
Abstract
Hypertension in the elderly substantially increases both the risk of vascular cognitive impairment (VCI) and Alzheimer’s disease (AD); however, the underlying mechanisms are not completely understood. This review discusses the effects of hypertension on structural and functional integrity of cerebral microcirculation, including hypertension-induced alterations in neurovascular coupling responses, cellular and molecular mechanisms involved in microvascular damage (capillary rarefaction, blood-brain barrier disruption), and the genesis of cerebral microhemorrhages and their potential role in exacerbation of cognitive decline associated with AD. Understanding and targeting the hypertension-induced cerebromicrovascular alterations that are involved in the onset and progression of AD and contribute to cognitive impairment are expected to have a major role in preserving brain health in high-risk older individuals.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hypertension in the elderly substantially increases the risk of Alzheimer’s disease (AD); however, the underlying mechanisms are not completely understood (Forette et al. 1998; Launer et al. 2000; Israeli-Korn et al. 2010; Guo et al. 2001; Marr and Hafez 2014; Petrovitch et al. 2000; van Dijk et al. 2004; Joas et al. 2012). In this review (published as part of the “Translational Geroscience” initiative of the journal (Callisaya et al. 2017; Kane et al. 2017; Kim et al. 2017; Liu et al. 2017; Meschiari et al. 2017; Perrott et al. 2017; Shobin et al. 2017; Ashpole et al. 2017; Bennis et al. 2017; Deepa et al. 2017; Grimmig et al. 2017; Hancock et al. 2017; Konopka et al. 2017; Podlutsky et al. 2017; Sierra and Kohanski 2017; Tenk et al. 2017; Ungvari et al. 2017a; Urfer et al. 2017a, b)), the effects of hypertension on structural and functional integrity of the cerebral microcirculation are considered, with a primary focus on cellular and molecular mechanisms involved in microvascular damage (capillary rarefaction, BBB disruption), neurovascular uncoupling, and the genesis of cerebral microhemorrhages and their potential role in exacerbation of cognitive decline associated with AD.
Hypertension and the pathogenesis of Alzheimer’s disease: microvascular deposition of Aβ and tauopathy
Alzheimer’s disease is the most common cause for dementia in the elderly and the sixth leading cause of death in the USA. An estimated 5.5 million Americans are living with AD in 2017, and it is projected that this number will double in the next 20 years. Since the original formulation of the vascular hypothesis of AD in the 1990s, evidence has been fast accumulating which strongly suggests that early stage of AD is indeed primarily a microvascular disorder (Zlokovic 2011). Consistent with this hypothesis, cerebrovascular dysfunction may be the earliest and most abnormal biomarker of AD progression. Large cross-sectional and longitudinal population-based studies consistently showed that a relationship exists between several vascular risk factors and incidence and progression of AD (de la Torre 2010, 2012, 2013, 2017). Among the vascular risk factors for AD, hypertension emerges as a critically important one, as it is known to double the risk for AD in the elderly (Forette et al. 1998; Launer et al. 2000; Israeli-Korn et al. 2010; Guo et al. 2001; Marr and Hafez 2014; Petrovitch et al. 2000; van Dijk et al. 2004; Joas et al. 2012). This observation led to several modifications and expansions of the original vascular hypothesis of AD, invoking hypertension-induced microvascular injury in various pathological manifestations of AD, from cerebral microhemorrhages (Ungvari et al. 2017b) to blood-brain barrier disruption and consequent neuroinflammation (Zlokovic 2011, 2008).
The amyloid cascade hypothesis has been the focus of AD research for two decades, and despite recent challenges, it still remains a frequently invoked hypothesis to explain the molecular cause of AD. The amyloid cascade hypothesis posits that altered production, processing, and deposition of amyloid β-peptide (Aβ) both in the neuropil and around cerebral microvessels plays a central role in the development of AD, a concept that is supported by substantial genetic and biochemical data. Consistent with the deposition of Aβ in the neurovascular unit, there is strong clinical and experimental evidence that increased levels of Aβ in AD associate with progressive, multifaceted cerebromicrovascular impairment, which contributes to both the development of early-stage pre-plaque cognitive dysfunction as well as subsequent progression of the disease (Zlokovic 2011; Gorelick et al. 2011; Iadecola et al. 2009; Niwa et al. 2001, 2002a, b; Park et al. 2005; Girouard and Iadecola 2006; Iadecola 2004; Park et al. 2014, 2013; Fotuhi et al. 2009; Skoog and Gustafson 2006). In agreement with the prediction based on both the vascular hypothesis and the amyloid hypothesis of AD, hypertension exacerbates Aβ-induced cerebromicrovascular impairment in AD, worsening the disease and accelerating its progression. In recent years, a series of important studies have provided critical insights into the pathophysiological mechanistic links among hypertension, Aβ deposition, and the development of AD (Iadecola et al. 2009; Niwa et al. 2002a; Capone et al. 2012; Girouard et al. 2006; Kazama et al. 2004; Faraco et al. 2016; Carnevale and Lembo 2011; Carnevale et al. 2012a, b; Diaz-Ruiz et al. 2009; Farkas et al. 2000; Hsu et al. 2013; Sparks et al. 1995). Using transgenic mouse models and angiotensin II infusion, Faraco and coworkers confirmed the association between hypertension and AD by showing that prolonged hypertension increases microvascular amyloid deposition in Tg2576 mice and enhanced beta-secretase APP cleavage (Faraco et al. 2016). Similar results were reported by other laboratories as well (Diaz-Ruiz et al. 2009). Hypertension induced by transverse aortic coarctation was also reported to exacerbate Aβ deposition in the mouse brain, promoting cognitive decline (Carnevale and Lembo 2011; Carnevale et al. 2012a, b). Further, there are studies extant showing that interaction of hypertension and aging promotes amyloidogenic gene expression in the mouse brain (Csiszar et al. 2013a). Importantly, the effects of high blood pressure on Aβ deposits in the mouse brain are manifested within 4 weeks after induction of hypertension (Carnevale and Lembo 2011; Carnevale et al. 2012a, b), suggesting that early hypertension-induced cerebromicrovascular impairment is sufficient to trigger molecular processes contributing to the pathogenesis of AD. There are studies suggesting that RAGE activation in the cerebral microvessels is a crucial mechanism by which hypertension promotes AD pathologies (Carnevale et al. 2012b). However, it is quite likely that several other mechanisms play equally important roles (Nicolakakis et al. 2008; Tong et al. 2012) and that inhibiting one or more of these molecular targets can limit the onset of microvascular-related AD deficits.
In addition to Aβ pathology, tau pathology is considered an important driver of disease progression in AD (Raskin et al. 2015). Albeit still not as extensively investigated as Aβ, recent studies have provided critical insights into the mechanistic links between hypertension and tau hyperphosphorylation and misfolding in AD. It has been shown that experimentally induced hypertension aggravates tau-related motor dysfunction in a mouse model of pure tauopathy (Diaz-Ruiz et al. 2009). Lobar microbleeds were independently associated with a higher likelihood of having an abnormal CSF phosphorylated tau 181 protein level (P = .004). Studies in a cohort of patients from the Alzheimer’s Disease Neuroimaging Initiative showed that, after adjusting for levels of CSF Aβ, lobar microbleeds associated with hypertension were correlated with accelerated longitudinal cognitive decline and with a higher likelihood of having abnormal CSF levels of phosphorylated tau181 (Chiang et al. 2017). Intraneuronal tau hyperphosphorylation preceded by Aβ deposition was demonstrated in a hypertensive rat model, non-transgenic spontaneously hypertensive stroke-prone rats (SHRSP), that displays cerebral small vessel disease (CSVD) as a consequence of hypertension, suggesting that CSVD associated with hypertension leads to increased brain Aβ and subsequent intraneuronal tau hyperphosphorylation (Schreiber et al. 2014). In an independent study, Kurata and collaborators showed that both low- and high-dose telmisartan decreased numbers of Aβ- and phospho-tau-positive neurons and decreased markers of neuroinflammation (Kurata et al. 2014). Interestingly, a potential direct role of hyperphosphorylated and misfolded tau in cerebromicrovascular dysfunction was suggested by a recent report of tau oligomer accumulation in cerebrovasculature of AD and progressive supranuclear palsy (PSP) patients, suggesting that aberrantly misfolded tau may accumulate in cells of cerebromicrovasculature in AD and in “pure” tauopathies (Castillo-Carranza et al. 2017).
