell. 2015;14:1034–44.
Koehler RC, Gebremedhin D, Harder DR. Role of astrocytes in cerebrovascular regulation. J Appl Physiol (1985). 2006;100:307–17.
Blanco VM, Stern JE, Filosa JA. Tone-dependent vascular responses to astrocyte-derived signals. Am J Physiol Heart Circ Physiol. 2008;294:H2855–63.
Girouard H, Bonev AD, Hannah RM, Meredith A, Aldrich RW, Nelson MT. Astrocytic endfoot Ca2+ and BK channels determine both arteriolar dilation and constriction. Proc Natl Acad Sci U S A. 2010;107:3811–6.
Cauli B, Hamel E. Brain perfusion and astrocytes. Trends Neurosci. 2018;41:409–13.
Rosenegger DG, Gordon GR. A slow or modulatory role of astrocytes in neurovascular coupling. Microcirculation. 2015;22:197–203.
Longden TA, Dunn KM, Draheim HJ, Nelson MT, Weston AH, Edwards G. Intermediate-conductance calcium-activated potassium channels participate in neurovascular coupling. Br J Pharmacol. 2011;164:922–33.
Liu X, Li C, Falck JR, Harder DR, Koehler RC. Relative contribution of cyclooxygenases, epoxyeicosatrienoic acids, and pH to the cerebral blood flow response to vibrissal stimulation. Am J Physiol Heart Circ Physiol. 2012;302:H1075–85.
Sonntag WE, Deak F, Ashpole N, Toth P, Csiszar A, Freeman W, et al. Insulin-like growth factor-1 in CNS and cerebrovascular aging. Front Aging Neurosci. 2013;5:27.
Ungvari Z, Csiszar A. The emerging role of IGF-1 deficiency in cardiovascular aging: recent advances. J Gerontol A Biol Sci Med Sci. 2012;67:599–610.
Fan X, Wheatley EG, Villeda SA. Mechanisms of hippocampal aging and the potential for rejuvenation. Annu Rev Neurosci. 2017;40:251–72.
Katsimpardi L, Litterman NK, Schein PA, Miller CM, Loffredo FS, Wojtkiewicz GR, et al. Vascular and neurogenic rejuvenation of the aging mouse brain by young systemic factors. Science. 2014;344:630–4.
Rebo J, Mehdipour M, Gathwala R, Causey K, Liu Y, Conboy MJ, et al. A single heterochronic blood exchange reveals rapid inhibition of multiple tissues by old blood. Nat Commun. 2016;7:13363.
Smith LK, He Y, Park JS, Bieri G, Snethlage CE, Lin K, et al. beta2-microglobulin is a systemic pro-aging factor that impairs cognitive function and neurogenesis. Nat Med. 2015;21:932–7.
Villeda SA, Plambeck KE, Middeldorp J, Castellano JM, Mosher KI, Luo J, et al. Young blood reverses age-related impairments in cognitive function and synaptic plasticity in mice. Nat Med. 2014;20:659–63.
Higashi Y, Gautam S, Delafontaine P, Sukhanov S. IGF-1 and cardiovascular disease. Growth Hormon IGF Res. 2019;45:6–16.
Farias Quipildor GE, Mao K, Hu Z, Novaj A, Cui MH, Gulinello M, et al. Central IGF-1 protects against features of cognitive and sensorimotor decline with aging in male mice. Geroscience. 2019;41:185–208.
Fulop GA, Ramirez-Perez FI, Kiss T, Tarantini S, Valcarcel Ares MN, Toth P, Yabluchanskiy A, Conley SM, Ballabh P, Martinez-Lemus LA, Ungvari Z and Csiszar A. IGF-1 deficiency promotes pathological remodeling of cerebral arteries: a potential mechanism contributing to the pathogenesis of intracerebral hemorrhages in aging. J Gerontol A Biol Sci Med Sci. 2018.
Tarantini S, Valcarcel-Ares NM, Yabluchanskiy A, Springo Z, Fulop GA, Ashpole N, et al. Insulin-like growth factor 1 deficiency exacerbates hypertension-induced cerebral microhemorrhages in mice, mimicking the aging phenotype. Aging Cell. 2017;16:469–79.
Tarantini S, Tucsek Z, Valcarcel-Ares MN, Toth P, Gautam T, Giles CB, et al. Circulating IGF-1 deficiency exacerbates hypertension-induced microvascular rarefaction in the mouse hippocampus and retrosplenial cortex: implications for cerebromicrovascular and brain aging. Age (Dordr). 2016;38:273–89.
