
Abstract
Whole brain irradiation (WBI, also known as whole brain radiation therapy or WBRT) is a mainstream therapy for patients with identifiable brain metastases and as a prophylaxis for microscopic malignancies. WBI accelerates brain aging, causing progressive cognitive dysfunction in ~ 50% of surviving patients, thus compromising quality of life. The mechanisms responsible for this WBI side effect remain obscure, and there are no effective treatments or prevention strategies. Here, we test the hypothesis that WBI induces astrocyte senescence, which contributes to impaired astrocytic neurovascular coupling (NVC) responses and the genesis of cognitive decline. To achieve this goal, we used transgenic p16-3MR mice, which allows the detection and selective elimination of senescent cells. We subjected these mice to a clinically relevant protocol of fractionated WBI (5 Gy twice weekly for 4 weeks). WBI-treated and control mice were tested for spatial memory performance (radial arm water maze), astrocyte-dependent NVC responses (whisker-stimulation-induced increases in cerebral blood flow, assessed by laser speckle contrast imaging), NVC-related gene expression, astrocytic release of eicosanoid gliotransmitters and the presence of senescent astrocytes (by flow cytometry, immunohistochemistry and gene expression profiling) at 6 months post-irradiation. WBI induced senescence in astrocytes, which associated with NVC dysfunction and impaired performance on cognitive tasks. To establish a causal relationship between WBI-induced senescence and NVC dysfunction, senescent cells were depleted from WBI-treated animals (at 3 months post-WBI) by genetic (ganciclovir treatment) or pharmacological (treatment with the BCL-2/BCL-xL inhibitor ABT263/Navitoclax, a known senolytic drug) means. In WBI-treated mice, both treatments effectively eliminated senescent astrocytes, rescued NVC responses, and improved cognitive performance. Our findings suggest that the use of senolytic drugs can be a promising strategy for preventing the cognitive impairment associated with WBI.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Despite the development of new therapeutic interventions such as stereotaxic radiosurgery, whole brain irradiation (WBI) remains a mainstream treatment option for patients with both identifiable brain metastases and as a prophylaxis for microscopic malignancies (Gaspar et al. 2010; Patil et al. 2012). Over 200,000 tumor patients with brain involvement receive either partial large field or WBI every year in the USA (Lee et al. 2012). Although WBI is clinically effective in eliminating dividing tumor cells, it also results in a range of serious unwanted side-effects in the healthy brain tissue. In particular, WBI can cause significant functional impairment in the cortex, hippocampus, and white matter months to years after treatment (Khuntia et al. 2006; Welzel et al. 2008a; Welzel et al. 2008b). As a consequence, up to 50% of patients who survive long-term after WBI treatment experience progressive cognitive decline, which severely compromises their quality of life (Lee et al. 2012). Preclinical studies show that WBI also leads to a progressive impairment of cognitive function in rodent models, mimicking the clinical condition of WBI-treated patients (Welzel et al. 2008a; Welzel et al. 2008b; Lamproglou et al. 2001; Shi et al. 2006; Soussain et al. 2009; Warrington et al. 2012; Ungvari et al. 2017). At present, there are no effective strategies to prevent or reverse WBI-induced cognitive impairment.
In light of the relatively new field of geroscience, our understanding of age-related dysregulation in cerebral blood flow (CBF) has undergone a paradigm shift such that many researchers now consider cerebrovascular dysfunction to be a critical contributor. In the past decade, a new concept has emerged, suggesting that WBI-induced cognitive decline is a clinically relevant case of accelerated cerebrovascular aging. Importantly, there is strong preclinical evidence that, similar to aging, the profound harmful effects of WBI on cognitive function are due, at least in part, to dysregulated CBF (Ungvari et al. 2017; Ashpole et al. 2014; Warrington et al. 2013).
Normal brain function is critically dependent on a continuous, tightly controlled supply of oxygen and nutrients through CBF. During periods of intense neuronal activity, to satisfy the high energetic demands of firing neurons, there is a requirement for rapid increases in oxygen and glucose delivery. This requirement is ensured by neurovascular coupling (NVC; functional hyperemia), a vital homeostatic mechanism that adjusts local CBF to the increased demands of the neurons in a moment-to-moment manner (Fulop et al. 2018; Attwell et al. 2010; Tarantini et al. 2015; Tarantini et al. 2016; Toth et al. 2016; Toth et al. 2015a; Toth et al. 2017; Toth et al. 2015b; Toth et al. 2014; Tucsek et al. 2014; Csiszar et al. 2019; Tarantini et al. 2019a; Wiedenhoeft et al. 2019; Csipo et al. 2019a; Farias Quipildor et al. 2019). NVC depends on an intact functional network of neurons, astrocytes, and cerebral microvessels (Toth et al. 2017; Tarantini et al. 2017a). A critical cellular mechanism that underlies neurovascular coupling is astrocytic production of vasodilator metabolites of arachidonic acid, including epoxygenase-derived epoxyeicosatrienoic acids (EETs) (Attwell et al. 2010; Toth et al. 2015a). Previous studies showed that pharmacological inhibition of the production of vasodilator eicosanoid gliotransmitters results in neurovascular dysfunction and cognitive impairment (Tarantini et al. 2015; Tarantini et al. 2017b). There is also strong evidence that impaired NVC is causally related to cognitive decline in models of aging (Tarantini et al. 2015; Tarantini et al. 2016) and pathophysiological conditions associated with accelerated neurovascular aging (Tucsek et al. 2014; Csiszar et al. 2017; Tarantini et al. 2018a; Girouard and Iadecola 2006; Gorelick et al. 2011; Kazama et al. 2004). Importantly, recent studies demonstrate that a clinically relevant protocol of fractionated WBI in mice results in persistent impairment of NVC responses, which predicts cognitive decline (Ungvari et al. 2017), mimicking the effects of aging (Toth et al. 2017; Toth et al. 2014; Tucsek et al. 2014; Tarantini et al. 2017a; Tarantini et al. 2018a; Lipecz et al. 2019; Tarantini et al. 2019b; Tarantini et al. 2018b). Importantly, restoration of NVC responses in rodent models of aging was shown to improve cognitive function (Toth et al. 2014; Tarantini et al. 2017a; Tarantini et al. 2019b; Tarantini et al. 2018b). Despite these advances, the cellular mechanisms by which WBI impairs NVC responses are unknown, and there are no interventions that rescue neurovascular function, thereby improving cognition, after WBI.
A critical mechanism by which radiation exerts its harmful effects on cellular and tissue function is the generation of persistent DNA damage, which induces the chronic stress response termed cellular senescence (Ungvari et al. 2013; Coppe et al. 2011; Rodier et al. 2009). Importantly, recent evidence suggest that astrocytes are particularly sensitive to DNA damaging agents and that cultured astrocytes readily acquire senescent phenotypes in response to DNA damage (Cohen and Torres 2019). Upon induction of cellular senescence in culture, astrocytes undergo cell cycle arrest and acquire a senescence-associated secretory phenotype (SASP), characterized by increased secretion of pro-inflammatory proteins and lipids (Cohen and Torres 2019; Freund et al. 2010). There is growing evidence that senescent cells and their SASPs contribute to the pathogenesis of many age-related diseases (Cohen and Torres 2019; Childs et al. 2016; Campisi 2016; Chinta et al. 2014; Tchkonia et al. 2013; Campisi 2013; Farr et al. 2017; Baker et al. 2011; Xu et al. 2018; Tchkonia and Kirkland 2018; Ogrodnik et al. 2018; Justice et al. 2018). Indeed, depletion of cells expressing the senescence marker cyclin-dependent kinase inhibitor p16INK4A in genetically modified mice prolongs median lifespan and improves overall health (Farr et al. 2017; Baker et al. 2011; Baker et al. 2016; Jeon et al. 2017; Abdul-Aziz et al. 2019; Kim et al. 2019; Patil et al. 2019; Xu et al. 2015; Roos et al. 2016; Baar et al. 2017), supporting a key role for cellular senescence in the process of aging. The role of irradiation-induced senescence in post-WBI neurovascular dysfunction and cognitive impairment remains unexplored.
Here, we test the hypothesis that WBI induces astrocyte senescence in vivo, which contributes to impaired astrocytic NVC responses and the genesis of cognitive decline. To achieve this goal, we used transgenic p16-3MR mice, which allows the detection and selective elimination of senescent cells (Demaria et al. 2014). The mice were subjected to a clinically relevant protocol of fractionated WBI, and then WBI-treated and control mice were tested for spatial memory performance, astrocyte-dependent NVC responses, NVC-related gene expression, astrocytic release of eicosanoid gliotransmitters and the presence of senescent astrocytes 6 months after irradiation. To establish a causal relationship between WBI-induced senescence and neurovascular dysfunction, senescent cells were depleted from WBI-treated animals 3 months after WBI using the p16-3MR transgene (Demaria et al. 2014) or a pharmacological agent known to target senescent cells (Chang et al. 2016).
Materials and methods
Experimental animals and experimental design
To identify and eliminate senescent cells, we used a novel transgenic mouse model (p16-3MR mice (Demaria et al. 2014)) that carry a trimodal fusion protein (3MR) under the control of the p16Ink4a promoter. 3MR contains functional fragments of Renilla luciferase, which allows us to detect senescent cells in living animals; monomeric red fluorescent protein (mRFP), which enables us to FACS sort senescent cells from tissues; and the herpes simplex virus thymidine kinase, which allows us to selectively kill p16-positive senescent cells by administering the prodrug ganciclovir (GCV). Previous studies have extensively characterized this model (Demaria et al. 2017).