Hypertension-induced capillary rarefaction
The brain is the most metabolically active organ in the human body. While it only accounts for 2% of the body mass, it consumes 20–25% of the body’s total energy requirements. Since energy stores in the brain are scarce, adequate supply of nutrients is crucial to support normal cerebral function. The brain relies on a dense cerebral microcirculatory network (600 km total length) for continuous supply of nutrients and O2 and for effective washout of metabolic waste products. It is generally considered that a decline in capillarization in the brain tissue (i.e., cerebromicrovascular rarefaction) contributes to a decline in cerebral blood flow that reduces metabolic support for neural signaling, thereby exacerbating neuronal dysfunction (Khan et al. 2002; Riddle et al. 2003; Sonntag et al. 1997; Troen et al. 2008; Tucsek et al. 2014). In that regard, it is important that increased deposition of Aβ has been shown to profoundly affect brain microvasculature, resulting in degeneration and disappearance of capillaries (Roher et al. 1993) and small vessels (Yamaguchi et al. 1992). Capillary loss ultimately also leads to an impaired clearance of Aβ, which further promotes vascular damage (Faraco and Iadecola 2013). Capillary loss can reach up to 30% in AD aged population when compared to control subjects (Fischer et al. 1990; Buee et al. 1994) and is strongly associated with a reduced cerebral perfusion (Buee et al. 1994). A reduction in cerebral perfusion occurs early in the development of AD before the brain atrophy, and the most severe changes occur in the areas with Aβ and tau pathology (Johnson et al. 2005; Hirao et al. 2005). Patients with mild cognitive impairment also exhibit hypoperfusion in the areas most affected in AD (Johnson et al. 2005; Brown and Thore 2011). Importantly, hypertension per se promotes cerebromicrovascular rarefaction (Feihl et al. 2009; Sokolova et al. 1985; Suzuki et al. 2003; Tarantini et al. 2016a; Toth et al. 2015a) and this effect is significantly exacerbated in old age (Toth et al. 2015a). On the basis of the existing evidence, we posit that the effects of hypertension, AD and old age are highly synergistic, and that hypertension in elderly AD patients results in an exacerbated microvascular structural damage. The mechanisms underlying hypertension-induced microvascular rarefaction are likely multifaceted and may involve endothelial impairment, endothelial apoptosis, decreased NO bioavailiability, oxidative stress, and an imbalance in humoral pro- and anti-angiogenic factors (Tarantini et al. 2016a). The cerebral microcirculation is subject to continuous dynamic structural adaptation, a concept that implies a high plasticity of the cerebral microvascular network (Riddle et al. 2003). Our current understanding is that there is a dynamic balance between capillary regression and growth and that both destruction of capillaries and impaired angiogenesis, due to dysregulated production of autocrine/paracrine regulators of angiogenic processes, is a critical mechanism involved in cerebromicrovascular rarefaction (Tucsek et al. 2014; Ungvari et al. 2010). It is likely that both in AD and in hypertension, capillary regression/destruction is a primary cause for capillary rarefaction, which is exacerbated by secondary impairment of capillary regrowth. Importantly, comparative analysis of cerebral capillary ultrastructure reveals similar cerebral capillary damage in AD and hypertension (Farkas et al. 2000). Previous studies provide evidence that in AD, capillaries indeed die. The histological footprint of this increased capillary loss is the formation of “string vessels” (“ghost vessels”), which are thin, acellular connective tissue strands, remnants of capillaries, with a lack of endothelial cells (Brown 2010). Capillary regression occurs by endothelial apoptosis, a process initiated by cellular damage, a decline in pro-survival factors, and/or increased presence of pro-apoptotic extracellular signals (including the toxic effects of Aβ) (Fonseca et al. 1832; Lyros et al. 2014; Religa et al. 2013; Wilhelmus et al. 2007). Recent evidence shows that macrophages also play a key role in capillary regression. Further studies are warranted to better understand the synergistic roles for AD, hypertension, and aging processes in regulation of microvascular regression. AD patients are typically old, and there is strong evidence that aging itself is associated with dysregulation of multiple aspects of the angiogenic process, including induction of endothelial cell proliferation, migration, and tube formation (Viana et al. 2015). In experimental mouse models, hypertension also results in dysregulated expression of pro- and anti-angiogenic genes (Tarantini et al. 2016a). Age- (Csiszar et al. 2014; Ungvari et al. 2011a, b) and perhaps AD- (Joshi et al. 2015; Kanninen et al. 2009) related mechanisms responsible for impairment of endothelial angiogenic capacity likely also include dysregulation of Nrf2, a newly discovered regulator of endothelial angiogenic processes (Valcarcel-Ares et al. 2012). There is also strong evidence that a causal link exists among decreased bioavailability of NO, impaired angiogenesis, and microvascular rarefaction (Ungvari et al. 2010). Endothelium-derived NO is both a downstream mediator of pro-angiogenic VEGF and IGF-1 signaling and a critical regulator of microvascular endothelial cell viability. There is ample evidence that AD/microvascular deposition of Aβ (Zlokovic 2011; Girouard and Iadecola 2006; Tong et al. 2012; Austin et al. 2013; Di Marco et al. 2015; Kimbrough et al. 2015), hypertension (Girouard and Iadecola 2006; Girouard et al. 2006; Chrissobolis et al. 2012; Pires et al. 2013; Girouard et al. 2007), and old age (Csiszar et al. 2014; Banki et al. 2015; Csiszar et al. 2002, 2004; Tarantini et al. 2016b; Ungvari et al. 2007; Modrick et al. 2009; Pena Silva et al. 2012) per se promotes microvascular endothelial dysfunction. Thus, it is highly likely that impaired cerebromicrovascular NO production significantly contributes to microvascular rarefaction in old hypertensive AD patients. Several additional mechanisms may also be considered to contribute to structural microvascular rarefaction in hypertensive AD patients, including pericyte damage (Toth et al. 2013a), increased pre-capillary arteriolar constriction and cessation of capillary blood flow, increased susceptibility to microemboli, platelet adhesion, and macrophage activation. Further, the mechanisms underlying the exacerbation of microvascular injury in hypertensive AD patients are also likely to include hemodynamic factors.
There is strong evidence that under normal conditions, pressure-induced myogenic constriction of proximal cerebral arteries, as a critical homeostatic mechanism that assures that increased systemic arterial pressure, cannot penetrate the distal portion of the cerebral microcirculation and cause damage to the thin-walled arteriolar and capillary microvessels (Toth et al. 2013a, 2014a; Kontos et al. 1978; Harper and Bohlen 1984). Young cerebral arteries in the absence of AD exhibit functional and structural adaptation to hypertension, including an augmented myogenic constriction at high pressures and autoregulatory adaptation, which protects the cerebral microcirculation from pressure-induced injury (Toth et al. 2013a, 2014a). In contrast, autoregulatory adaptation to hypertension in compromised both in aging (Toth et al. 2013a, b, 2014a; Springo et al. 2015a) and in AD (Iadecola et al. 2009; Niwa et al. 2002a; Toth et al. 2017; Brickman et al. 2015; den Abeelen et al. 2014; Tarumi et al. 2014; Iadecola 2014). Pathological loss of autoregulatory protection in old hypertensive AD patients likely allows high blood pressure to penetrate the distal, injury-prone portion of the cerebral microcirculation, leading to significant downstream damage (Toth et al. 2017).
Hypertension-induced blood-brain barrier disruption
In recent years, overwhelming evidence has accumulated demonstrating that blood-brain barrier (BBB) breakdown contributes to the onset and progression of the pathological processes associated with AD (Zlokovic 2011, 2008; Carnevale et al. 2012b; Sagare et al. 2013a; Mackic et al. 2002, 1998; Montagne et al. 2015; Sagare et al. 2013b; Winkler et al. 2015; Bell and Zlokovic 2009; Halliday et al. 2016; Nelson et al. 1862; Zlokovic 2013). Hypertension, especially in aging, is known to significantly increase BBB permeability (Toth et al. 2013a, 2014a; Mueller and Heistad 1980; Zhang et al. 2010), which has been linked to the hypertension-induced exacerbation of AD pathologies in mouse models (Carnevale and Lembo 2011; Carnevale et al. 2012a, b). The mechanisms of hypertension-related BBB disruption are likely multifaceted and may involve increased endothelial oxidative stress and endothelial injury, pericyte damage, and changes in tight junctions, which form an essential structural component of the BBB (Toth et al. 2013a; Takemori et al. 2013). Pericytes are important cellular constituents of the BBB (Zlokovic 2008), and recent studies demonstrate that pericyte deficiency in Pdgfrβ +/− mice leads to significant impairment of BBB function (Bell et al. 2010) and that pericyte loss exacerbates AD-like neurodegeneration in mice (Sagare et al. 2013b). Importantly, hypertension in aging was shown to promote pericyte loss (Toth et al. 2013a), which may contribute to BBB disruption, exacerbating AD pathogenesis. Pericytes have also key roles in preservation of the structural integrity of the cerebral microcirculation (Winkler et al. 2011); thus, loss of pericytes is also likely to contribute to hypertension-induced microvascular rarefaction in brain (Tarantini et al. 2016a; Toth et al. 2013a). Through the damaged BBB, plasma constituents, including IgG, thrombin, and fibrinogen, enter the brain parenchyma (Toth et al. 2013a), which induce of neuroinflammation by activating microglia (Bruce-Keller et al. 2010; Pistell et al. 2010; White et al. 2009; Davalos et al. 2012; Carreno-Muller et al. 2003). There is substantial evidence implicating oxidative stress, microglia activation, and neuroinflammation in the development of AD-like pathologies in hypertensive mice (Carnevale and Lembo 2011; Carnevale et al. 2012a, b).
Hypertension-induced neurovascular uncoupling
Energy and O2 demand of the brain tissue vary both spatially and temporally with changes in neuronal activity, which require prompt adjustments of blood flow by regulating arteriolar resistance in a highly controlled fashion to maintain cellular homeostasis and function (Mathiesen et al. 1998; Enager et al. 2009). This is accomplished through a process termed neurovascular coupling (or “functional hyperemia”), which is orchestrated by an inter-cellular signaling network comprised of neurons and astrocytes, as well as smooth muscle cells and endothelial cells of cerebral microvessels (Petzold and Murthy 2011; Stobart et al. 2013; Wells et al. 2015; Chen et al. 2014). There is compelling evidence that AD patients exhibit significant impairment of neurovascular coupling responses (Hock et al. 1997; Rombouts et al. 2000). These findings accord with the conclusions of pre-clinical studies demonstrating that in rodent models of AD, functional hyperemia is also significantly impaired (Rancillac et al. 2012; Shin et al. 2007), at least in part, due to increased oxidative stress (Park et al. 2005, 2008; Nicolakakis et al. 2008). Experimental studies support a causal link between impaired neurovascular coupling and cognitive impairment (Tarantini et al. 2015). Indeed, recent evidence suggests that pharmacological interventions that rescue neurovascular coupling responses result in improved cognitive function in mice with AD pathologies (Nicolakakis et al. 2008; Tong et al. 2012). In that regard, it is significant that hypertension also causes marked neurovascular dysfunction (Girouard and Iadecola 2006; Capone et al. 2012; Girouard et al. 2006; Kazama et al. 2004; Faraco et al. 2016; Kazama et al. 2003). Hypertension-induced neurovascular uncoupling superimposed on amyloid pathologies is likely to significantly exacerbate dysregulation of CBF and cognitive decline. Indeed, in AβPPswe/PS1dE9 mice angiotensin II-induced hypertension was reported to exacerbate impairment of cerebral blood flow regulation (Wiesmann et al. 2016). There is substantial evidence obtained both in pre-clinical and in clinical studies demonstrating that aging per se impairs neurovascular coupling responses (Tong et al. 2012; Balbi et al. 2015; Fabiani et al. 2013; Sorond et al. 2013; Toth et al. 2014b; Zaletel et al. 2005; Park et al. 2007), suggesting that combination of old age, amyloid pathologies, and hypertension likely results in a critical mismatch between supply and demand of oxygen and metabolic substrates in functioning cerebral tissue (Iadecola et al. 2009).