Toth P, Tucsek Z, Tarantini S, Sosnowska D, Gautam T, Mitschelen M, et al. IGF-1 deficiency impairs cerebral myogenic autoregulation in hypertensive mice. J Cereb Blood Flow Metab. 2014;34:1887–97.
Dong X, Chang G, Ji XF, Tao DB, Wang YX. The relationship between serum insulin-like growth factor I levels and ischemic stroke risk. PLoS One. 2014;9:e94845.
Troncoso R, Vicencio JM, Parra V, Nemchenko A, Kawashima Y, Del Campo A, et al. Energy-preserving effects of IGF-1 antagonize starvation-induced cardiac autophagy. Cardiovasc Res. 2012;93:320–9.
Higashi Y, Sukhanov S, Anwar A, Shai SY, Delafontaine P. Aging, atherosclerosis, and IGF-1. J Gerontol A Biol Sci Med Sci. 2012;67:626–39.
von der Thusen JH, Borensztajn KS, Moimas S, van Heiningen S, Teeling P, van Berkel TJ, et al. IGF-1 has plaque-stabilizing effects in atherosclerosis by altering vascular smooth muscle cell phenotype. Am J Pathol. 2011;178:924–34.
Shai SY, Sukhanov S, Higashi Y, Vaughn C, Rosen CJ, Delafontaine P. Low circulating insulin-like growth factor i increases atherosclerosis in apoe-deficient mice. Am J Physiol Heart Circ Physiol. 2011;300:H1898–906.
Prabhu D, Khan SM, Blackburn K, Marshall JP, Ashpole NM. Loss of insulin-like growth factor-1 signaling in astrocytes disrupts glutamate handling. J Neurochem. 2019;151:689–702.
Logan S, Pharaoh GA, Marlin MC, Masser DR, Matsuzaki S, Wronowski B, et al. Insulin-like growth factor receptor signaling regulates working memory, mitochondrial metabolism, and amyloid-beta uptake in astrocytes. Mol Metab. 2018;9:141–55.
Littlejohn EL, Scott D, Saatman KE. Insulin-like growth factor-1 overexpression increases long-term survival of posttrauma-born hippocampal neurons while inhibiting ectopic migration following traumatic brain injury. Acta Neuropathol Commun. 2020;8:46.
Garwood CJ, Ratcliffe LE, Morgan SV, Simpson JE, Owens H, Vazquez-Villasenor I, et al. Insulin and IGF1 signalling pathways in human astrocytes in vitro and in vivo; characterisation, subcellular localisation and modulation of the receptors. Mol Brain. 2015;8:51.
Pardo J, Uriarte M, Console GM, Reggiani PC, Outeiro TF, Morel GR, et al. Insulin-like growth factor-I gene therapy increases hippocampal neurogenesis, astrocyte branching and improves spatial memory in female aging rats. Eur J Neurosci. 2016;44:2120–8.
Labandeira-Garcia JL, Costa-Besada MA, Labandeira CM, Villar-Cheda B, Rodriguez-Perez AI. Insulin-like growth factor-1 and neuroinflammation. Front Aging Neurosci. 2017;9:365.
Okoreeh AK, Bake S, Sohrabji F. Astrocyte-specific insulin-like growth factor-1 gene transfer in aging female rats improves stroke outcomes. Glia. 2017;65:1043–58.
Piriz J, Muller A, Trejo JL, Torres-Aleman I. IGF-I and the aging mammalian brain. Exp Gerontol. 2011;46:96–9.
Fernandez AM, Torres-Aleman I. The many faces of insulin-like peptide signalling in the brain. Nat Rev Neurosci. 2012;13:225–39.
Muller AP, Fernandez AM, Haas C, Zimmer E, Portela LV, Torres-Aleman I. Reduced brain insulin-like growth factor I function during aging. Mol Cell Neurosci. 2012;49:9–12.
Trueba-Saiz A, Cavada C, Fernandez AM, Leon T, Gonzalez DA, Fortea Ormaechea J, et al. Loss of serum IGF-I input to the brain as an early biomarker of disease onset in Alzheimer mice. Transl Psychiatry. 2013;3:e330.
Ascenzi F, Barberi L, Dobrowolny G, Villa Nova Bacurau A, Nicoletti C, Rizzuto E, et al. Effects of IGF-1 isoforms on muscle growth and sarcopenia. Aging Cell. 2019;18:e12954.
Williamson TT, Ding B, Zhu X, Frisina RD. Hormone replacement therapy attenuates hearing loss: mechanisms involving estrogen and the IGF-1 pathway. Aging Cell. 2019;18:e12939.