Three-month-old male p16-3MR mice were housed 3 per cage in the specific pathogen-free animal facility at the University of Oklahoma Health Sciences Center (OUHSC). Animals were kept on a 12 h light/dark cycle and fed standard rodent chow and water ad libitum, following standard husbandry techniques. One week before irradiation, mice were transferred to the conventional animal facility of the UOHSC and housed under similar conditions. Mice were anesthetized and subjected to WBI (n = 60, 5 Gy twice weekly for a total cumulative dose of 40 Gy) or sham irradiated as a control group (n = 20). Mice were then left to recover for 3 months in the original environment. WBI-treated mice were assigned randomly to three groups. Two groups received the senolytic drug ABT263 (Navitoclax, intraperitoneal (i.p.) injection, 1.5 mg/kg/daily in DMSO/70% ethanol) (Chang et al. 2016; Zhu et al. 2016) or ganciclovir (i.p. 25 mg/kg/daily in PBS) for 5 days and for 2 cycles with a 2-week interval between cycles. The third group served as vehicle controls. To test for non-specific effects of senolytic treatments, a separate control cohort of p16-3MR mice animals received GCV or ABT263 without WBI. At the end of the recovery period, 6 months after the WBI protocol, mice were tested for cognitive function and NVC responses, then euthanized for tissue collection. All animal protocols were approved by the Institutional Animal Care and Use Committee of the OUHSC.
Fractionated whole brain irradiation protocol
After acclimating to the conventional facility for 1 week, mice were randomly assigned to either control or irradiated groups. Animals were weighed and anesthetized via i.p. injection of ketamine and xylazine (100/15 mg per kg). Mice in the irradiated group were subjected to clinically relevant WBI (5 Gy twice weekly for a total cumulative dose of 40 Gy). Radiation was administered using a 137Cesium gamma irradiator (GammaCell 40, Nordion International). A Cerrobend® shield was utilized to minimize exposure outside the brain. The radiation dose received by the mice was verified using film dosimetry, as described (Warrington et al. 2012; Ungvari et al. 2017; Warrington et al. 2011).
Radial arms water maze testing
To determine how senescence induced by WBI and depletion of senescent cells affect cognitive function, spatial memory and long-term memory were tested by assessing performance in the radial arms water maze at 6 months post-WBI, following our published protocols (Ungvari et al. 2017; Tarantini et al. 2019b; Tarantini et al. 2018b). The maze consists of 8 arms with a submerged escape platform at the end of one arm. Food coloring was added to the water to make it opaque. The maze was surrounded by privacy blinds with extramaze visual cues. Intramaze visual cues were placed at the end of the arms. Mice were monitored by video tracking directly above the maze, and parameters were measured using Ethovision software (Noldus Information Technology Inc., Leesburg, VA, USA). Experimenters were blinded to the experimental conditions of the mice. Each day, mice were given the opportunity to learn the location of the submerged platform during 2 session blocks, each consisting of 4 consecutive acquisition trials. On each trial, the mouse was started in an arm not containing the platform and allowed to wade for up to 1 min to find the escape platform. All mice spent 30 s on the platform following each trial before beginning the next trial. The platform was located in the same arm on each trial. Over 3 days of training, mice gradually improved performance. Upon entering an incorrect arm (all 4 paws within the distal half of the arm), the mouse was charged an error. Learning was assessed by comparing performance on days 2 and 3 of the learning period. When both groups learned the task, mice were placed in their home cage for 7 days. Then, they were given a recall trial on day 10. On day 11 (extinction), mice were tested for ability to relearn the task when the platform had been moved to a different arm not adjacent or diametrically positioned to the previous location. Mice were tested for 2 session blocks; the second, consisting of 4 trials, was used for comparison.
Assessment of neurovascular coupling responses
After behavioral testing, NVC responses were assessed as described previously (Tarantini et al. 2018b; Tarantini et al. 2017c). In brief, mice in each group were anesthetized with isoflurane (2% induction and 1% maintenance), endotracheally intubated and ventilated (MousVent G500; Kent Scientific Co, Torrington, CT). A thermostatic heating pad (Kent Scientific Co, Torrington, CT) was used to maintain rectal temperature at 37 °C (Toth et al. 2014). End-tidal CO2 was controlled between 3.2 and 3.7% to keep blood gas values within the physiological range, as described (Toth et al. 2015a; Tarantini et al. 2015). The right femoral artery was cannulated for arterial blood pressure measurement (Living Systems Instrumentations, Burlington, VT) (Toth et al. 2014). Blood pressure was within the physiological range throughout the experiments (90–110 mmHg). Mice were immobilized and placed on a stereotaxic frame (Leica Microsystems, Buffalo Grove, IL), the scalp and periosteum were pulled aside, and the skull was gently thinned using a dental drill while cooled with dripping buffer. A laser speckle contrast imager (Perimed, Järfälla, Sweden) was placed 10 cm above the thinned skull. To achieve the highest CBF response the right whiskers were stimulated for 30 s at 5 Hz from side to side. Differential perfusion maps of the brain surface were captured. Changes in CBF were assessed above the left barrel cortex in six trials in each group, separated by 5-min intervals. To assess the role of EET mediation, CBF responses to whisker stimulation were repeated after administrating the epoxigenase inhibitor MSSPOH (N-(methylsulfonyl)-2-(2-propynyloxy)-benzenehexanamide; Cayman Chemicals; 20 mg/kg, dissolved in DMSO and diluted to final concentration with 45% cyclodextrin, Cayman Chemicals) which inhibits EET production. Changes in CBF were averaged and expressed as percent (%) increase from the baseline value. Experiments lasted < 1 h/mouse, which permitted stable physiological parameters to be obtained. In each study the experimenter was blinded to the treatment of the animals. At the end of the experiments the animals (with the exception of those assigned to brain lipidomics studies) were transcardially perfused with ice-cold PBS and decapitated. Animals assigned to brain lipidomics studies were decapitated without perfusion to avoid wash-out of lipid mediators. The brains were immediately removed and samples were collected for subsequent studies. Whole brains were collected for FACS analysis. Acute brain slices were collected for lipidomics studies (EET release). Half brains were immersion fixed in 4% paraformaldehyde for 24 h, transferred to sucrose gradients and embedded and cut for immunohistochemistry. Pieces of the somatosensory and motor cortex as well as the hippocampi were isolated and frozen for subsequent analysis (qPCR and lipidomics).
Measurement of glutamate-induced release of EETs from acute hippocampal slices
To determine how WBI affects synthesis of eicosanoid gliotransmitters, horizontal hippocampal slices of 325 μm thickness from mice in each cohort were prepared using a HM650V vibrating microtome (Thermo Scientific) in ice cold solution containing (in mmol/L) sucrose 110, NaCl 60, KCl 3, NaH2PO4 1.25, NaHCO3 28, sodium ascorbate acid 0.6, glucose 5, MgCl2 7, and CaCl2 0.5 using a HM650V vibrating microtome (Thermo Scientific). Slices were then transferred to a holding chamber containing oxygenated artificial cerebrospinal fluid (aCSF) of the following composition (in mM): NaCl 126, KCl 2.5, NaH2PO4 1.25, MgCl2 2, CaCl2 2, NaHCO3 26, glucose 10, pyruvic acid 2, ascorbic acid 0.4. Slices were left to recover for at least 60 min at room temperature prior to experimentation, then were transferred to a 24-well plate containing oxygenated aCSF, 2 slices per plate. Five minutes later, 500 μL of aCSF was removed, mixed with 1 mL of LC-MS grade methanol (ThermoFisher, A456–1), snap frozen, and used for control purposes. To activate astrocytes glutamate (3 × 10−4 mol/L, for 5 min) was added to the chamber. Then, the aCSF was removed, mixed with 1 mL of LC-MS grade methanol, and snap-frozen for analyses. Brain slices were snap frozen for protein concentration analyses to normalize the lipidomics data.
Identification and quantification of EETs (5,6-,8,9-, 11,12- and 14,15-EET), gliotransmitters involved in NVC responses, by LC-MS/MS was performed by the Schwartzman laboratory using a Shimadzu Triple Quadrupole Mass Spectrometer LCMS-8050 using multiple reaction monitoring mode (Garcia et al. 2015). Protein concentrations from frozen brain slices were used to normalize the data.
Analysis of lipid mediator content in the mouse brain
To determine how WBI affects levels of key lipid mediators, high-throughput mass spectrometric analysis of eicosanoids in whole brain samples was performed on an ABI/Sciex 6500 Q-TRAP according to published protocols (Wang et al. 2014; Valcarcel-Ares et al. 2018).
qPCR: senescence markers and neurovascular coupling-related gene expression
Quantitative real-time PCR (qPCR) was used to analyze mRNA levels of senescence markers, RFP and herpes simplex virus thymidine kinase, and genes relevant for neurovascular impairment in cortical and hippocampal samples using validated TaqMan probes (Applied Biosystems) and a Strategen MX3000 platform, as previously reported (Toth et al. 2015a; Toth et al. 2014; Tucsek et al. 2017). Targets included enzymes involved in synthesis of eicosanoid gliotransmitters and other lipid mediators as well as pro- and antioxidative factors. In brief, total RNA was isolated with a Mini RNA Isolation Kit (Zymo Research, Orange, CA) and was reverse transcribed using Superscript III RT (Invitrogen) as described previously (Toth et al. 2015a). Quantification was performed using the ΔΔCq method. The relative quantities of the reference genes Hprt, Ywhaz, B2m, Actb, and S18 were determined, and a normalization factor was calculated based on the geometric mean for internal normalization.
Detection of senescent astrocytes by immunohistochemistry
The effect of WBI on the prevalence of senescent cells in the brain was also assessed by immunohistochemistry. Brains were perfusion-fixed (4% ice-cold paraformaldehyde; at 100 mmHg). Frozen OCT-embedded sagittal sections (35 μm) were cut and stored free-floating in cryoprotectant solution (25% glycerol, 25% ethylene glycol, 25% of 0.1 m phosphate buffer, and 25% water) at − 20 °C. Sections were rinsed with Tris-buffered saline (TBS), permeabilized with TBS with 0.05% Tween-20. Antigen retrieval was achieved using 10 mM citrate buffer (10 mm sodium citrate and 0.05% Tween 20, pH 6.0) at 90 °C for 20 min followed by 3× washes with TBST. After blocking with 5% BSA and 1% fish gelatin in TBS at room temperature for 2 h, sections were immunostained for astrocytes using goat anti-mouse GFAP (1:100, Abcam, AB53554) overnight at 4 °C. Sections were washed for 5 min (3×) with TBST followed by incubation with donkey anti-goat Alexa Fluor 488 secondary antibody (1:200, Abcam, ab150129) for 2 h at room temperature. The sections were then washed with TBST for 5 min (3×), followed by nuclei staining with DAPI (5 mg/mL, Invitrogen, 1:10,000) for 5 min. Finally, the sections were washed and mounted on to slides using Prolong antifade mounting medium. Confocal images of were obtained using Leica SP8 MP confocal laser scanning microscope. Senescent astrocytes were identified by the co-expression of GFAP and RFP in the brain sections.