Hypertension-induced cerebral microhemorrhages
Cerebral microhemorrhages (CMHs; also described as microbleeds) are small chronic intracerebral hemorrhages (< 5 to 10 mm in diameter), which develop due to the rupture of small arteries, arterioles, and/or capillaries (Ungvari et al. 2017b). Hypertension (Jeerakathil et al. 2004; Cordonnier et al. 2007; Romero et al. 2014; Roob et al. 1999; Sveinbjornsdottir et al. 2008; Vernooij et al. 2008), advanced age (Jeerakathil et al. 2004; Chai et al. 2016; Caunca et al. 2016), and cerebral amyloid angiopathy and AD (Yates et al. 2011, 2014; Pettersen et al. 2008; Benedictus et al. 2013) are the major risk factors for CMHs. The prevalence of CMHs reaches 50% in patients at risk (Ungvari et al. 2017b). CMHs are clinically important as they may exacerbate cognitive decline in AD patients. There is strong experimental evidence that aging or vascular AD pathologies exacerbate the effects of hypertension on the pathogenesis of CMHs (Ungvari et al. 2017b; Toth et al. 2015a, 2017; Tarantini et al. 2017), worsening the clinical outcome. The cellular and molecular mechanisms by which hypertension promotes CMHs include induction of oxidative stress and MMP activation in the vascular wall, breakdown of the extracellular matrix, pathological structural adaptation to high blood pressure, and/or impairment of myogenic autoregulatory protection, which allows high blood pressure to penetrate the vulnerable distal portion of the cerebral microcirculation (Toth et al. 2015a, 2017; Tarantini et al. 2017; Wakisaka et al. 2008, 2010). Future studies are needed to identify effective strategies for microvascular protection to prevent the development of CMHs, thereby delaying cognitive decline in AD patients.
Conclusion
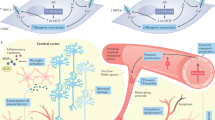
Strong epidemiological and experimental evidence indicate that hypertension in the elderly promotes the pathogenesis of AD and/or exacerbates cognitive decline by inducing capillary rarefaction, BBB disruption, and consequential neuroinflammation, by impairing neurovascular coupling responses and promoting the genesis of cerebral microhemorrhages. Because clinical trials with Aβ targeting antibodies (i.e., bapineuzumab and solanezumab) and γ-secretase inhibitors (semagacestat) failed (Laske 2014; Doody et al. 2013), research into treatments that exert microvascular protection and thereby may prevent development/progression of the disease became a high priority. In light of recent pre-clinical and clinical trials showing that anti-hypertensive drugs (including diuretics, angiotensin I receptor blockers, and angiotensin-converting enzyme inhibitors) may have beneficial effects in AD, further studies of the interaction between hypertension and AD pathophysiology are highly warranted. In addition to repurposing existing drugs with microvascular protective effects (Nicolakakis et al. 2008; Papadopoulos et al. 2014, 2016), geroscience research has identified promising novel molecular targets involved in the regulation of cellular aging processes that can be targeted to improve neurovascular health. Among them, the mTOR inhibitor rapamycin (Urfer et al. 2017a) shows great promise in treating AD, as it was shown to rescue cerebromicrovascular function and improve cognition in pre-clinical models of the disease (Lin et al. 2013, 2017; Galvan and Hart 2016; Richardson et al. 2015). Many other geronic factors involved in aging processes, including IGF-1 (Ashpole et al. 2017; Podlutsky et al. 2017), Nrf2 (Ungvari et al. 2011a, b; Valcarcel-Ares et al. 2012; Pearson et al. 2008; Ungvari et al. 2011c; Bailey-Downs et al. 2012), and factors involved in redox regulation (Deepa et al. 2017; Grimmig et al. 2017; Konopka et al. 2017), were shown to influence microvascular health, regulating neurovascular coupling responses (Tarantini et al. 2016b; Toth et al. 2014b, 2015b, c), angiogenesis and capillary rarefaction (Tarantini et al. 2016a; Valcarcel-Ares et al. 2012; Banki et al. 2015; Sonntag et al. 2013; Csiszar et al. 2013b; Ungvari et al. 2013), and the pathogenesis of cerebral microhemorrhages (Ungvari et al. 2017b; Toth et al. 2013a, b, 2014a, 2015a, 2017; Springo et al. 2015a; Tarantini et al. 2017). New developments in our understanding of these microvascular mechanisms and their pathophysiological roles may lead to novel interventions for delaying the progression of AD (Fig. 1).

Cerebromicrovascular alterations by which hypertension promotes the pathogenesis of vascular cognitive impairment and AD. Hypertension promotes microvascular oxidative stress and microvascular injury, which are exacerbated in aging (Girouard et al. 2006; Toth et al. 2015a, 2013a; Springo et al. 2015b). In patients with AD pathologies, hypertension increases the deposition of Aβ and exacerbates cerebromicrovascular dysfunction induced by Aβ. Microvascular injury, aggravated by structural and functional microvascular maladaptation to hypertension, leads to blood-brain barrier disruption promoting neuroinflammation, microhemorrhages, and microvascular rarefaction. Hypertension-induced, oxidative stress-mediated neurovascular uncoupling further compromises the blood supply to the brain. The model predicts that these hypertension-induced structural and functional microvascular alterations critically contribute to cognitive decline in high-risk elderly patients
References
Ashpole NM, Logan S, Yabluchanskiy A, Mitschelen MC, Yan H, Farley JA, Hodges EL, Ungvari Z, Csiszar A, Chen S, Georgescu C, Hubbard GB, Ikeno Y, Sonntag WE (2017) IGF-1 has sexually dimorphic, pleiotropic, and time-dependent effects on healthspan, pathology, and lifespan. Geroscience
Austin SA, Santhanam AV, Hinton DJ, Choi DS, Katusic ZS (2013) Endothelial nitric oxide deficiency promotes Alzheimer’s disease pathology. J Neurochem 127:691–700
Bailey-Downs LC, Mitschelen M, Sosnowska D, Toth P, Pinto JT, Ballabh P, Valcarcel-Ares MN, Farley J, Koller A, Henthorn JC, Bass C, Sonntag WE, Ungvari Z, Csiszar A (2012) Liver-specific knockdown of IGF-1 decreases vascular oxidative stress resistance by impairing the Nrf2-dependent antioxidant response: a novel model of vascular aging. J Gerontol Biol Med Sci 67:313–329
Balbi M, Ghosh M, Longden TA, Jativa Vega M, Gesierich B, Hellal F, Lourbopoulos A, Nelson MT, Plesnila N (2015) Dysfunction of mouse cerebral arteries during early aging. J Cereb Blood Flow Metab 35:1445–1453
Banki E, Sosnowska D, Tucsek Z, Gautam T, Toth P, Tarantini S, Tamas A, Helyes Z, Reglodi D, Sonntag WE, Csiszar A, Ungvari Z (2015) Age-related decline of autocrine pituitary adenylate cyclase-activating polypeptide impairs angiogenic capacity of rat cerebromicrovascular endothelial cells. J Gerontol A Biol Sci Med Sci 70:665–674
Bell RD, Zlokovic BV (2009) Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer’s disease. Acta Neuropathol 118:103–113
Bell RD, Winkler EA, Sagare AP, Singh I, LaRue B, Deane R, Zlokovic BV (2010) Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron 68:409–427
Benedictus MR, Goos JD, Binnewijzend MA, Muller M, Barkhof F, Scheltens P, Prins ND, van der Flier WM (2013) Specific risk factors for microbleeds and white matter hyperintensities in Alzheimer’s disease. Neurobiol Aging 34:2488–2494
Bennis MT, Schneider A, Victoria B, Do A, Wiesenborn DS, Spinel L, Gesing A, Kopchick JJ, Siddiqi SA, Masternak MM (2017) The role of transplanted visceral fat from the long-lived growth hormone receptor knockout mice on insulin signaling. Geroscience 39:51–59
Brickman AM, Guzman VA, Gonzalez-Castellon M, Razlighi Q, Gu Y, Narkhede A, Janicki S, Ichise M, Stern Y, Manly JJ, Schupf N, Marshall RS (2015) Cerebral autoregulation, beta amyloid, and white matter hyperintensities are interrelated. Neurosci Lett 592:54–58
Brown WR (2010) A review of string vessels or collapsed, empty basement membrane tubes. J Alzheimers Dis 21:725–739
Brown WR, Thore CR (2011) Review: cerebral microvascular pathology in ageing and neurodegeneration. Neuropathol Appl Neurobiol 37:56–74