Sonntag WE, Ramsey M, Carter CS. Growth hormone and insulin-like growth factor-1 (IGF-1) and their influence on cognitive aging. Ageing Res Rev. 2005;4:195–212.
O’Connor KG, Tobin JD, Harman SM, Plato CC, Roy TA, Sherman SS, et al. Serum levels of insulin-like growth factor-I are related to age and not to body composition in healthy women and men. J Gerontol A Biol Sci Med Sci. 1998;53:M176–82.
Pavlov EP, Harman SM, Merriam GR, Gelato MC, Blackman MR. Responses of growth hormone (GH) and somatomedin-C to GH-releasing hormone in healthy aging men. J Clin Endocrinol Metab. 1986;62:595–600.
Ameri P, Canepa M, Fabbi P, Leoncini G, Milaneschi Y, Mussap M, et al. Vitamin D modulates the association of circulating insulin-like growth factor-1 with carotid artery intima-media thickness. Atherosclerosis. 2014;236:418–25.
Tarantini S, Hertelendy P, Tucsek Z, Valcarcel-Ares MN, Smith N, Menyhart A, et al. Pharmacologically-induced neurovascular uncoupling is associated with cognitive impairment in mice. J Cereb Blood Flow Metab. 2015;35:1871–81.
Tarantini S, Valcarcel-Ares MN, Toth P, Yabluchanskiy A, Tucsek Z, Kiss T, et al. Nicotinamide mononucleotide (NMN) supplementation rescues cerebromicrovascular endothelial function and neurovascular coupling responses and improves cognitive function in aged mice. Redox Biol. 2019;24:101192.
Tarantini S, Valcarcel-Ares NM, Yabluchanskiy A, Fulop GA, Hertelendy P, Gautam T, et al. Treatment with the mitochondrial-targeted antioxidant peptide SS-31 rescues neurovascular coupling responses and cerebrovascular endothelial function and improves cognition in aged mice. Aging Cell. 2018;17:e12731.
Shi Y, Liu X, Gebremedhin D, Falck JR, Harder DR, Koehler RC. Interaction of mechanisms involving epoxyeicosatrienoic acids, adenosine receptors, and metabotropic glutamate receptors in neurovascular coupling in rat whisker barrel cortex. J Cereb Blood Flow Metab. 2008;28:111–25.
Imig JD, Falck JR, Inscho EW. Contribution of cytochrome P450 epoxygenase and hydroxylase pathways to afferent arteriolar autoregulatory responsiveness. Br J Pharmacol. 1999;127:1399–405.
Wang MH, Brand-Schieber E, Zand BA, Nguyen X, Falck JR, Balu N, et al. Cytochrome P450-derived arachidonic acid metabolism in the rat kidney: characterization of selective inhibitors. J Pharmacol Exp Ther. 1998;284:966–73.
Kazama K, Anrather J, Zhou P, Girouard H, Frys K, Milner TA, et al. Angiotensin II impairs neurovascular coupling in neocortex through NADPH oxidase-derived radicals. Circ Res. 2004;95:1019–26.
Yabluchanskiy A, Tarantini S, Balasubramanian P, Kiss T, Csipo T, Fulop GA, et al. Pharmacological or genetic depletion of senescent astrocytes prevents whole brain irradiation-induced impairment of neurovascular coupling responses protecting cognitive function in mice. Geroscience. 2020;42:409–28.
Garcia V, Cheng J, Weidenhammer A, Ding Y, Wu CC, Zhang F, et al. Androgen-induced hypertension in angiotensinogen deficient mice: role of 20-HETE and EETS. Prostaglandins Other Lipid Mediat. 2015;116-117:124–30.
Haydon PG, Carmignoto G. Astrocyte control of synaptic transmission and neurovascular coupling. Physiol Rev. 2006;86:1009–31.
Shen J, Wang D, Wang X, Gupta S, Ayloo B, Wu S, et al. Neurovascular coupling in the dentate gyrus regulates adult hippocampal neurogenesis. Neuron. 2019;103:878–90 e3.
Ratcliffe LE, Vazquez Villasenor I, Jennings L, Heath PR, Mortiboys H, Schwartzentruber A, et al. Loss of IGF1R in human astrocytes alters complex i activity and support for neurons. Neuroscience. 2018;390:46–59.
Genis L, Davila D, Fernandez S, Pozo-Rodrigalvarez A, Martinez-Murillo R, Torres-Aleman I. Astrocytes require insulin-like growth factor I to protect neurons against oxidative injury. F1000Res. 2014;3:–28.