Quantification of senescent cell burden by flow cytometric analysis
We obtained single-cell suspensions from the brain samples. After perfusion with ice-cold PBS, the brains were gently diced and digested in the mix of collagenase (Sigma), dispase I (D4818), Hyaluronidase (Sigma, H4272), and elastase (Sigma, E0127) in PBS for 30 min at 37 °C with gentle, continuous agitation. Further, brain tissue was mechanically dissociated and run through 100 and 30 μm nylon mesh (Miltenyi) to prepare single-cell suspensions. The cells were fixed in 2% PBS for 5 min, washed once with ice-cold PBS, and stained with RFP-Booster (Atto-647, 1:1000) (ChromoTek GmbH) for Fluorescent Activated Cell Sorting (FACS) using FACSAria Fusion. Sorted cell population enriched for RFP+ cells were subsequently stained with anti-GLAST-PE monoclonal antibody (Miltenyi Biotech) for flow-cytometric analysis. The RFP-Booster allows for the detection of senescent cells that express RFP-containing 3MR construct under the control of the p16Ink4a promoter. The anti-GLAST monoclonal antibody specifically detects an extracellular epitope of the astrocyte-specific l-glutamate/l-aspartate transporter GLAST (EAAT1, Slc1a3). Previous studies showed that it labels virtually all astrocytes, whereas oligodendrocytes, microglia, neurons, and neuronal progenitors are GLAST negative (Jungblut et al. 2012). First, the percentage of p16-RFP positive cells was determined in the single cell suspensions from whole brain lysates using flow cytometry. Then, the ratio of RFP+/GLAST+ senescent astrocytes as a percentage of all GLAST+ astrocytes was determined. Next, FACS was utilized to enrich RFP+ senescent cells. The ratio of GLAST+/RFP+ senescent astrocytes as a percentage of all RFP+ senescent cells was determined. Data was acquired on a FACSCalibur™ flow cytometer (BD Biosciences). Cell debris was excluded from the analysis.
Irradiation-induced senescence in cultured astrocytes
Cellular senescence is characterized by expression of senescence-associated β-galactosidase (SA-β-gal) activity. To assess the sensitivity of cultured primary astrocytes to γ-irradiation-induced senescence, we measured SA-β-gal activity in irradiated rat brain hippocampal astrocytes. These cells were purchased from Lonza (www.lonza.com; Lonza No. R-HiAs-521, passage 1) and cultured in six-well plates in astrocyte basal medium (Lonza No. CC-3187) supplemented with AGM-SingleQuots (Lonza No. CC-4123) according to the vendor’s guidelines and as previously described (Ungvari et al. 2013). γ-Irradiation (6 Gy) was administered as described above. Astrocytes were cultured for 7 days after irradiation in complete growth media. To assess the sensitivity of astrocytes to γ-irradiation-induced senescence, SA-β-gal activity was compared in irradiated rat astrocytes and non-irradiated controls. On day 8 after irradiation, histochemical staining for SA-β-gal activity was performed using the Sigma-Senescence Cells Histochemical Staining Kit (Sigma, No. CS0030, St. Louis, MO), following the manufacturer’s guidelines. To analyze the ratio of senescent cells in each well, microscopic images of the stained astrocyte cultures were captured (at × 10 magnification, 30 random fields per group). The percentage of β-galactosidase-positive cells (blue cytoplasmic staining) was calculated by a naïve observer.
In separate experiments the effect of irradiation-induced senescence on secretion of eicosanoid mediators was assessed. To stimulate astrocytes glutamate (3 × 10−4 mol/L, for 5 min) was added to the control astrocytes and cells with irradiation-induced senescence. Then, the medium was removed, mixed with 1 mL of LC-MS grade methanol, and snap-frozen for analyses. To determine the effects of irradiation-induced senescence on release of EETs and the oxidative stress marker 8-iso-PGF2α, mass spectrometric analysis of the supernatant for these eicosanoids was performed on an ABI/Sciex 6500 Q-TRAP according to published protocols (Wang et al. 2014; Valcarcel-Ares et al. 2018).
Statistical analysis
Depending on the experiment, statistical analyses were carried out by unpaired t test, one-way or two-way ANOVA using Prism 7.0 for Windows (Graphpad Software, La Jolla, CA), as appropriate. A P value of < 0.05 was considered statistically significant. Data are expressed as mean ± SEM.
Results
WBI induces astrocyte senescence: protective effects of senolytic treatments
To determine the effect of WBI on cellular senescence we analyzed senescence markers in the brain samples by qPCR. The expression of Cdkn2a (p16Ink4a; Fig. 1a) as well as other markers of senescence, including inflammatory factors characteristic of the SASP increased significantly after WBI both in the cortex (Fig. 1b) and the hippocampus (data not shown). WBI also resulted in significant expression of RFP and herpes simplex virus thymidine kinase in p16-3MR mice (Fig. 1a), confirming that irradiation promotes cellular senescence in the brain.

Transcriptional footprint of increased cellular senescence in the cortex of WBI-treated mice. Panel a Taqman qPCR data showing that WBI results in increased expression of the senescence marker Cdkn2a (p16Ink4a) and the senescence indicators RFP and herpes simplex virus thymidine kinase (HStk) in the cortices of WBI p16-3MR mice as compared with those in control p16-3MR mice. Data are mean ± SEM (n = 9–10 for each data point). *P < 0.05 vs. control. Brains were analyzed 6 months post-WBI. Panel b Heat-map depicting normalized log2-fold changes in mRNA expression of senescence markers and components of the senescence-associated secretory phenotype in the cortices of control and WBI-treated mice. The WBI-induced changes in the expression of each gene depicted is statistically significant
To determine the efficacy of senolytic treatments, we used flow cytometry to detect p16-RFP+ senescent cells 6 months after WBI. The number of p16-RFP+ cells in the mouse brain after WBI (Fig. 2a, b). In contrast, both GCV and ABT263 treatment reduced p16-RFP+ senescent cell burden in the irradiated mouse brains (Fig. 2a, b). We also found that expression of markers of cellular senescence, including that of RFP (Fig. 2c), was significantly decreased by both GCV and ABT263 treatment, consistent with the successful elimination of senescent cells.

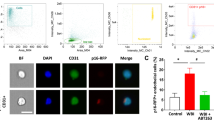
Successful elimination of senescent cells in WBI-treated p16-3MR mice by GCV and ABT263. Panel a Flow cytometric detection of senescent RFP+ cells in single-cell suspensions obtained from the brains of control and WBI-treated p16-3MR mice that received vehicle, ganciclovir (GCV), or the senolytic drug ABT263. Dot plots of RFP-Booster Atto647N fluorescence (which correlates with p16-3MR expression) versus autofluorescence (583/26 nm) depict the percentage of senescent cells with bright fluorescence. Brains were analyzed 6 months post-WBI. Summary data are shown in panel b. Note that WBI significantly increases the presence of senescent cells in the mouse brain, which is reversed by both GCV and ABT263 treatment. Data are mean ± SEM (n = 4 for each data point). *P < 0.05 vs. control, #P < 0.05 vs. WBI. Panel c qPCR data showing expression of the senescence indicator p16-3MR component RFP in the cortices of control and WBI-treated p16-3MR mice that received vehicle, GCV, or ABT263. Data are mean ± SEM (n = 4–8 for each data point). *P < 0.05 vs. control, #P < 0.05 vs. WBI
To demonstrate how WBI affects astrocytes, samples enriched for senescent p16-RFP+ cells were co-sorted for the astrocyte marker GLAST+ (Fig. 3a). Among the p16-RFP positive cells 41 ± 8% were GLAST+/p16-RFP+, indicating that WBI induces significant astrocyte senescence (Fig. 3b). Using immunohistochemistry we confirmed that nearly 40% of all p16-RFP+ cells in the brains of WBI-treated mice were GFAP+ astrocytes (Fig. 3c). Labeling for NeuN and Olig2 confirmed that neurons and oligodendrocytes do not exhibit significant WBI-induced 3MR expression (data not shown). We found that ~ 10% of all GFAP+ cells were p16-RFP+.

GCV and ABT263 treatment successfully deplete senescent astrocytes from WBI-treated p16-3MR mice. Panel a Flow cytometric detection of RFP+/GLAST+ senescent astrocytes and RFP+/GLAST-senescent non-astrocytic cells in single-cell suspension obtained from the brain of a WBI-treated p16-3MR mouse, which was enriched for RFP+ cells by FACS. Dot plot of RFP-Booster Atto647N fluorescence (which correlates with p16-3MR expression) versus GLAST-PE (phycoerythrin) fluorescence depicts the percentages of senescent astrocytes with bright fluorescence in both channels and that of RFP+/GLAST-senescent non-astrocytic cells. Panel b Pie chart showing the percentage of GLAST+ senescent astrocytes as a percentage of all senescent (RFP+) cells. Panel c Fluorescence micrographs of p16-RFP detection in GFAP+ astrocytes in the hippocampus of a WBI-treated p16-3MR mouse. Note that ~ 10% of GFAP+ astrocytes exhibit red RFP-Booster Atto647N fluorescence. Panel d Flow cytometric detection of RFP+/GLAST+ senescent astrocytes in single-cell suspensions obtained from the brains of control p16-3MR mice and WBI-treated p16-3MR mice that received vehicle, ganciclovir (GCV), or the senolytic drug ABT263. Dot plots of RFP-Booster Atto647N fluorescence versus GLAST-PE fluorescence depict the ratio of senescent astrocytes as a percentage of all GLAST+ astrocytic cells. Brains were analyzed 6 months post-WBI. Summary data are shown in panel d. Note that WBI significantly increases the presence of senescent astrocytes in the mouse brain, which is reversed by both GCV and ABT263 treatment. Data are mean ± SEM (n = 4 for each data point). *P < 0.05 vs. control, #P < 0.05 vs. WBI
Further, FACS analysis confirmed that in response to WBI close to ~ 10% of GLAST+ astrocytes express the 3MR protein (Fig. 3d, e). Both GCV and ABT263 treatment significantly reduced p16-RFP+/GLAST+ senescent astrocytes in the irradiated mouse brains (Fig. 3d, e).