Bruce-Keller AJ, White CL, Gupta S, Knight AG, Pistell PJ, Ingram DK, Morrison CD, Keller JN (2010) NOX activity in brain aging: exacerbation by high fat diet. Free Radic Biol Med 49:22–30
Buee L, Hof PR, Bouras C, Delacourte A, Perl DP, Morrison JH, Fillit HM (1994) Pathological alterations of the cerebral microvasculature in Alzheimer’s disease and related dementing disorders. Acta Neuropathol 87:469–480
Callisaya ML, Launay CP, Srikanth VK, Verghese J, Allali G, Beauchet O (2017) Cognitive status, fast walking speed and walking speed reserve—the Gait and Alzheimer Interactions Tracking (GAIT) study. Geroscience 39:231–239
Capone C, Faraco G, Peterson JR, Coleman C, Anrather J, Milner TA, Pickel VM, Davisson RL, Iadecola C (2012) Central cardiovascular circuits contribute to the neurovascular dysfunction in angiotensin II hypertension. J Neurosci 32:4878–4886
Carnevale D, Lembo G (2011) ‘Alzheimer-like’ pathology in a murine model of arterial hypertension. Biochem Soc Trans 39:939–944
Carnevale D, Mascio G, Ajmone-Cat MA, D'Andrea I, Cifelli G, Madonna M, Cocozza G, Frati A, Carullo P, Carnevale L, Alleva E, Branchi I, Lembo G, Minghetti L (2012a) Role of neuroinflammation in hypertension-induced brain amyloid pathology. Neurobiol Aging 33:205 e19–205 e29
Carnevale D, Mascio G, D'Andrea I, Fardella V, Bell RD, Branchi I, Pallante F, Zlokovic B, Yan SS, Lembo G (2012b) Hypertension induces brain beta-amyloid accumulation, cognitive impairment, and memory deterioration through activation of receptor for advanced glycation end products in brain vasculature. Hypertension 60:188–197
Carreno-Muller E, Herrera AJ, de Pablos RM, Tomas-Camardiel M, Venero JL, Cano J, Machado A (2003) Thrombin induces in vivo degeneration of nigral dopaminergic neurones along with the activation of microglia. J Neurochem 84:1201–1214
Castillo-Carranza DL, Nilson AN, Van Skike CE, Jahrling JB, Patel K, Garach P, Gerson JE, Sengupta U, Abisambra J, Nelson P, Troncoso J, Ungvari Z, Galvan V, Kayed R (2017) Cerebral microvascular accumulation of tau oligomers in Alzheimer’s disease and related tauopathies. Aging Dis 8:257–266
Caunca MR, Del Brutto V, Gardener H, Shah N, Dequatre-Ponchelle N, Cheung YK, Elkind MS, Brown TR, Cordonnier C, Sacco RL, Wright CB (2016) Cerebral microbleeds, vascular risk factors, and magnetic resonance imaging markers: the Northern Manhattan Study. J Am Heart Assoc 5(9). https://doi.org/10.1161/JAHA.116.003477
Chai C, Wang Z, Fan L, Zhang M, Chu Z, Zuo C, Liu L, Mark Haacke E, Guo W, Shen W, Xia S (2016) Increased number and distribution of cerebral microbleeds is a risk factor for cognitive dysfunction in hemodialysis patients: a longitudinal study. Medicine (Baltimore) 95:e2974
Chen BR, Kozberg MG, Bouchard MB, Shaik MA, Hillman EM (2014) A critical role for the vascular endothelium in functional neurovascular coupling in the brain. J Am Heart Assoc 3:e000787
Chiang GC, Mao X, Kang G, Chang E, Pandya S, Vallabhajosula S, Isaacson R, Ravdin LD, Shungu DC (2017) Relationships among cortical glutathione levels, brain amyloidosis, and memory in healthy older adults investigated in vivo with 1H-MRS and Pittsburgh compound-B PET. AJNR Am J Neuroradiol 38:1130–1137
Chrissobolis S, Banfi B, Sobey CG, Faraci FM (2012) Role of Nox isoforms in angiotensin II-induced oxidative stress and endothelial dysfunction in brain. J Appl Physiol (1985) 113:184–191
Cordonnier C, Al-Shahi Salman R, Wardlaw J (2007) Spontaneous brain microbleeds: systematic review, subgroup analyses and standards for study design and reporting. Brain 130:1988–2003
Csiszar A, Ungvari Z, Edwards JG, Kaminski PM, Wolin MS, Koller A, Kaley G (2002) Aging-induced phenotypic changes and oxidative stress impair coronary arteriolar function. Circ Res 90:1159–1166
Csiszar A, Ungvari Z, Koller A, Edwards JG, Kaley G (2004) Proinflammatory phenotype of coronary arteries promotes endothelial apoptosis in aging. Physiol Genomics 17:21–30
Csiszar A, Tucsek Z, Toth P, Sosnowska D, Gautam T, Koller A, Deak F, Sonntag WE, Ungvari Z (2013a) Synergistic effects of hypertension and aging on cognitive function and hippocampal expression of genes involved in beta-amyloid generation and Alzheimer’s disease. Am J Physiol Heart Circ Physiol 305:H1120–H1130
Csiszar A, Sosnowska D, Tucsek Z, Gautam T, Toth P, Losonczy G, Colman RJ, Weindruch R, Anderson RM, Sonntag WE, Ungvari Z (2013b) Circulating factors induced by caloric restriction in the nonhuman primate Macaca mulatta activate angiogenic processes in endothelial cells. J Gerontol A Biol Sci Med Sci 68:235–249
Csiszar A, Gautam T, Sosnowska D, Tarantini S, Banki E, Tucsek Z, Toth P, Losonczy G, Koller A, Reglodi D, Giles CB, Wren JD, Sonntag WE, Ungvari Z (2014) Caloric restriction confers persistent anti-oxidative, pro-angiogenic, and anti-inflammatory effects and promotes anti-aging miRNA expression profile in cerebromicrovascular endothelial cells of aged rats. Am J Physiol Heart Circ Physiol 307:H292–H306
Davalos D, Ryu JK, Merlini M, Baeten KM, Le Moan N, Petersen MA, Deerinck TJ, Smirnoff DS, Bedard C, Hakozaki H, Gonias Murray S, Ling JB, Lassmann H, Degen JL, Ellisman MH, Akassoglou K (2012) Fibrinogen-induced perivascular microglial clustering is required for the development of axonal damage in neuroinflammation. Nat Commun 3:1227
de la Torre JC (2010) Vascular risk factor detection and control may prevent Alzheimer’s disease. Ageing Res Rev 9:218–225
de la Torre JC (2012) Cerebral hemodynamics and vascular risk factors: setting the stage for Alzheimer’s disease. J Alzheimers Dis 32:553–567
de la Torre JC (2013) Vascular risk factors: a ticking time bomb to Alzheimer’s disease. Am J Alzheimers Dis Other Demen 28:551–559
de la Torre JC (2017) Are major dementias triggered by poor blood flow to the brain? Theoretical considerations. J Alzheimers Dis 57:353–371
Deepa SS, Bhaskaran S, Espinoza S, Brooks SV, McArdle A, Jackson MJ, Van Remmen H, Richardson A (2017) A new mouse model of frailty: the Cu/Zn superoxide dismutase knockout mouse. Geroscience 39:187–198
den Abeelen AS, Lagro J, van Beek AH, Claassen JA (2014) Impaired cerebral autoregulation and vasomotor reactivity in sporadic Alzheimer’s disease. Curr Alzheimer Res 11:11–17
Di Marco LY, Venneri A, Farkas E, Evans PC, Marzo A, Frangi AF (2015) Vascular dysfunction in the pathogenesis of Alzheimer’s disease—a review of endothelium-mediated mechanisms and ensuing vicious circles. Neurobiol Dis 82:593–606
Diaz-Ruiz C, Wang J, Ksiezak-Reding H, Ho L, Qian X, Humala N, Thomas S, Martinez-Martin P, Pasinetti GM (2009) Role of hypertension in aggravating Abeta neuropathology of AD type and tau-mediated motor impairment. Cardiovasc Psychiatry Neurol 2009:107286
Doody RS, Raman R, Farlow M, Iwatsubo T, Vellas B, Joffe S, Kieburtz K, He F, Sun X, Thomas RG, Aisen PS, Alzheimer’s Disease Cooperative Study Steering C, Siemers E, Sethuraman G, Mohs R, Semagacestat Study G (2013) A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N Engl J Med 369:341–350
Enager P, Piilgaard H, Offenhauser N, Kocharyan A, Fernandes P, Hamel E, Lauritzen M (2009) Pathway-specific variations in neurovascular and neurometabolic coupling in rat primary somatosensory cortex. J Cereb Blood Flow Metab 29:976–986
Fabiani M, Gordon BA, Maclin EL, Pearson MA, Brumback-Peltz CR, Low KA, McAuley E, Sutton BP, Kramer AF, Gratton G (2013) Neurovascular coupling in normal aging: a combined optical, ERP and fMRI study. Neuroimage 85(Pt 1):592–607
Faraco G, Iadecola C (2013) Hypertension: a harbinger of stroke and dementia. Hypertension 62:810–817
Faraco G, Park L, Zhou P, Luo W, Paul SM, Anrather J, Iadecola C (2016) Hypertension enhances Abeta-induced neurovascular dysfunction, promotes beta-secretase activity, and leads to amyloidogenic processing of APP. J Cereb Blood Flow Metab 36:241–252
Farkas E, De Jong GI, Apro E, De Vos RA, Steur EN, Luiten PG (2000) Similar ultrastructural breakdown of cerebrocortical capillaries in Alzheimer’s disease, Parkinson’s disease, and experimental hypertension. What is the functional link? Ann N Y Acad Sci 903:72–82