Davila D, Fernandez S, Torres-Aleman I. Astrocyte resilience to oxidative stress induced by insulin-like growth factor I (IGF-I) involves preserved AKT (protein kinase B) activity. J Biol Chem. 2016;291:2510–23.
Fernandez AM, Hernandez-Garzon E, Perez-Domper P, Perez-Alvarez A, Mederos S, Matsui T, et al. Insulin regulates astrocytic glucose handling through cooperation with IGF-I. Diabetes. 2017;66:64–74.
Hernandez-Garzon E, Fernandez AM, Perez-Alvarez A, Genis L, Bascunana P, de la Rosa Fernandez R, et al. The insulin-like growth factor I receptor regulates glucose transport by astrocytes. Glia. 2016;64:1962–71.
Fernandez AM, Hernandez E, Guerrero-Gomez D, Miranda-Vizuete A, Torres AI. A network of insulin peptides regulate glucose uptake by astrocytes: potential new druggable targets for brain hypometabolism. Neuropharmacology. 2018;136:216–22.
Suzuki K, Ikegaya Y, Matsuura S, Kanai Y, Endou H, Matsuki N. Transient upregulation of the glial glutamate transporter GLAST in response to fibroblast growth factor, insulin-like growth factor and epidermal growth factor in cultured astrocytes. J Cell Sci. 2001;114:3717–25.
Shih EK, Robinson MB. Role of astrocytic mitochondria in limiting ischemic brain injury? Physiology (Bethesda). 2018;33:99–112.
Nishijima T, Piriz J, Duflot S, Fernandez AM, Gaitan G, Gomez-Pinedo U, et al. Neuronal activity drives localized blood-brain-barrier transport of serum insulin-like growth factor-I into the CNS. Neuron. 2010;67:834–46.
Ashpole NM, Logan S, Yabluchanskiy A, Mitschelen MC, Yan H, Farley JA, et al. IGF-1 has sexually dimorphic, pleiotropic, and time-dependent effects on healthspan, pathology, and lifespan. Geroscience. 2017;39:129–45.
Ashpole NM, Herron JC, Estep PN, Logan S, Hodges EL, Yabluchanskiy A, et al. Differential effects of IGF-1 deficiency during the life span on structural and biomechanical properties in the tibia of aged mice. Age (Dordr). 2016;38:38.
Moloney AM, Griffin RJ, Timmons S, O’Connor R, Ravid R, O’Neill C. Defects in IGF-1 receptor, insulin receptor and IRS-1/2 in Alzheimer’s disease indicate possible resistance to IGF-1 and insulin signalling. Neurobiol Aging. 2010;31:224–43.
Klammt J, Kiess W, Pfaffle R. IGF1R mutations as cause of SGA. Best Pract Res Clin Endocrinol Metab. 2011;25:191–206.
Ester WA, Hokken-Koelega AC. Polymorphisms in the IGF1 and IGF1R genes and children born small for gestational age: results of large population studies. Best Pract Res Clin Endocrinol Metab. 2008;22:415–31.
Lohaugen GC, Ostgard HF, Andreassen S, Jacobsen GW, Vik T, Brubakk AM, et al. Small for gestational age and intrauterine growth restriction decreases cognitive function in young adults. J Pediatr. 2013;163:447–53.
Funding
This work was supported by grants from the Oklahoma Center for the Advancement of Science and Technology (to AC, AY, ZU), the National Institute on Aging (R01-AG047879; R01-AG055395; R0-AG068295; and K99AG056662 to SL), the National Institute of Neurological Disorders and Stroke (NINDS; R01-NS100782), the National Cancer Institute (NCI; R01-CA255840-01), the National Institute of General Medical Sciences Oklahoma Shared Clinical and Translational Resources (OSCTR) (GM104938, to AY), the Presbyterian Health Foundation, the NIA-supported Geroscience Training Program in Oklahoma (T32AG052363), the Oklahoma Nathan Shock Center (P30AG050911), the Cellular and Molecular GeroScience CoBRE (1P20GM125528, sub#5337), the American Federation for Aging Research (Irene/Diamond Postdoctoral Transition Award to PB), the Hungarian Center for Excellence in Molecular Medicine, and the NKFIH (Nemzeti Szivlabor). The funding sources had no role in the study design; in the collection, analysis and interpretation of data; in the writing of the report; and in the decision to submit the article for publication.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
About this article

Cite this article
Tarantini, S., Balasubramanian, P., Yabluchanskiy, A. et al. IGF1R signaling regulates astrocyte-mediated neurovascular coupling in mice: implications for brain aging. GeroScience 43, 901–911 (2021). https://doi.org/10.1007/s11357-021-00350-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11357-021-00350-0