WBI impairs eicosanoid gliotransmitter-mediated NVC responses: protective effects of senolytic treatments
To determine whether astrocyte senescence impairs astrocyte-mediated NVC responses, we assessed functional hyperemia in the whisker barrel cortex in mice at 6 months after WBI. CBF responses in the whisker barrel cortex elicited by contralateral whisker stimulation were significantly decreased in WBI-treated mice compared with control animals indicating impaired NVC (representative laser speckle contrast images and CBF tracings are shown in Fig. 4a, b; summary data are shown in Fig. 4c), extending our recent findings (Ungvari et al. 2017). Consistent with an important role for EETs in mediating functional hyperemia (Toth et al. 2015a), administration of the epoxygenase inhibitor MSPPOH significantly decreased NVC responses in control animals (representative CBF tracings are shown in Fig. 4b; summary data are shown in Fig. 4c). In contrast, in WBI-treated animals NVC responses were unaffected by MSPPOH, indicating that WBI impairs NVC responses mediated by astrocyte-derived EETs (Fig. 4b, c). In WBI-treated mice removal of senescent astrocytes by GCV or ABT263 significantly increased CBF responses induced by contralateral whisker stimulation, restoring NVC to levels observed in control mice (Fig. 4b, c). In GCV- and ABT263-treated WBI mice MSPPOH significantly decreased CBF responses elicited by whisker stimulation (Fig. 4b, c), suggesting that removal of senescent astrocytes restored mediation of NVC responses mediated by astrocyte-derived EETs. NVC responses in non-irradiated mice were not significantly affected by GCV or ABT263 (data not shown).

Elimination of senescent cells improves astrocyte-mediated neurovascular coupling responses in WBI-treated mice. Panel a Representative pseudocolor laser speckle flowmetry maps of baseline CBF (upper panels) and CBF changes in the whisker barrel field relative to baseline during contralateral whisker stimulation (lower panels, right oval, 30 s, 5 Hz) in control and WBI-treated p16-3MR mice that received vehicle, ganciclovir (GCV), or the senolytic drug ABT263. NVC responses were assessed 6 month post-WBI (see “Materials and methods”). The EET-mediated component of the NVC response was assessed by treatment with the epoxygenase inhibitor MSPPOH. Color bar represents CBF as percent change from baseline. Panel b shows the time-course of CBF changes after the start of contralateral whisker stimulation (horizontal bars). Summary data are shown in panel c. Data are mean ± SEM (n = 7–21 in each group), *P < 0.05 vs. control, #P < 0.05 vs. vehicle treated WBI. &P < 0.05 vs. no MSPPOH
WBI impairs production of eicosanoid gliotransmitters
LC/MS/MS measurements demonstrated that WBI resulted in a diminished production of the potent vasodilator EET regioisomers 5,6-EET (Fig. 5a), 8,9-EET (Fig. 5b), 11,12-EET (Fig. 5c), and 14,15-EET (Fig. 5d) in response to glutamate stimulation of brain slices, extending the findings of the physiological studies.

WBI treatment of mice and irradiation-induced senescence in cultured astrocytes impair synthesis/release of eicosanoid gliotransmitters. Panels a–d Production of the eicosanoid gliotrasmitters 5,6 EET (a), 8,9 EET (b), 11,12 EET (c), and 14,15 EET (d) in glutamate-activated brain slices from control and WBI-treated mice, measured by liquid chromatography/mass spectrometry (LC/MS) (n = 6 in each group; *P < 0.05 vs. control; see “Materials and methods”). Panel e WBI-induced changes in lipid mediator content in the mouse cortex (x axis) and hippocampus (y axis). Lipid mediators were quantified in fresh-frozen, non-perfused brain samples using LC/MS (see “Materials and methods”). Some lipid mediators with significant (P < 0.05) and consistent WBI-induced changes are highlighted. Summary data (n = 6 for each data point). Panel f γ-Irradiation induces cellular senescence in cultured rat astrocytes. Astrocytes were irradiated (6 Gy) and stained for senescence-associated (SA)-β-galactosidase activity 7 days post irradiation. Panels g–f Irradiated, senescent astrocytes exhibit increased level of the oxidative stress marker 8-iso-PGF2α (panel g) and decreased glutamate-induced release of EET gliotransmitters (panel h). *P < 0.05 vs. non-irradiated controls. Data are mean ± SEM (n = 6 for each data point)
To further assess the effect of WBI on the level of lipid mediators, we identified consistent WBI-induced differences defined as significant lipid mediator concentration differences between control and WBI-treated mice that are prevalent in both the hippocampus and cortex (Fig. 5e). The analysis was based on lipid mediator measurements from six animals per group in both tissues. WBI consistently increased the levels of 8-isoprostanes (including 8-iso-PGF2α and 8-iso-15-keto-PGF2α), which are vasoconstrictor metabolites produced by non-specific lipid peroxidation and are considered markers of oxidative stress. WBI also increased levels of 16,17-EpDPE (16,17-epoxy-docosapentaenoic acid), which is produced from cytochrome P450 epoxygenase action on docosahexaenoic acid (DHA) and may exert anti-angiogenic (Zhang et al. 2013) and vasoactive (Sato et al. 2014; Wang et al. 2011) effects and modulate inflammation (Ulu et al. 2013). WBI also increased 5-oxo-ETE (5-oxo-eicosatetraenoic acid), a potent pro-inflammatory member of the 5-HETE family of autocrine/paracrine mediators, which is preferentially produced under increased oxidative stress conditions and in senescent cells. WBI also increased 5-oxo-EDE (15-oxo-eicosadienoic acid), a related eicosanoid produced by the oxidation of 15-HEDE and 5-HETrE (5-hydroxy-eicosatrienoic acid), which is an arachidonic acid metabolite that promotes platelet aggregation and is readily converted to the pro-inflammatory leukotriene A3 via the 5-lipoxygenase-leukotriene pathway. In contrast, WBI resulted in significant decreases in the anti-inflammatory eicosanoid PGJ2 and the putative vasodilators 11β-PGE2 and 14(15)-EpETE (14,15-epoxy eicosatetraenoic acid), which is the ω-3 homolog of 14,15-EET, derived via CYP450-dependent epoxidation of eicosapentaenoic acid.
γ-Irradiation induces senescence in cultured astrocytes: effects eicosanoid gliotransmitter production
To provide evidence that irradiation induces senescence in astrocytes, we performed SA-β-gal staining of irradiated and nonirradiated astrocyte cultures. We found that γ-irradiation increased the ratio of astrocytes with positive SA-β-gal staining. At the 6 Gy irradiation over ~ 70% of irradiated astrocytes were β-galactosidase positive on day 8 post-irradiation (Fig. 5f).
LC/MS/MS measurements demonstrated that γ-irradiation-induced senescence resulted in an increased release of the oxidative stress marker 8-iso-PGF2α from the astrocytes (Fig. 5g). γ-Irradiation-induced senescence also resulted in diminished astrocytic production of vasodilator EETs (Fig. 5h).
WBI-induced persistent changes in neurovascular coupling-related gene expression
To better understand the gene expression footprint of WBI-induced senescence we quantified by qPCR mRNA expression of genes that are implicated in astrocyte dysfunction and neurovascular impairment, including enzymes involved in synthesis of eicosanoid gliotransmitters and other lipid mediators as well as pro- and antioxidative factors. We found that WBI resulted in significant changes in the expression of several cytochrome P450 enzymes both in the cortex (Fig. 6a) and hippocampus (Fig. 6b). The superoxide generating enzyme NADPH oxidase 2 (Nox2/Cybb) and arachidonate 5-lipoxygenase (Alox5), which produces 5-oxo-ETE, were also consistently upregulated by WBI in both tissues.