Feihl F, Liaudet L, Waeber B (2009) The macrocirculation and microcirculation of hypertension. Curr Hypertens Rep 11:182–189
Fischer VW, Siddiqi A, Yusufaly Y (1990) Altered angioarchitecture in selected areas of brains with Alzheimer’s disease. Acta Neuropathol 79:672–679
Fonseca AC, Ferreiro E, Oliveira CR, Cardoso SM, Pereira CF (1832) Activation of the endoplasmic reticulum stress response by the amyloid-beta 1-40 peptide in brain endothelial cells. Biochim Biophys Acta 2013:2191–2203
Forette F, Seux ML, Staessen JA, Thijs L, Birkenhager WH, Babarskiene MR, Babeanu S, Bossini A, Gil-Extremera B, Girerd X, Laks T, Lilov E, Moisseyev V, Tuomilehto J, Vanhanen H, Webster J, Yodfat Y, Fagard R (1998) Prevention of dementia in randomised double-blind placebo-controlled Systolic Hypertension in Europe (Syst-Eur) trial. Lancet 352:1347–1351
Fotuhi M, Hachinski V, Whitehouse PJ (2009) Changing perspectives regarding late-life dementia. Nat Rev Neurol 5:649–658
Galvan V, Hart MJ (2016) Vascular mTOR-dependent mechanisms linking the control of aging to Alzheimer’s disease. Biochim Biophys Acta 1862:992–1007
Girouard H, Iadecola C (2006) Neurovascular coupling in the normal brain and in hypertension, stroke, and Alzheimer disease. J Appl Physiol (1985) 100:328–335
Girouard H, Park L, Anrather J, Zhou P, Iadecola C (2006) Angiotensin II attenuates endothelium-dependent responses in the cerebral microcirculation through nox-2-derived radicals. Arterioscler Thromb Vasc Biol 26:826–832
Girouard H, Park L, Anrather J, Zhou P, Iadecola C (2007) Cerebrovascular nitrosative stress mediates neurovascular and endothelial dysfunction induced by angiotensin II. Arterioscler Thromb Vasc Biol 27:303–309
Gorelick PB, Scuteri A, Black SE, Decarli C, Greenberg SM, Iadecola C, Launer LJ, Laurent S, Lopez OL, Nyenhuis D, Petersen RC, Schneider JA, Tzourio C, Arnett DK, Bennett DA, Chui HC, Higashida RT, Lindquist R, Nilsson PM, Roman GC, Sellke FW, Seshadri S (2011) Vascular contributions to cognitive impairment and dementia: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 42:2672–2713
Grimmig B, Kim SH, Nash K, Bickford PC, Douglas Shytle R (2017) Neuroprotective mechanisms of astaxanthin: a potential therapeutic role in preserving cognitive function in age and neurodegeneration. Geroscience 39:19–32
Guo Z, Qiu C, Viitanen M, Fastbom J, Winblad B, Fratiglioni L (2001) Blood pressure and dementia in persons 75+ years old: 3-year follow-up results from the Kungsholmen Project. J Alzheimers Dis 3:585–591
Halliday MR, Rege SV, Ma Q, Zhao Z, Miller CA, Winkler EA, Zlokovic BV (2016) Accelerated pericyte degeneration and blood-brain barrier breakdown in apolipoprotein E4 carriers with Alzheimer’s disease. J Cereb Blood Flow Metab 36:216–227
Hancock SE, Friedrich MG, Mitchell TW, Truscott RJ, Else PL (2017) The phospholipid composition of the human entorhinal cortex remains relatively stable over 80 years of adult aging. Geroscience 39:73–82
Harper SL, Bohlen HG (1984) Microvascular adaptation in the cerebral cortex of adult spontaneously hypertensive rats. Hypertension 6:408–419
Hirao K, Ohnishi T, Hirata Y, Yamashita F, Mori T, Moriguchi Y, Matsuda H, Nemoto K, Imabayashi E, Yamada M, Iwamoto T, Arima K, Asada T (2005) The prediction of rapid conversion to Alzheimer’s disease in mild cognitive impairment using regional cerebral blood flow SPECT. NeuroImage 28:1014–1021
Hock C, Villringer K, Muller-Spahn F, Wenzel R, Heekeren H, Schuh-Hofer S, Hofmann M, Minoshima S, Schwaiger M, Dirnagl U, Villringer A (1997) Decrease in parietal cerebral hemoglobin oxygenation during performance of a verbal fluency task in patients with Alzheimer’s disease monitored by means of near-infrared spectroscopy (NIRS)—correlation with simultaneous rCBF-PET measurements. Brain Res 755:293–303
Hsu CY, Huang CC, Chan WL, Huang PH, Chiang CH, Chen TJ, Chung CM, Lin SJ, Chen JW, Leu HB (2013) Angiotensin-receptor blockers and risk of Alzheimer’s disease in hypertension population. Circ J 77:405–410
Iadecola C (2004) Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat Rev Neurosci 5:347–360
Iadecola C (2014) Hypertension and dementia. Hypertension 64:3–5
Iadecola C, Park L, Capone C (2009) Threats to the mind: aging, amyloid, and hypertension. Stroke 40:S40–S44
Israeli-Korn SD, Masarwa M, Schechtman E, Abuful A, Strugatsky R, Avni S, Farrer LA, Friedland RP, Inzelberg R (2010) Hypertension increases the probability of Alzheimer’s disease and of mild cognitive impairment in an Arab community in northern Israel. Neuroepidemiology 34:99–105
Jeerakathil T, Wolf PA, Beiser A, Hald JK, Au R, Kase CS, Massaro JM, DeCarli C (2004) Cerebral microbleeds: prevalence and associations with cardiovascular risk factors in the Framingham Study. Stroke 35:1831–1835
Joas E, Backman K, Gustafson D, Ostling S, Waern M, Guo X, Skoog I (2012) Blood pressure trajectories from midlife to late life in relation to dementia in women followed for 37 years. Hypertension 59:796–801
Johnson NA, Jahng GH, Weiner MW, Miller BL, Chui HC, Jagust WJ, Gorno-Tempini ML, Schuff N (2005) Pattern of cerebral hypoperfusion in Alzheimer disease and mild cognitive impairment measured with arterial spin-labeling MR imaging: initial experience. Radiology 234:851–859
Joshi G, Gan KA, Johnson DA, Johnson JA (2015) Increased Alzheimer’s disease-like pathology in the APP/PS1DeltaE9 mouse model lacking Nrf2 through modulation of autophagy. Neurobiol Aging 36:664–679
Kane AE, Gregson E, Theou O, Rockwood K, Howlett SE (2017) The association between frailty, the metabolic syndrome, and mortality over the lifespan. Geroscience 39:221–229
Kanninen K, Heikkinen R, Malm T, Rolova T, Kuhmonen S, Leinonen H, Yla-Herttuala S, Tanila H, Levonen AL, Koistinaho M, Koistinaho J (2009) Intrahippocampal injection of a lentiviral vector expressing Nrf2 improves spatial learning in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A 106:16505–16510
Kazama K, Wang G, Frys K, Anrather J, Iadecola C (2003) Angiotensin II attenuates functional hyperemia in the mouse somatosensory cortex. Am J Physiol Heart Circ Physiol 285:H1890–H1899
Kazama K, Anrather J, Zhou P, Girouard H, Frys K, Milner TA, Iadecola C (2004) Angiotensin II impairs neurovascular coupling in neocortex through NADPH oxidase-derived radicals. Circ Res 95:1019–1026
Khan AS, Sane DC, Wannenburg T, Sonntag WE (2002) Growth hormone, insulin-like growth factor-1 and the aging cardiovascular system. Cardiovasc Res 54:25–35
Kim S, Myers L, Wyckoff J, Cherry KE, Jazwinski SM (2017) The frailty index outperforms DNA methylation age and its derivatives as an indicator of biological age. Geroscience 39:83–92
Kimbrough IF, Robel S, Roberson ED, Sontheimer H (2015) Vascular amyloidosis impairs the gliovascular unit in a mouse model of Alzheimer’s disease. Brain 138:3716–3733
Konopka AR, Laurin JL, Musci RV, Wolff CA, Reid JJ, Biela LM, Zhang Q, Peelor FF 3rd, Melby CL, Hamilton KL, Miller BF (2017) Influence of Nrf2 activators on subcellular skeletal muscle protein and DNA synthesis rates after 6 weeks of milk protein feeding in older adults. Geroscience 39:175–186
Kontos HA, Wei EP, Navari RM, Levasseur JE, Rosenblum WI, Patterson JL Jr (1978) Responses of cerebral arteries and arterioles to acute hypotension and hypertension. Am J Phys 234:H371–H383
Kurata T, Lukic V, Kozuki M, Wada D, Miyazaki K, Morimoto N, Ohta Y, Deguchi K, Ikeda Y, Kamiya T, Abe K (2014) Telmisartan reduces progressive accumulation of cellular amyloid beta and phosphorylated tau with inflammatory responses in aged spontaneously hypertensive stroke resistant rat. J Stroke Cerebrovasc Dis 23:2580–2590
Laske C (2014) Phase 3 trials of solanezumab and bapineuzumab for Alzheimer’s disease. N Engl J Med 370:1459
Launer LJ, Ross GW, Petrovitch H, Masaki K, Foley D, White LR, Havlik RJ (2000) Midlife blood pressure and dementia: the Honolulu-Asia aging study. Neurobiol Aging 21:49–55