WBI elicits complex alterations in neurovascular coupling-related gene expression in the mouse brain. qPCR data showing the effect of WBI treatment on cortical (panel a) and hippocampal (panel b) mRNA levels of targets that play a role in synthesis/release of eicosanoid gliotransmitters and regulation of NVC responses in the mouse brain. The y axis represents the average log2 fold change in mRNA levels in cortical (a) and hippocampal (b) samples from WBI-treated p16-3MR mice relative to the corresponding controls. Significant (P < 0.05) changes are highlighted. The x axis indicates the mRNA rank from the most upregulated to the most downregulated. n = 10 for each data point. Gene expression was analyzed using a custom-designed microfluidic Taqman qPCR array
WBI impairs cognitive performance in mice: protective effects of senolytic treatments
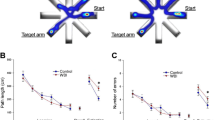
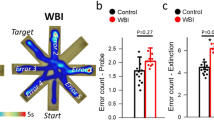
We demonstrated that C57BL/6 mice exhibit significant impairment of hippocampal encoded functions of learning and memory at 3 to 5 months after WBI treatment (Ungvari et al. 2017; Warrington et al. 2013). To gain insight into the mechanism underlying WBI-induced cognitive impairment, we assessed the effects of senolytic treatments on learning and memory function in mice 6 months after WBI using the radial-arm water maze (Fig. 7a). We compared the learning performance of mice in each experimental group by analyzing the day-to-day changes in the combined error rate, calculated by adding 1 error for each incorrect arm entry as well as for every 15 s spent not exploring the arms. The error rate for each mouse is then averaged among each experimental group in every trial block. During acquisition, mice from all groups showed a decrease in the path length (not shown) and the combined error rate across days, indicating learning of the task. We confirmed that learning was significantly impaired when tested 6 months after WBI (Fig. 7b). Memory recall 7 days later was also impaired in WBI-treated mice, which also showed impaired extinction ability on day 11 (Fig. 7b). The extinction blocks describe the ability of the mouse to forget and re-learn the task with a different platform location. These findings confirm that both learning plasticity and memory are impaired after WBI treatment. These data are consistent with the progressive development of cognitive impairment post-WBI. Restoration of neurovascular function after administration of senolytic treatments positively affected cognitive function in WBI-treated mice. Figure 7b shows that both GCV and ABT263 treatment resulted in a decreased error rate in WBI-treated mice. Mice treated with GCV after WBI showed better memory recall on day 7, whereas the effect of ABT263 treatment did not reach significance. Both GCV- and ABT263-treated mice exhibited improved extinction ability on day 11 and better relearning (Fig. 7b). Cognitive performance in non-irradiated mice was not significantly affected by GCV or ABT263 (data not shown). Analyses of noncognitive parameters revealed no improvement in swimming speed or non-exploratory behavior (cumulative time mice spent not actively looking for the platform, e.g., floating) in WBI-treated mice that were given senolytics (data not shown). Together, the results suggest that removal of senescent cells after WBI improved performance in the radial-arm water maze, which results from enhanced hippocampal-dependent spatial learning and memory and not from changes in motor or motivational processes.

Rescue of NVC responses by elimination of senescent cells improves performance of WBI-treated p16-3MR mice in the radial-arm water maze (RAWM). Control p16-3MR mice and WBI-treated p16-3MR mice that received vehicle, ganciclovir (GCV), or ABT263 were tested in the RAWM. a Representative probe test search path of a randomly selected animal from each group is shown with the target position highlighted in green. Note the WBI-treated mice that received vehicle required more time and a longer path length in order to find the hidden escape platform than both control animals and GCV- or ABT263-treated WBI mice. This particular WBI-treated mouse did not even find the platform within the allowed time. The WBI-treated mouse also re-entered a previously visited arm multiple times. b WBI-treated animals have higher combined error rates throughout day 2 and 3 of the learning phase, and the retrieval day 10 as compared with control animals. WBI-treated animals also make significantly more errors during probe reversal than control animals. In contrast, WBI-treated mice treated with GCV or ABT263 perform this task significantly better than untreated irradiated mice. Combined error rate is calculated by adding 1 error for each incorrect arm entry as well as for every 15 s spent not exploring the arms. All data are shown as mean ± SEM (n = 20 for each data point). *P < 0.05 vs. control, #P < 0.05 vs. vehicle treated WBI
Discussion
The key finding of his study is that both pharmacological (ABT263/Navitoclax) and genetic depletion of senescent astrocytes rescue WBI-induced impairment of NVC responses and improve cognitive function.
Our studies demonstrate that a clinical regimen of WBI induces senescence in astrocytes. Our cell culture findings taken together with our previous studies confirm that cultured astrocytes exhibit significant DNA damage in response to γ-irradiation comparable to that used in the WBI protocol (Ungvari et al. 2013), which results in induction of cellular senescence. The clinical relevance of our studies is confirmed by a recent report demonstrating that in irradiated human brain tissues astrocytes express the senescence markers p16INK4A and Hp1γ(Turnquist et al. 2019). The pathophysiological consequences of WBI-induced astrocyte senescence are likely multifaceted.
An important function of astrocytes is to mediate NVC responses (Toth et al. 2015a; Filosa and Iddings 2013; Tran and Gordon 2015; Dunn et al. 2013; Girouard et al. 2010). We provide critical evidence that astrocyte senescence impairs release/synthesis of vasodilator EETs, which contribute to WBI-induced impairment of NVC responses. A critical role for senescent astrocytes in impaired gliovascular coupling is demonstrated by the findings that removal of senescent astrocytes results in near complete recovery of the astrocytic component of functional hyperemia in WBI-treated animals.
We posit that restoration of NVC responses contributes to the cognitive benefits offered by senolytic treatments in WBI-treated mice. Several lines of evidence support this concept. First, clinical studies show that impaired NVC responses predict a decline in higher cortical functions in humans (Sorond et al. 2013; Sorond et al. 2011). Second, there is strong experimental evidence showing that NVC responses positively correlate with cognitive function in rodents (Febo and Foster 2016), that pharmacological inhibition of gliovascular coupling impairs cognition (Tarantini et al. 2017b; Tarantini et al. 2015), and that pharmacological improvement of functional hyperemia results in cognitive benefits in mouse models of aging (Toth et al. 2014; Tarantini et al. 2019b; Tarantini et al. 2018b; Oomen et al. 2009). Importantly, astrocyte senescence occurs in other pathological conditions associated with cognitive decline (e.g., chemotherapy-induced cognitive decline, Alzheimer’s disease, Parkinson’s disease, aging) (Csipo et al. 2019b; Lye et al. 2019),(Cohen and Torres 2019; Bhat et al. 2012; Chinta et al. 2018; Bussian et al. 2018). Future studies should also investigate the mechanistic role of senescent astrocytes in neurovascular dysfunction and cognitive impairment in these conditions as well. Importantly, in addition to EETs astrocyte senescence also alters the synthesis of a wide range of other vasoactive lipid mediators (Chinta et al. 2018), which can also contribute to CBF dysregulation (e.g., by decreasing basal tone of resistance arterioles) (Rosenegger and Gordon 2015; Mehina et al. 2017). The mechanisms by which induction of senescence pathways alter production of lipid mediators in astrocytes are not well understood and likely involve both dysregulation of the expression and activity of the enzymes involved in the synthesis of the lipid mediators (e.g., cytochrome P450 enzymes). It is likely that there is also a bi-directional link between increased cellular oxidative stress and production of eicosanoid mediators. We propose that changes in lipid mediators are critical components of the SASP in astrocytes, which, in addition to their role in neurovascular impairment, could contribute to a number of age-related diseases (e.g., by modulating neuroinflammation).
Astrocytes also support the micro-architecture of the brain parenchyma; store (e.g., glycogen fuel reserve), metabolize, and distribute energy substrates (e.g., cerebral gluconeogenesis, lactate production); maintain brain homeostasis (including regulation of ion concentration in the extracellular space); regulate transmitter uptake and release; maintain the blood-brain barrier; regulate the myelinating activity of oligodendrocytes; control synaptogenesis, synaptic maintenance, and long-term potentiation; and contribute to neural repair. Senescent astrocytes are known to undergo complex phenotypic changes (Cohen and Torres 2019; Chinta et al. 2018), including metabolic reprogramming, morphological alterations, changes in the gene expression profile, and critical changes in their secretome. All of these senescence-related phenotypic changes likely fundamentally affect the diverse physiological roles of astrocytes, contributing to alterations in brain function. As astrocytes are extremely heterogeneous, in future studies it will be interesting to test whether astrocytes in different brain regions are similarly affected by WBI. Our current findings demonstrating similar transcriptome and lipidome changes in the hippocampus and the cortex provide preliminary evidence in support of this view. Unlike the neuronal circuits, astrocytes are connected via gap junctions and form a functional syncytium. They share cytoplasmic signals (e.g., intracellular Ca2+ waves can easily propagate through the astrocyte network), and thus, one senescent astrocyte can directly modulate both directly and indirectly (through paracrine factors) the function of a number of neighboring astrocytes. Thus, increased presence of senescent astrocytes in this network is expected to alter the function of a number of neighboring astrocytes and thereby the function of the astrocyte network as a whole. Further studies are evidently needed to experimentally dissect the specific mechanistic roles of senescence-related changes in the aforementioned astrocyte functions in the pathogenesis of WBI-induced cognitive impairment.
Our studies suggest that ~ 40% of senescent cells in the brain after WBI are senescent astrocytes. This is somewhat higher than the ratio of astrocytic glial cells in the murine brain (Keller et al. 2018). We posit that astrocytes are more sensitive to γ-radiation-induced senescence than many other cell types (e.g., neuronal senescence could not be detected after WBI). Other cell types that are likely affected by WBI-induced senescence include cerebromicrovascular endothelial cells (Ungvari et al. 2013), microglia, and pericytes. Further studies are needed to assess senescence-related changes in these cells after WBI and elucidate their role in the pathogenesis of WBI-induced cognitive impairment.
In conclusion, our results demonstrate that senolytic treatments exert neurovascular protective effects in WBI-treated mice. Depletion of senescent cells by genetic means or by treatment with Navitoclax (ABT263) rescued NVC responses in WBI-treated mice, which likely contributes to improvements in higher cortical function. Our findings point to the potential use of senolytic drugs as therapies to prevent cognitive impairment associated with WBI. Our findings have important clinical relevance. Navitoclax (ABT263) is used as an FDA-approved, orally active anti-cancer drug in humans, which does not have significant off-target effects. Thus, future clinical trials using Navitoclax or other senolytics are feasible and hold promise for preserving neurovascular function and cognitive outcomes in WBI-treated patients.
References
Abdul-Aziz AM, Sun Y, Hellmich C, Marlein CR, Mistry J, Forde E, Piddock RE, Shafat MS, Morfakis A, Mehta T, Di Palma F, Macaulay I, Ingham CJ, Haestier A, Collins A, Campisi J, Bowles KM, Rushworth SA (2019) Acute myeloid leukemia induces protumoral p16INK4a-driven senescence in the bone marrow microenvironment. Blood 133:446–456
Ashpole NM, Warrington JP, Mitschelen MC, Yan H, Sosnowska D, Gautam T, Farley JA, Csiszar A, Ungvari Z, Sonntag WE (2014) Systemic influences contribute to prolonged microvascular rarefaction after brain irradiation: a role for endothelial progenitor cells. Am J Physiol Heart Circ Physiol 307:H858–H868
Attwell D, Buchan AM, Charpak S, Lauritzen M, Macvicar BA, Newman EA (2010) Glial and neuronal control of brain blood flow. Nature. 468:232–243
Baar MP, Brandt RMC, Putavet DA, Klein JDD, Derks KWJ, Bourgeois BRM, Stryeck S, Rijksen Y, van Willigenburg H, Feijtel DA, van der Pluijm I, Essers J, van Cappellen WA, IWF v, Houtsmuller AB, Pothof J, de Bruin RWF, Madl T, JHJ H, Campisi J, de Keizer PLJ (2017) Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell 169:132–147 e16
Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, van Deursen JM (2011) Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 479:232–236
Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, Saltness RA, Jeganathan KB, Verzosa GC, Pezeshki A, Khazaie K, Miller JD, van Deursen JM (2016) Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 530:184–189
Bhat R, Crowe EP, Bitto A, Moh M, Katsetos CD, Garcia FU, Johnson FB, Trojanowski JQ, Sell C, Torres C (2012) Astrocyte senescence as a component of Alzheimer’s disease. PLoS One 7:e45069
Bussian TJ, Aziz A, Meyer CF, Swenson BL, van Deursen JM, Baker DJ (2018) Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature 562:578–582
Campisi J (2013) Aging, cellular senescence, and cancer. Annu Rev Physiol 75:685–705
Campisi J (2016) Cellular senescence and lung function during aging. Yin and Yang. Ann Am Thorac Soc 13:S402–S406
Chang J, Wang Y, Shao L, Laberge RM, Demaria M, Campisi J, Janakiraman K, Sharpless NE, Ding S, Feng W, Luo Y, Wang X, Aykin-Burns N, Krager K, Ponnappan U, Hauer-Jensen M, Meng A, Zhou D (2016) Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med 22:78–83
Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J, van Deursen JM (2016) Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science. 354:472–477
Chinta SJ, Woods G, Rane A, Demaria M, Campisi J, Andersen JK (2015) Cellular senescence and the aging brain. Exp Gerontol 68:3–7. https://doi.org/10.1016/j.exger.2014.09.018
Chinta SJ, Woods G, Demaria M, Rane A, Zou Y, McQuade A, Rajagopalan S, Limbad C, Madden DT, Campisi J, Andersen JK (2018) Cellular senescence is induced by the environmental neurotoxin paraquat and contributes to neuropathology linked to Parkinson’s disease. Cell Rep 22:930–940
Cohen J, Torres C (2019) Astrocyte senescence: evidence and significance. Aging Cell 18:e12937
Coppe JP, Rodier F, Patil CK, Freund A, Desprez PY, Campisi J (2011) Tumor suppressor and aging biomarker p16(INK4a) induces cellular senescence without the associated inflammatory secretory phenotype. J Biol Chem 286:36396–36403
Csipo T, Mukli P, Lipecz A, Tarantini S, Bahadli D, Abdulhussein O, Owens C, Kiss T, Balasubramanian P, Nyul-Toth A, Hand RA, Yabluchanska V, Sorond FA, Csiszar A, Ungvari Z, Yabluchanskiy A (2019a) Assessment of age-related decline of neurovascular coupling responses by functional near-infrared spectroscopy (fNIRS) in humans. Geroscience. 41:495–509
Csipo T, Lipecz A, Ashpole NM, Balasubramanian P, Tarantini S (2019) Astrocyte senescence contributes to cognitive decline. Geroscience. https://doi.org/10.1007/s11357-019-00140-9
Csiszar A, Tarantini S, Fulop GA, Kiss T, Valcarcel-Ares MN, Galvan V, Ungvari Z, Yabluchanskiy A (2017) Hypertension impairs neurovascular coupling and promotes microvascular injury: role in exacerbation of Alzheimer’s disease. Geroscience 39(4):359–372. https://doi.org/10.1007/s11357-017-9991-9
Csiszar A, Yabluchanskiy A, Ungvari A, Ungvari Z, Tarantini S (2019) Overexpression of catalase targeted to mitochondria improves neurovascular coupling responses in aged mice. Geroscience. 41:609–617
Demaria M, Ohtani N, Youssef SA, Rodier F, Toussaint W, Mitchell JR, Laberge RM, Vijg J, Van Steeg H, Dolle ME, Hoeijmakers JH, de Bruin A, Hara E, Campisi J (2014) An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell 31:722–733
Demaria M, O'Leary MN, Chang J, Shao L, Liu S, Alimirah F, Koenig K, Le C, Mitin N, Deal AM, Alston S, Academia EC, Kilmarx S, Valdovinos A, Wang B, de Bruin A, Kennedy BK, Melov S, Zhou D, Sharpless NE, Muss H, Campisi J (2017) Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discov 7:165–176
Dunn KM, Hill-Eubanks DC, Liedtke WB, Nelson MT (2013) TRPV4 channels stimulate Ca2+−induced Ca2+ release in astrocytic endfeet and amplify neurovascular coupling responses. Proc Natl Acad Sci U S A 110:6157–6162
Farias Quipildor GE, Mao K, Hu Z, Novaj A, Cui MH, Gulinello M, Branch CA, Gubbi S, Patel K, Moellering DR, Tarantini S, Kiss T, Yabluchanskiy A, Ungvari Z, Sonntag WE, Huffman DM (2019) Central IGF-1 protects against features of cognitive and sensorimotor decline with aging in male mice. Geroscience. 41:185–208
Farr JN, Xu M, Weivoda MM, Monroe DG, Fraser DG, Onken JL, Negley BA, Sfeir JG, Ogrodnik MB, Hachfeld CM, LeBrasseur NK, Drake MT, Pignolo RJ, Pirtskhalava T, Tchkonia T, Oursler MJ, Kirkland JL, Khosla S (2017) Targeting cellular senescence prevents age-related bone loss in mice. Nat Med 23:1072–1079
Febo M, Foster TC (2016) Preclinical magnetic resonance imaging and spectroscopy studies of memory, aging, and cognitive decline. Front Aging Neurosci 8:158
Filosa JA, Iddings JA (2013) Astrocyte regulation of cerebral vascular tone. Am J Physiol Heart Circ Physiol 305:H609–H619
Freund A, Orjalo AV, Desprez PY, Campisi J (2010) Inflammatory networks during cellular senescence: causes and consequences. Trends Mol Med 16:238–246
Fulop GA, Kiss T, Tarantini S, Balasubramanian P, Yabluchanskiy A, Farkas E, Bari F, Ungvari Z, Csiszar A (2018) Nrf2 deficiency in aged mice exacerbates cellular senescence promoting cerebrovascular inflammation. Geroscience. 40:513–521
Garcia V, Cheng J, Weidenhammer A, Ding Y, Wu CC, Zhang F, Gotlinger K, Falck JR, Schwartzman ML (2015) Androgen-induced hypertension in angiotensinogen deficient mice: role of 20-HETE and EETS. Prostaglandins Other Lipid Mediat 116–117:124–130
Gaspar LE, Mehta MP, Patchell RA, Burri SH, Robinson PD, Morris RE, Ammirati M, Andrews DW, Asher AL, Cobbs CS, Kondziolka D, Linskey ME, Loeffler JS, McDermott M, Mikkelsen T, Olson JJ, Paleologos NA, Ryken TC, Kalkanis SN (2010) The role of whole brain radiation therapy in the management of newly diagnosed brain metastases: a systematic review and evidence-based clinical practice guideline. J Neuro-Oncol 96:17–32
Girouard H, Iadecola C (2006) Neurovascular coupling in the normal brain and in hypertension, stroke, and Alzheimer disease. J Appl Physiol (1985) 100:328–335
Girouard H, Bonev AD, Hannah RM, Meredith A, Aldrich RW, Nelson MT (2010) Astrocytic endfoot Ca2+ and BK channels determine both arteriolar dilation and constriction. Proc Natl Acad Sci U S A 107:3811–3816
Gorelick PB, Scuteri A, Black SE, Decarli C, Greenberg SM, Iadecola C, Launer LJ, Laurent S, Lopez OL, Nyenhuis D, Petersen RC, Schneider JA, Tzourio C, Arnett DK, Bennett DA, Chui HC, Higashida RT, Lindquist R, Nilsson PM, Roman GC, Sellke FW, Seshadri S (2011) Vascular contributions to cognitive impairment and dementia: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 42:2672–2713
Jeon OH, Kim C, Laberge RM, Demaria M, Rathod S, Vasserot AP, Chung JW, Kim DH, Poon Y, David N, Baker DJ, van Deursen JM, Campisi J, Elisseeff JH (2017) Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat Med 23:775–781
Jungblut M, Tiveron MC, Barral S, Abrahamsen B, Knobel S, Pennartz S, Schmitz J, Perraut M, Pfrieger FW, Stoffel W, Cremer H, Bosio A (2012) Isolation and characterization of living primary astroglial cells using the new GLAST-specific monoclonal antibody ACSA-1. Glia 60:894–907
Justice JN, Gregory H, Tchkonia T, LeBrasseur NK, Kirkland JL, Kritchevsky SB, Nicklas BJ (2018) Cellular senescence biomarker p16INK4a+ cell burden in thigh adipose is associated with poor physical function in older women. J Gerontol A Biol Sci Med Sci 73:939–945
Kazama K, Anrather J, Zhou P, Girouard H, Frys K, Milner TA, Iadecola C (2004) Angiotensin II impairs neurovascular coupling in neocortex through NADPH oxidase-derived radicals. Circ Res 95:1019–1026