Lin AL, Zheng W, Halloran JJ, Burbank RR, Hussong SA, Hart MJ, Javors M, Shih YY, Muir E, Solano Fonseca R, Strong R, Richardson AG, Lechleiter JD, Fox PT, Galvan V (2013) Chronic rapamycin restores brain vascular integrity and function through NO synthase activation and improves memory in symptomatic mice modeling Alzheimer’s disease. J Cereb Blood Flow Metab 33:1412–1421
Lin AL, Jahrling JB, Zhang W, DeRosa N, Bakshi V, Romero P, Galvan V, Richardson A (2017) Rapamycin rescues vascular, metabolic and learning deficits in apolipoprotein E4 transgenic mice with pre-symptomatic Alzheimer’s disease. J Cereb Blood Flow Metab 37:217–226
Liu X, Bhatt T, Wang S, Yang F, Pai YC (2017) Retention of the “first-trial effect” in gait-slip among community-living older adults. Geroscience 39:93–102
Lyros E, Bakogiannis C, Liu Y, Fassbender K (2014) Molecular links between endothelial dysfunction and neurodegeneration in Alzheimer’s disease. Curr Alzheimer Res 11:18–26
Mackic JB, Weiss MH, Miao W, Kirkman E, Ghiso J, Calero M, Bading J, Frangione B, Zlokovic BV (1998) Cerebrovascular accumulation and increased blood-brain barrier permeability to circulating Alzheimer’s amyloid beta peptide in aged squirrel monkey with cerebral amyloid angiopathy. J Neurochem 70:210–215
Mackic JB, Bading J, Ghiso J, Walker L, Wisniewski T, Frangione B, Zlokovic BV (2002) Circulating amyloid-beta peptide crosses the blood-brain barrier in aged monkeys and contributes to Alzheimer’s disease lesions. Vasc Pharmacol 38:303–313
Marr RA, Hafez DM (2014) Amyloid-beta and Alzheimer’s disease: the role of neprilysin-2 in amyloid-beta clearance. Front Aging Neurosci 6:187
Mathiesen C, Caesar K, Akgoren N, Lauritzen M (1998) Modification of activity-dependent increases of cerebral blood flow by excitatory synaptic activity and spikes in rat cerebellar cortex. J Physiol 512(Pt 2):555–566
Meschiari CA, Ero OK, Pan H, Finkel T, Lindsey ML (2017) The impact of aging on cardiac extracellular matrix. Geroscience 39:7–18
Modrick ML, Didion SP, Sigmund CD, Faraci FM (2009) Role of oxidative stress and AT1 receptors in cerebral vascular dysfunction with aging. Am J Physiol Heart Circ Physiol 296:H1914–H1919
Montagne A, Barnes SR, Sweeney MD, Halliday MR, Sagare AP, Zhao Z, Toga AW, Jacobs RE, Liu CY, Amezcua L, Harrington MG, Chui HC, Law M, Zlokovic BV (2015) Blood-brain barrier breakdown in the aging human hippocampus. Neuron 85:296–302
Mueller SM, Heistad DD (1980) Effect of chronic hypertension on the blood-brain barrier. Hypertension 2:809–812
Nelson AR, Sweeney MD, Sagare AP, Zlokovic BV (1862) Neurovascular dysfunction and neurodegeneration in dementia and Alzheimer’s disease. Biochim Biophys Acta 2016:887–900
Nicolakakis N, Aboulkassim T, Ongali B, Lecrux C, Fernandes P, Rosa-Neto P, Tong XK, Hamel E (2008) Complete rescue of cerebrovascular function in aged Alzheimer’s disease transgenic mice by antioxidants and pioglitazone, a peroxisome proliferator-activated receptor gamma agonist. J Neurosci 28:9287–9296
Niwa K, Porter VA, Kazama K, Cornfield D, Carlson GA, Iadecola C (2001) A beta-peptides enhance vasoconstriction in cerebral circulation. Am J Physiol Heart Circ Physiol 281:H2417–H2424
Niwa K, Kazama K, Younkin L, Younkin SG, Carlson GA, Iadecola C (2002a) Cerebrovascular autoregulation is profoundly impaired in mice overexpressing amyloid precursor protein. Am J Physiol Heart Circ Physiol 283:H315–H323
Niwa K, Kazama K, Younkin SG, Carlson GA, Iadecola C (2002b) Alterations in cerebral blood flow and glucose utilization in mice overexpressing the amyloid precursor protein. Neurobiol Dis 9:61–68
Papadopoulos P, Tong XK, Hamel E (2014) Selective benefits of simvastatin in bitransgenic APPSwe, Ind/TGF-beta1 mice. Neurobiol Aging 35:203–212
Papadopoulos P, Tong XK, Imboden H, Hamel E (2016) Losartan improves cerebrovascular function in a mouse model of Alzheimer’s disease with combined overproduction of amyloid-beta and transforming growth factor-beta1. J Cereb Blood Flow Metab. 2016:271678X16658489
Park L, Anrather J, Zhou P, Frys K, Pitstick R, Younkin S, Carlson GA, Iadecola C (2005) NADPH-oxidase-derived reactive oxygen species mediate the cerebrovascular dysfunction induced by the amyloid beta peptide. J Neurosci 25:1769–1777
Park L, Anrather J, Girouard H, Zhou P, Iadecola C (2007) Nox2-derived reactive oxygen species mediate neurovascular dysregulation in the aging mouse brain. J Cereb Blood Flow Metab 27:1908–1918
Park L, Zhou P, Pitstick R, Capone C, Anrather J, Norris EH, Younkin L, Younkin S, Carlson G, McEwen BS, Iadecola C (2008) Nox2-derived radicals contribute to neurovascular and behavioral dysfunction in mice overexpressing the amyloid precursor protein. Proc Natl Acad Sci U S A 105:1347–1352
Park L, Zhou P, Koizumi K, El Jamal S, Previti ML, Van Nostrand WE, Carlson G, Iadecola C (2013) Brain and circulating levels of Abeta1-40 differentially contribute to vasomotor dysfunction in the mouse brain. Stroke 44:198–204
Park L, Koizumi K, El Jamal S, Zhou P, Previti ML, Van Nostrand WE, Carlson G, Iadecola C (2014) Age-dependent neurovascular dysfunction and damage in a mouse model of cerebral amyloid angiopathy. Stroke 45:1815–1821
Pearson KJ, Lewis KN, Price NL, Chang JW, Perez E, Cascajo MV, Tamashiro KL, Poosala S, Csiszar A, Ungvari Z, Kensler TW, Yamamoto M, Egan JM, Longo DL, Ingram DK, Navas P, de Cabo R (2008) Nrf2 mediates cancer protection but not prolongevity induced by caloric restriction. Proc Natl Acad Sci U S A 105:2325–2330
Pena Silva RA, Chu Y, Miller JD, Mitchell IJ, Penninger JM, Faraci FM, Heistad DD (2012) Impact of ACE2 deficiency and oxidative stress on cerebrovascular function with aging. Stroke 43:3358–3363
Perrott KM, Wiley CD, Desprez PY, Campisi J (2017) Apigenin suppresses the senescence-associated secretory phenotype and paracrine effects on breast cancer cells. Geroscience 39:161–173
Petrovitch H, White LR, Izmirilian G, Ross GW, Havlik RJ, Markesbery W, Nelson J, Davis DG, Hardman J, Foley DJ, Launer LJ (2000) Midlife blood pressure and neuritic plaques, neurofibrillary tangles, and brain weight at death: the HAAS. Honolulu-Asia aging Study. Neurobiol Aging 21:57–62
Pettersen JA, Sathiyamoorthy G, Gao FQ, Szilagyi G, Nadkarni NK, St George-Hyslop P, Rogaeva E, Black SE (2008) Microbleed topography, leukoaraiosis, and cognition in probable Alzheimer disease from the Sunnybrook dementia study. Arch Neurol 65:790–795
Petzold GC, Murthy VN (2011) Role of astrocytes in neurovascular coupling. Neuron 71:782–797
Pires PW, Dams Ramos CM, Matin N, Dorrance AM (2013) The effects of hypertension on the cerebral circulation. Am J Physiol Heart Circ Physiol 304:H1598–H1614
Pistell PJ, Morrison CD, Gupta S, Knight AG, Keller JN, Ingram DK, Bruce-Keller AJ (2010) Cognitive impairment following high fat diet consumption is associated with brain inflammation. J Neuroimmunol 219:25–32
Podlutsky A, Valcarcel-Ares MN, Yancey K, Podlutskaya V, Nagykaldi E, Gautam T, Miller RA, Sonntag WE, Csiszar A, Ungvari Z (2017) The GH/IGF-1 axis in a critical period early in life determines cellular DNA repair capacity by altering transcriptional regulation of DNA repair-related genes: implications for the developmental origins of cancer. Geroscience 39:147–160
Rancillac A, Geoffroy H, Rossier J (2012) Impaired neurovascular coupling in the APPxPS1 mouse model of Alzheimer’s disease. Curr Alzheimer Res 9:1221–1230
Raskin J, Cummings J, Hardy J, Schuh K, Dean RA (2015) Neurobiology of Alzheimer’s disease: integrated molecular, physiological, anatomical, biomarker, and cognitive dimensions. Curr Alzheimer Res 12:712–722
Religa P, Cao R, Religa D, Xue Y, Bogdanovic N, Westaway D, Marti HH, Winblad B, Cao Y (2013) VEGF significantly restores impaired memory behavior in Alzheimer’s mice by improvement of vascular survival. Sci Rep 3:2053
Richardson A, Galvan V, Lin AL, Oddo S (2015) How longevity research can lead to therapies for Alzheimer’s disease: the rapamycin story. Exp Gerontol 68:51–58
Riddle DR, Sonntag WE, Lichtenwalner RJ (2003) Microvascular plasticity in aging. Ageing Res Rev 2:149–168