Keller D, Ero C, Markram H (2018) Cell densities in the mouse brain: a systematic review. Front Neuroanat 12:83
Khuntia D, Brown P, Li J, Mehta MP (2006) Whole-brain radiotherapy in the management of brain metastasis. J Clin Oncol 24:1295–1304
Kim HN, Chang J, Iyer S, Han L, Campisi J, Manolagas SC, Zhou D, Almeida M (2019) Elimination of senescent osteoclast progenitors has no effect on the age-associated loss of bone mass in mice. Aging Cell 18:e12923
Lamproglou I, Martin S, Diserbo M, Multon E, Petiet A, Colas-Linhart N, Bok B, Martin C (2001) Total body 4.5 Gy gamma irradiation-induced early delayed learning and memory dysfunction in the rat. Cell Mol Biol (Noisy-le-grand) 47:453–457
Lee YW, Cho HJ, Lee WH, Sonntag WE (2012) Whole brain radiation-induced cognitive impairment: pathophysiological mechanisms and therapeutic targets. Biomol Ther (Seoul) 20:357–370
Lipecz A, Csipo T, Tarantini S, Hand RA, Ngo BN, Conley S, Nemeth G, Tsorbatzoglou A, Courtney DL, Yabluchanska V, Csiszar A, Ungvari ZI, Yabluchanskiy A (2019) Age-related impairment of neurovascular coupling responses: a dynamic vessel analysis (DVA)-based approach to measure decreased flicker light stimulus-induced retinal arteriolar dilation in healthy older adults. Geroscience 41(3):341–349. https://doi.org/10.1007/s11357-019-00078-y
Lye JJ, Latorre E, Lee BP, Bandinelli S, Holley JE, Gutowski NJ, Ferrucci L, Harries LW (2019) Astrocyte senescence may drive alterations in GFAPalpha, CDKN2A p14(ARF), and TAU3 transcript expression and contribute to cognitive decline. Geroscience 41:561–573
Mehina EMF, Murphy-Royal C, Gordon GR (2017) Steady-state free Ca(2+) in astrocytes is decreased by experience and impacts arteriole tone. J Neurosci 37:8150–8165
Ogrodnik M, Zhu Y, Langhi LGP, Tchkonia T, Kruger P, Fielder E, Victorelli S, Ruswhandi RA, Giorgadze N, Pirtskhalava T, Podgorni O, Enikolopov G, Johnson KO, Xu M, Inman C, Schafer M, Weigl M, Ikeno Y, Burns TC, Passos JF, von Zglinicki T, Kirkland JL, Jurk D (2019) Obesity-induced cellular senescence drives anxiety and impairs neurogenesis. Cell Metab 29(5):1233. https://doi.org/10.1016/j.cmet.2019.01.013
Oomen CA, Farkas E, Roman V, van der Beek EM, Luiten PG, Meerlo P (2009) Resveratrol preserves cerebrovascular density and cognitive function in aging mice. Front Aging Neurosci 1:4
Patil CG, Pricola K, Sarmiento JM, Garg SK, Bryant A, Black KL (2012) Whole brain radiation therapy (WBRT) alone versus WBRT and radiosurgery for the treatment of brain metastases. Cochrane Database Syst Rev 9:CD006121
Patil P, Dong Q, Wang D, Chang J, Wiley C, Demaria M, Lee J, Kang J, Niedernhofer LJ, Robbins PD, Sowa G, Campisi J, Zhou D, Vo N (2019) Systemic clearance of p16(INK4a) -positive senescent cells mitigates age-associated intervertebral disc degeneration. Aging Cell 18:e12927
Rodier F, Coppe JP, Patil CK, Hoeijmakers WA, Munoz DP, Raza SR, Freund A, Campeau E, Davalos AR, Campisi J (2009) Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol 11:973–979
Roos CM, Zhang B, Palmer AK, Ogrodnik MB, Pirtskhalava T, Thalji NM, Hagler M, Jurk D, Smith LA, Casaclang-Verzosa G, Zhu Y, Schafer MJ, Tchkonia T, Kirkland JL, Miller JD (2016) Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell 15:973–977
Rosenegger DG, Gordon GR (2015) A slow or modulatory role of astrocytes in neurovascular coupling. Microcirculation 22:197–203
Sato K, Chino D, Nishioka N, Kanai K, Aoki M, Obara K, Miyauchi S, Tanaka Y (2014) Pharmacological evidence showing significant roles for potassium channels and CYP epoxygenase metabolites in the relaxant effects of docosahexaenoic acid on the rat aorta contracted with U46619. Biol Pharm Bull 37:394–403
Shi L, Adams MM, Long A, Carter CC, Bennett C, Sonntag WE, Nicolle MM, Robbins M, D'Agostino R, Brunso-Bechtold JK (2006) Spatial learning and memory deficits after whole-brain irradiation are associated with changes in NMDA receptor subunits in the hippocampus. Radiat Res 166:892–899
Sorond FA, Kiely DK, Galica A, Moscufo N, Serrador JM, Iloputaife I, Egorova S, Dell’Oglio E, Meier DS, Newton E, Milberg WP, Guttmann CR, Lipsitz LA (2011) Neurovascular coupling is impaired in slow walkers: the MOBILIZE Boston study. Ann Neurol 70:213–220
Sorond FA, Hurwitz S, Salat DH, Greve DN, Fisher ND (2013) Neurovascular coupling, cerebral white matter integrity, and response to cocoa in older people. Neurology 81(10):904–909. https://doi.org/10.1212/WNL.0b013e3182a351aa
Soussain C, Ricard D, Fike JR, Mazeron JJ, Psimaras D, Delattre JY (2009) CNS complications of radiotherapy and chemotherapy. Lancet. 374:1639–1651
Tarantini S, Hertelendy P, Tucsek Z, Valcarcel-Ares MN, Smith N, Menyhart A, Farkas E, Hodges E, Towner R, Deak F, Sonntag WE, Csiszar A, Ungvari Z, Toth P (2015) Pharmacologically-induced neurovascular uncoupling is associated with cognitive impairment in mice. J Cereb Blood Flow Metab 35(11):1871–1881. https://doi.org/10.1038/jcbfm.2015.162
Tarantini S, Tran CH, Gordon GR, Ungvari Z, Csiszar A (2016) Impaired neurovascular coupling in aging and Alzheimer's disease: Contribution of astrocyte dysfunction and endothelial impairment to cognitive decline. Exp Gerontol. https://doi.org/10.1016/j.exger.2016.11.004
Tarantini S, Tran CHT, Gordon GR, Ungvari Z, Csiszar A (2017a) Impaired neurovascular coupling in aging and Alzheimer’s disease: contribution of astrocyte dysfunction and endothelial impairment to cognitive decline. Exp Gerontol 94:52–58. https://doi.org/10.1016/j.exger.2016.11.004
Tarantini S, Yabluchanksiy A, Fulop GA, Hertelendy P, Valcarcel-Ares MN, Kiss T, Bagwell JM, O'Connor D, Farkas E, Sorond F, Csiszar A, Ungvari Z (2017b) Pharmacologically induced impairment of neurovascular coupling responses alters gait coordination in mice. GeroScience 39(5–6):601–614. https://doi.org/10.1007/s11357-017-0003-x
Tarantini S, Fulop GA, Kiss T, Farkas E, Zolei-Szenasi D, Galvan V, Toth P, Csiszar A, Ungvari Z, Yabluchanskiy A (2017c) Demonstration of impaired neurovascular coupling responses in TG2576 mouse model of Alzheimer’s disease using functional laser speckle contrast imaging. Geroscience 39(4):465–473. https://doi.org/10.1007/s11357-017-9980-z
Tarantini S, Valcarcel-Ares MN, Yabluchanskiy A, Tucsek Z, Hertelendy P, Kiss T, Gautam T, Zhang XA, Sonntag WE, de Cabo R, Farkas E, Elliott ME, Kinter MT, Deak F, Ungvari Z, Csiszar A 2018a) Nrf2 deficiency exacerbates obesity-induced oxidative stress, neurovascular dysfunction, blood brain barrier disruption, neuroinflammation, amyloidogenic gene expression and cognitive decline in mice, mimicking the aging phenotype. J Gerontol A Biol Sci Med Sci. in press
Tarantini S, Valcarcel-Ares NM, Yabluchanskiy A, Fulop GA, Hertelendy P, Gautam T, Farkas E, Perz A, Rabinovitch PS, Sonntag WE, Csiszar A, Ungvari Z (2018b) Treatment with the mitochondrial-targeted antioxidant peptide SS-31 rescues neurovascular coupling responses and cerebrovascular endothelial function and improves cognition in aged mice. Aging Cell 17
Tarantini S, Yabluchanskiy A, Csipo T, Fulop G, Kiss T, Balasubramanian P, DelFavero J, Ahire C, Ungvari A, Nyul-Toth A, Farkas E, Benyo Z, Toth A, Csiszar A, Ungvari Z (2019a) Treatment with the poly(ADP-ribose) polymerase inhibitor PJ-34 improves cerebromicrovascular endothelial function, neurovascular coupling responses and cognitive performance in aged mice, supporting the NAD+ depletion hypothesis of neurovascular aging. Geroscience. 41:533–542
Tarantini S, Valcarcel-Ares MN, Toth P, Yabluchanskiy A, Tucsek Z, Kiss T, Hertelendy P, Kinter M, Ballabh P, Sule Z, Farkas E, Baur JA, Sinclair DA, Csiszar A, Ungvari Z (2019b) Nicotinamide mononucleotide (NMN) supplementation rescues cerebromicrovascular endothelial function and neurovascular coupling responses and improves cognitive function in aged mice. Redox Biol 24:101192
Tchkonia T, Kirkland JL (2018) Aging, cell senescence, and chronic disease: emerging therapeutic strategies. JAMA 320:1319–1320
Tchkonia T, Zhu Y, van Deursen J, Campisi J, Kirkland JL (2013) Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest 123:966–972
Toth P, Tarantini S, Tucsek Z, Ashpole NM, Sosnowska D, Gautam T, Ballabh P, Koller A, Sonntag WE, Csiszar A, Ungvari ZI (2014) Resveratrol treatment rescues neurovascular coupling in aged mice:role of improved cerebromicrovascular endothelial function and down-regulation of NADPH oxidas. Am J Physiol Heart Circ Physiol 306:H299–H308
Toth P, Tarantini S, Ashpole NM, Tucsek Z, Milne GL, Valcarcel-Ares NM, Menyhart A, Farkas E, Sonntag WE, Csiszar A, Ungvari Z (2015a) IGF-1 deficiency impairs neurovascular coupling in mice: implications for cerebromicrovascular aging. Aging Cell 14:1034–1044