Roher AE, Lowenson JD, Clarke S, Woods AS, Cotter RJ, Gowing E, Ball MJ (1993) beta-Amyloid-(1-42) is a major component of cerebrovascular amyloid deposits: implications for the pathology of Alzheimer disease. Proc Natl Acad Sci U S A 90:10836–10840
Rombouts SA, Barkhof F, Veltman DJ, Machielsen WC, Witter MP, Bierlaagh MA, Lazeron RH, Valk J, Scheltens P (2000) Functional MR imaging in Alzheimer’s disease during memory encoding. AJNR Am J Neuroradiol 21:1869–1875
Romero JR, Preis SR, Beiser A, DeCarli C, Viswanathan A, Martinez-Ramirez S, Kase CS, Wolf PA, Seshadri S (2014) Risk factors, stroke prevention treatments, and prevalence of cerebral microbleeds in the Framingham Heart Study. Stroke 45:1492–1494
Roob G, Schmidt R, Kapeller P, Lechner A, Hartung HP, Fazekas F (1999) MRI evidence of past cerebral microbleeds in a healthy elderly population. Neurology 52:991–994
Sagare AP, Bell RD, Zlokovic BV (2013a) Neurovascular defects and faulty amyloid-beta vascular clearance in Alzheimer’s disease. J Alzheimers Dis 33(Suppl 1):S87–100
Sagare AP, Bell RD, Zhao Z, Ma Q, Winkler EA, Ramanathan A, Zlokovic BV (2013b) Pericyte loss influences Alzheimer-like neurodegeneration in mice. Nat Commun 4:2932
Schreiber S, Drukarch B, Garz C, Niklass S, Stanaszek L, Kropf S, Bueche C, Held F, Vielhaber S, Attems J, Reymann KG, Heinze HJ, Carare RO, Wilhelmus MM (2014) Interplay between age, cerebral small vessel disease, parenchymal amyloid-beta, and tau pathology: longitudinal studies in hypertensive stroke-prone rats. J Alzheimers Dis 42(Suppl 3):S205–S215
Shin HK, Jones PB, Garcia-Alloza M, Borrelli L, Greenberg SM, Bacskai BJ, Frosch MP, Hyman BT, Moskowitz MA, Ayata C (2007) Age-dependent cerebrovascular dysfunction in a transgenic mouse model of cerebral amyloid angiopathy. Brain 130:2310–2319
Shobin E, Bowley MP, Estrada LI, Heyworth NC, Orczykowski ME, Eldridge SA, Calderazzo SM, Mortazavi F, Moore TL, Rosene DL (2017) Microglia activation and phagocytosis: relationship with aging and cognitive impairment in the rhesus monkey. Geroscience 39:199–220
Sierra F, Kohanski R (2017) Geroscience and the trans-NIH Geroscience Interest Group, GSIG. Geroscience 39:1–5
Skoog I, Gustafson D (2006) Update on hypertension and Alzheimer’s disease. Neurol Res 28:605–611
Sokolova IA, Manukhina EB, Blinkov SM, Koshelev VB, Pinelis VG, Rodionov IM (1985) Rarefication of the arterioles and capillary network in the brain of rats with different forms of hypertension. Microvasc Res 30:1–9
Sonntag WE, Lynch CD, Cooney PT, Hutchins PM (1997) Decreases in cerebral microvasculature with age are associated with the decline in growth hormone and insulin-like growth factor 1. Endocrinology 138:3515–3520
Sonntag WE, Deak F, Ashpole N, Toth P, Csiszar A, Freeman W, Ungvari Z (2013) Insulin-like growth factor-1 in CNS and cerebrovascular aging. Front Aging Neurosci 5:27
Sorond FA, Hurwitz S, Salat DH, Greve DN, Fisher ND (2013) Neurovascular coupling, cerebral white matter integrity, and response to cocoa in older people. Neurology 81:904–909
Sparks DL, Scheff SW, Liu H, Landers TM, Coyne CM, Hunsaker JC 3rd (1995) Increased incidence of neurofibrillary tangles (NFT) in non-demented individuals with hypertension. J Neurol Sci 131:162–169
Springo Z, Toth P, Tarantini S, Ashpole NM, Tucsek Z, Sonntag WE, Csiszar A, Koller A, Ungvari ZI (2015a) Aging impairs myogenic adaptation to pulsatile pressure in mouse cerebral arteries. J Cereb Blood Flow Metab 35:527–530
Springo Z, Tarantini S, Toth P, Tucsek Z, Koller A, Sonntag WE, Csiszar A, Ungvari Z (2015b) Aging exacerbates pressure-induced mitochondrial oxidative stress in mouse cerebral arteries. J Gerontol A Biol Sci Med Sci 70:1355–1359
Stobart JL, Lu L, Anderson HD, Mori H, Anderson CM (2013) Astrocyte-induced cortical vasodilation is mediated by D-serine and endothelial nitric oxide synthase. Proc Natl Acad Sci U S A 110:3149–3154
Suzuki K, Masawa N, Sakata N, Takatama M (2003) Pathologic evidence of microvascular rarefaction in the brain of renal hypertensive rats. J Stroke Cerebrovasc Dis 12:8–16
Sveinbjornsdottir S, Sigurdsson S, Aspelund T, Kjartansson O, Eiriksdottir G, Valtysdottir B, Lopez OL, van Buchem MA, Jonsson PV, Gudnason V, Launer LJ (2008) Cerebral microbleeds in the population based AGES-Reykjavik study: prevalence and location. J Neurol Neurosurg Psychiatry 79:1002–1006
Takemori K, Murakami T, Kometani T, Ito H (2013) Possible involvement of oxidative stress as a causative factor in blood-brain barrier dysfunction in stroke-prone spontaneously hypertensive rats. Microvasc Res 90:169–172
Tarantini S, Hertelendy P, Tucsek Z, Valcarcel-Ares MN, Smith N, Menyhart A, Farkas E, Hodges E, Towner R, Deak F, Sonntag WE, Csiszar A, Ungvari Z, Toth P (2015) Pharmacologically-induced neurovascular uncoupling is associated with cognitive impairment in mice. J Cereb Blood Flow Metab 35:1871–1881
Tarantini S, Tucsek Z, Valcarcel-Ares M, Toth P, Gautam T, Giles C, Ballabh P, Wei Y, Wren J, Ashpole N, Sonntag W, Ungvari Z, Csiszar A (2016a) Circulating IGF-1 deficiency exacerbates hypertension-induced microvascular rarefaction in the mouse hippocampus and retrosplenial cortex: implications for cerebromicrovascular and brain aging. Age (Dordr) 38:273–289
Tarantini S, Tran CH, Gordon GR, Ungvari Z, Csiszar A (2016b) Impaired neurovascular coupling in aging and Alzheimer’s disease: contribution of astrocyte dysfunction and endothelial impairment to cognitive decline. Exp Gerontol. https://doi.org/10.1016/j.exger.2016.11.004
Tarantini S, Valcarcel-Ares NM, Yabluchanskiy A, Springo Z, Fulop GA, Ashpole N, Gautam T, Giles CB, Wren JD, Sonntag WE, Csiszar A, Ungvari Z (2017) IGF-1 deficiency exacerbates hypertension-induced cerebral microhemorrhages in mice, mimicking the aging phenotype. Aging Cell 16:469–479
Tarumi T, Dunsky DI, Khan MA, Liu J, Hill C, Armstrong K, Martin-Cook K, Cullum CM, Zhang R (2014) Dynamic cerebral autoregulation and tissue oxygenation in amnestic mild cognitive impairment. J Alzheimers Dis 41(3):765–778
Tenk J, Rostas I, Furedi N, Miko A, Solymar M, Soos S, Gaszner B, Feller D, Szekely M, Petervari E, Balasko M (2017) Age-related changes in central effects of corticotropin-releasing factor (CRF) suggest a role for this mediator in aging anorexia and cachexia. Geroscience 39:61–72
Tong XK, Lecrux C, Rosa-Neto P, Hamel E (2012) Age-dependent rescue by simvastatin of Alzheimer’s disease cerebrovascular and memory deficits. J Neurosci 32:4705–4715
Toth P, Tucsek Z, Sosnowska D, Gautam T, Mitschelen M, Tarantini S, Deak F, Koller A, Sonntag WE, Csiszar A, Ungvari Z (2013a) Age-related autoregulatory dysfunction and cerebromicrovascular injury in mice with angiotensin II-induced hypertension. J Cereb Blood Flow Metab 33:1732–1742
Toth P, Csiszar A, Tucsek Z, Sosnowska D, Gautam T, Koller A, Schwartzman ML, Sonntag WE, Ungvari Z (2013b) Role of 20-HETE, TRPC channels, and BKCa in dysregulation of pressure-induced Ca2+ signaling and myogenic constriction of cerebral arteries in aged hypertensive mice. Am J Physiol Heart Circ Physiol 305:H1698–H1708
Toth P, Tucsek Z, Tarantini S, Sosnowska D, Gautam T, Mitschelen M, Koller A, Sonntag WE, Csiszar A, Ungvari Z (2014a) IGF-1 deficiency impairs cerebral myogenic autoregulation in hypertensive mice. J Cereb Blood Flow Metab 34:1887–1897
Toth P, Tarantini S, Tucsek Z, Ashpole NM, Sosnowska D, Gautam T, Ballabh P, Koller A, Sonntag WE, Csiszar A, Ungvari ZI (2014b) Resveratrol treatment rescues neurovascular coupling in aged mice: role of improved cerebromicrovascular endothelial function and down-regulation of NADPH oxidase. Am J Physiol Heart Circ Physiol 306:H299–H308
Toth P, Tarantini S, Springo Z, Tucsek Z, Gautam T, Giles CB, Wren JD, Koller A, Sonntag WE, Csiszar A, Ungvari Z (2015a) Aging exacerbates hypertension-induced cerebral microhemorrhages in mice: role of resveratrol treatment in vasoprotection. Aging Cell 14:400–408
Toth P, Tarantini S, Ashpole NM, Tucsek Z, Milne GL, Valcarcel-Ares NM, Menyhart A, Farkas E, Sonntag WE, Csiszar A, Ungvari Z (2015b) IGF-1 deficiency impairs neurovascular coupling in mice: implications for cerebromicrovascular aging. Aging Cell 14:1034–1044