Toth P, Tarantini S, Davila A, Valcarcel-Ares MN, Tucsek Z, Varamini B, Ballabh P, Sonntag WE, Baur JA, Csiszar A, Ungvari Z (2015b) Purinergic glio-endothelial coupling during neuronal activity: role of P2Y1 receptors and eNOS in functional hyperemia in the mouse somatosensory cortex. Am J Physiol Heart Circ Physiol 309:H1837–H1845
Toth P, Szarka N, Farkas E, Ezer E, Czeiter E, Amrein K, Ungvari Z, Hartings JA, Buki A, Koller A (2016) Traumatic brain injury-induced autoregulatory dysfunction and spreading depression-related neurovascular uncoupling: Pathomechanisms, perspectives, and therapeutic implications. Am J Physiol Heart Circ Physiol 311:H1118–H1131
Toth P, Tarantini S, Csiszar A, Ungvari Z (2017) Functional vascular contributions to cognitive impairment and dementia: mechanisms and consequences of cerebral autoregulatory dysfunction, endothelial impairment, and neurovascular uncoupling in aging. Am J Physiol Heart Circ Physiol 312:H1–H20
Tran CH, Gordon GR (2015) Astrocyte and microvascular imaging in awake animals using two-photon microscopy. Microcirculation 22:219–227
Tucsek Z, Toth P, Tarantini S, Sosnowska D, Gautam T, Warrington JP, Giles CB, Wren JD, Koller A, Ballabh P, Sonntag WE, Ungvari Z, Csiszar A (2014) Aging exacerbates obesity-induced cerebromicrovascular rarefaction, neurovascular uncoupling, and cognitive decline in mice. J Gerontol A Biol Sci Med Sci 69:1339–1352
Tucsek Z, Noa Valcarcel-Ares M, Tarantini S, Yabluchanskiy A, Fulop G, Gautam T, Orock A, Csiszar A, Deak F, Ungvari Z (2017) Hypertension-induced synapse loss and impairment in synaptic plasticity in the mouse hippocampus mimics the aging phenotype: implications for the pathogenesis of vascular cognitive impairment. Geroscience 39(4):385–406. https://doi.org/10.1007/s11357-017-9981-y
Turnquist C, Beck JA, Horikawa I, Obiorah IE, Von Muhlinen N, Vojtesek B, Lane DP, Grunseich C, Chahine JJ, Ames HM, Smart DD, Harris BT, Harris CC (2019) Radiation-induced astrocyte senescence is rescued by Delta133p53. Neuro-Oncology 21:474–485
Ulu A, Harris TR, Morisseau C, Miyabe C, Inoue H, Schuster G, Dong H, Iosif AM, Liu JY, Weiss RH, Chiamvimonvat N, Imig JD, Hammock BD (2013) Anti-inflammatory effects of omega-3 polyunsaturated fatty acids and soluble epoxide hydrolase inhibitors in angiotensin-II-dependent hypertension. J Cardiovasc Pharmacol 62:285–297
Ungvari Z, Podlutsky A, Sosnowska D, Tucsek Z, Toth P, Deak F, Gautam T, Csiszar A, Sonntag WE (2013) Ionizing radiation promotes the acquisition of a senescence-associated secretory phenotype and impairs angiogenic capacity in cerebromicrovascular endothelial cells: role of increased DNA damage and decreased DNA repair capacity in microvascular radiosensitivity. J Gerontol A Biol Sci Med Sci 68:1443–1457
Ungvari Z, Tarantini S, Hertelendy P, Valcarcel-Ares MN, Fulop GA, Logan S, Kiss T, Farkas E, Csiszar A, Yabluchanskiy A (2017) Cerebromicrovascular dysfunction predicts cognitive decline and gait abnormalities in a mouse model of whole brain irradiation-induced accelerated brain senescence. Geroscience. 39:33–42
Valcarcel-Ares MN, Tucsek Z, Kiss T, Giles CB, Tarantini S, Yabluchanskiy A, Balasubramanian P, Gautam T, Galvan V, Ballabh P, Richardson A, Freeman WM, Wren JD, Deak F, Ungvari Z, Csiszar A (2019) Obesity in aging exacerbates neuroinflammation, dysregulating synaptic function-related genes and altering eicosanoid synthesis in the mouse hippocampus: potential role in impaired synaptic plasticity and cognitive decline. J Gerontol A Biol Sci Med Sci 74(3):290–298. https://doi.org/10.1093/gerona/gly127
Wang RX, Chai Q, Lu T, Lee HC (2011) Activation of vascular BK channels by docosahexaenoic acid is dependent on cytochrome P450 epoxygenase activity. Cardiovasc Res 90:344–352
Wang Y, Armando AM, Quehenberger O, Yan C, Dennis EA (2014) Comprehensive ultra-performance liquid chromatographic separation and mass spectrometric analysis of eicosanoid metabolites in human samples. J Chromatogr A 1359:60–69
Warrington JP, Csiszar A, Johnson DA, Herman TS, Ahmad S, Lee YW, Sonntag WE (2011) Cerebral microvascular rarefaction induced by whole brain radiation is reversible by systemic hypoxia in mice. Am J Physiol Heart Circ Physiol 300:H736–H744
Warrington JP, Csiszar A, Mitschelen M, Lee YW, Sonntag WE (2012) Whole brain radiation-induced impairments in learning and memory are time-sensitive and reversible by systemic hypoxia. PLoS One 7:e30444
Warrington JP, Ashpole N, Csiszar A, Lee YW, Ungvari Z, Sonntag WE (2013) Whole brain radiation-induced vascular cognitive impairment: mechanisms and implications. J Vasc Res 50:445–457
Welzel G, Fleckenstein K, Mai SK, Hermann B, Kraus-Tiefenbacher U, Wenz F (2008a) Acute neurocognitive impairment during cranial radiation therapy in patients with intracranial tumors. Strahlenther Onkol 184:647–654
Welzel G, Fleckenstein K, Schaefer J, Hermann B, Kraus-Tiefenbacher U, Mai SK, Wenz F (2008b) Memory function before and after whole brain radiotherapy in patients with and without brain metastases. Int J Radiat Oncol Biol Phys 72:1311–1318
Wiedenhoeft T, Tarantini S, Nyul-Toth A, Yabluchanskiy A, Csipo T, Balasubramanian P, Lipecz A, Kiss T, Csiszar A, Csiszar A, Ungvari Z (2019) Fusogenic liposomes effectively deliver resveratrol to the cerebral microcirculation and improve endothelium-dependent neurovascular coupling responses in aged mice. Geroscience. 41:711–725
Xu M, Palmer AK, Ding H, Weivoda MM, Pirtskhalava T, White TA, Sepe A, Johnson KO, Stout MB, Giorgadze N, Jensen MD, LeBrasseur NK, Tchkonia T, Kirkland JL (2015) Targeting senescent cells enhances adipogenesis and metabolic function in old age. Elife 4:e12997. https://doi.org/10.7554/eLife.12997
Xu M, Pirtskhalava T, Farr JN, Weigand BM, Palmer AK, Weivoda MM, Inman CL, Ogrodnik MB, Hachfeld CM, Fraser DG, Onken JL, Johnson KO, Verzosa GC, Langhi LGP, Weigl M, Giorgadze N, LeBrasseur NK, Miller JD, Jurk D, Singh RJ, Allison DB, Ejima K, Hubbard GB, Ikeno Y, Cubro H, Garovic VD, Hou X, Weroha SJ, Robbins PD, Niedernhofer LJ, Khosla S, Tchkonia T, Kirkland JL (2018) Senolytics improve physical function and increase lifespan in old age. Nat Med 24:1246–1256
Zhang G, Panigrahy D, Mahakian LM, Yang J, Liu JY, Stephen Lee KS, Wettersten HI, Ulu A, Hu X, Tam S, Hwang SH, Ingham ES, Kieran MW, Weiss RH, Ferrara KW, Hammock BD (2013) Epoxy metabolites of docosahexaenoic acid (DHA) inhibit angiogenesis, tumor growth, and metastasis. Proc Natl Acad Sci U S A 110:6530–6535
Zhu Y, Tchkonia T, Fuhrmann-Stroissnigg H, Dai HM, Ling YY, Stout MB, Pirtskhalava T, Giorgadze N, Johnson KO, Giles CB, Wren JD, Niedernhofer LJ, Robbins PD, Kirkland JL (2016) Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell 15:428–435
Acknowledgments
The authors express their gratitude to Signe Dahlberg-Wright (University of California) for the expert help with the eicosanoid measurements and for Unity Biotechnology, Inc. for providing the p163MR mice model.
Funding
This work was supported by grants from the American Heart Association (ST), the Oklahoma Center for the Advancement of Science and Technology (to AC, AY, ZU), the National Institute on Aging (R01-AG047879; R01-AG038747; R01-AG055395), the National Institute of Neurological Disorders and Stroke (R01-NS056218 to AC, R01-NS100782 to ZU), the Oklahoma Shared Clinical and Translational Resources (OSCTR) program funded by the National Institute of General Medical Sciences (GM104938, to AY), the Presbyterian Health Foundation (to ZU, AC, AY), the NIA-supported Geroscience Training Program in Oklahoma (T32AG052363), the Oklahoma Nathan Shock Center (P30AG050911), and the Cellular and Molecular GeroScience CoBRE (1P20GM125528, sub#5337). The funding sources had no role in the study design; in the collection, analysis and interpretation of data; in the writing of the report; and in the decision to submit the article for publication.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
All animal protocols were approved by the Institutional Animal Care and Use Committee of the OUHSC.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
About this article

Cite this article
Yabluchanskiy, A., Tarantini, S., Balasubramanian, P. et al. Pharmacological or genetic depletion of senescent astrocytes prevents whole brain irradiation–induced impairment of neurovascular coupling responses protecting cognitive function in mice. GeroScience 42, 409–428 (2020). https://doi.org/10.1007/s11357-020-00154-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11357-020-00154-8