Toth P, Tarantini S, Davila A, Valcarcel-Ares MN, Tucsek Z, Varamini B, Ballabh P, Sonntag WE, Baur JA, Csiszar A, Ungvari Z (2015c) Purinergic glio-endothelial coupling during neuronal activity: role of P2Y1 receptors and eNOS in functional hyperemia in the mouse somatosensory cortex. Am J Physiol Heart Circ Physiol 309:H1837–H1845
Toth P, Tarantini S, Csiszar A, Ungvari Z (2017) Functional vascular contributions to cognitive impairment and dementia: mechanisms and consequences of cerebral autoregulatory dysfunction, endothelial impairment, and neurovascular uncoupling in aging. Am J Physiol Heart Circ Physiol 312:H1–H20
Troen AM, Shea-Budgell M, Shukitt-Hale B, Smith DE, Selhub J, Rosenberg IH (2008) B-vitamin deficiency causes hyperhomocysteinemia and vascular cognitive impairment in mice. Proc Natl Acad Sci U S A 105:12474–12479
Tucsek Z, Toth P, Tarantini S, Sosnowska D, Gautam T, Warrington JP, Giles CB, Wren JD, Koller A, Ballabh P, Sonntag WE, Ungvari Z, Csiszar A (2014) Aging exacerbates obesity-induced cerebromicrovascular rarefaction, neurovascular uncoupling, and cognitive decline in mice. J Gerontol A Biol Sci Med Sci 69:1339–1352
Ungvari Z, Orosz Z, Labinskyy N, Rivera A, Xiangmin Z, Smith K, Csiszar A (2007) Increased mitochondrial H2O2 production promotes endothelial NF-kappaB activation in aged rat arteries. Am J Physiol Heart Circ Physiol 293:H37–H47
Ungvari Z, Kaley G, de Cabo R, Sonntag WE, Csiszar A (2010) Mechanisms of vascular aging: new perspectives. J Gerontol A Biol Sci Med Sci 65:1028–1041
Ungvari Z, Bailey-Downs L, Gautam T, Sosnowska D, Wang M, Monticone RE, Telljohann R, Pinto JT, de Cabo R, Sonntag WE, Lakatta E, Csiszar A (2011a) Age-associated vascular oxidative stress, Nrf2 dysfunction and NF-kB activation in the non-human primate Macaca mulatta. J Gerontol A Biol Sci Med Sci 66:866–875
Ungvari Z, Bailey-Downs L, Sosnowska D, Gautam T, Koncz P, Losonczy G, Ballabh P, de Cabo R, Sonntag WE, Csiszar A (2011b) Vascular oxidative stress in aging: a homeostatic failure due to dysregulation of Nrf2-mediated antioxidant response. Am J Physiol Heart Circ Physiol 301:H363–H372
Ungvari ZI, Bailey-Downs L, Gautam T, Jimenez R, Losonczy G, Zhang C, Ballabh P, Recchia FA, Wilkerson DC, Sonntag WE, Pearson KJ, de Cabo R, Csiszar A (2011c) Adaptive induction of NF-E2-related factor-2-driven antioxidant genes in endothelial cells in response to hyperglycemia. Am J Physiol Heart Circ Physiol 300:H1133–H1140
Ungvari Z, Tucsek Z, Sosnowska D, Toth P, Gautam T, Podlutsky A, Csiszar A, Losonczy G, Valcarcel-Ares MN, Sonntag WE (2013) Aging-induced dysregulation of dicer1-dependent microRNA expression impairs angiogenic capacity of rat cerebromicrovascular endothelial cells. J Gerontol A Biol Sci Med Sci 68:877–891
Ungvari Z, Tarantini S, Hertelendy P, Valcarcel-Ares MN, Fulop GA, Logan S, Kiss T, Farkas E, Csiszar A, Yabluchanskiy A (2017a) Cerebromicrovascular dysfunction predicts cognitive decline and gait abnormalities in a mouse model of whole brain irradiation-induced accelerated brain senescence. Geroscience 39:33–42
Ungvari Z, Tarantini S, Kirkpatrick AC, Csiszar A, Prodan CI (2017b) Cerebral microhemorrhages: mechanisms, consequences and prevention. Am J Physiol Heart Circ Physiol in press
Urfer SR, Kaeberlein TL, Mailheau S, Bergman PJ, Creevy KE, Promislow DE, Kaeberlein M (2017a) A randomized controlled trial to establish effects of short-term rapamycin treatment in 24 middle-aged companion dogs. Geroscience 39:117–127
Urfer SR, Kaeberlein TL, Mailheau S, Bergman PJ, Creevy KE, Promislow DE, Kaeberlein M (2017b) Asymptomatic heart valve dysfunction in healthy middle-aged companion dogs and its implications for cardiac aging. Geroscience 39:43–50
Valcarcel-Ares MN, Gautam T, Warrington JP, Bailey-Downs L, Sosnowska D, de Cabo R, Losonczy G, Sonntag WE, Ungvari Z, Csiszar A (2012) Disruption of Nrf2 signaling impairs angiogenic capacity of endothelial cells: implications for microvascular aging. J Gerontol A Biol Sci Med Sci 67:821–829
van Dijk EJ, Breteler MM, Schmidt R, Berger K, Nilsson LG, Oudkerk M, Pajak A, Sans S, de Ridder M, Dufouil C, Fuhrer R, Giampaoli S, Launer LJ, Hofman A, Consortium C (2004) The association between blood pressure, hypertension, and cerebral white matter lesions: cardiovascular determinants of dementia study. Hypertension 44:625–630
Vernooij MW, van der Lugt A, Ikram MA, Wielopolski PA, Niessen WJ, Hofman A, Krestin GP, Breteler MM (2008) Prevalence and risk factors of cerebral microbleeds: the Rotterdam Scan Study. Neurology 70:1208–1214
Viana IM, de Almeida ME, Lins MP, dos Santos Reis MD, de Araujo Vieira LF, Smaniotto S (2015) Combined effect of insulin-like growth factor-1 and CC chemokine ligand 2 on angiogenic events in endothelial cells. PLoS One 10:e0121249
Wakisaka Y, Miller JD, Chu Y, Baumbach GL, Wilson S, Faraci FM, Sigmund CD, Heistad DD (2008) Oxidative stress through activation of NAD(P)H oxidase in hypertensive mice with spontaneous intracranial hemorrhage. J Cereb Blood Flow Metab 28:1175–1185
Wakisaka Y, Chu Y, Miller JD, Rosenberg GA, Heistad DD (2010) Spontaneous intracerebral hemorrhage during acute and chronic hypertension in mice. J Cereb Blood Flow Metab 30:56–69
Wells JA, Christie IN, Hosford PS, Huckstepp RT, Angelova PR, Vihko P, Cork SC, Abramov AY, Teschemacher AG, Kasparov S, Lythgoe MF, Gourine AV (2015) A critical role for purinergic signalling in the mechanisms underlying generation of BOLD fMRI responses. J Neurosci 35:5284–5292
White CL, Pistell PJ, Purpera MN, Gupta S, Fernandez-Kim SO, Hise TL, Keller JN, Ingram DK, Morrison CD, Bruce-Keller AJ (2009) Effects of high fat diet on Morris maze performance, oxidative stress, and inflammation in rats: contributions of maternal diet. Neurobiol Dis 35:3–13
Wiesmann M, Roelofs M, van der Lugt R, Heerschap A, Kiliaan AJ, Claassen JA (2016) Angiotensin II, hypertension, and angiotensin II receptor antagonism: roles in the behavioural and brain pathology of a mouse model of Alzheimer’s disease. J Cereb Blood Flow Metab 7(7):2396–2413
Wilhelmus MM, Otte-Holler I, van Triel JJ, Veerhuis R, Maat-Schieman ML, Bu G, de Waal RM, Verbeek MM (2007) Lipoprotein receptor-related protein-1 mediates amyloid-beta-mediated cell death of cerebrovascular cells. Am J Pathol 171:1989–1999
Winkler EA, Bell RD, Zlokovic BV (2011) Central nervous system pericytes in health and disease. Nat Neurosci 14:1398–1405
Winkler EA, Nishida Y, Sagare AP, Rege SV, Bell RD, Perlmutter D, Sengillo JD, Hillman S, Kong P, Nelson AR, Sullivan JS, Zhao Z, Meiselman HJ, Wenby RB, Soto J, Abel ED, Makshanoff J, Zuniga E, De Vivo DC, Zlokovic BV (2015) GLUT1 reductions exacerbate Alzheimer’s disease vasculo-neuronal dysfunction and degeneration. Nat Neurosci 18:521–530
Yamaguchi H, Yamazaki T, Lemere CA, Frosch MP, Selkoe DJ (1992) Beta amyloid is focally deposited within the outer basement membrane in the amyloid angiopathy of Alzheimer’s disease. An immunoelectron microscopic study. Am J Pathol 141:249–259
Yates PA, Sirisriro R, Villemagne VL, Farquharson S, Masters CL, Rowe CC (2011) Cerebral microhemorrhage and brain beta-amyloid in aging and Alzheimer disease. Neurology 77:48–54
Yates PA, Desmond PM, Phal PM, Steward C, Szoeke C, Salvado O, Ellis KA, Martins RN, Masters CL, Ames D, Villemagne VL, Rowe CC (2014) Incidence of cerebral microbleeds in preclinical Alzheimer disease. Neurology 82:1266–1273
Zaletel M, Strucl M, Pretnar-Oblak J, Zvan B (2005) Age-related changes in the relationship between visual evoked potentials and visually evoked cerebral blood flow velocity response. Funct Neurol 20:115–120
Zhang M, Mao Y, Ramirez SH, Tuma RF, Chabrashvili T (2010) Angiotensin II induced cerebral microvascular inflammation and increased blood-brain barrier permeability via oxidative stress. Neuroscience 171:852–858
Zlokovic BV (2008) The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 57:178–201
Zlokovic BV (2011) Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat Rev Neurosci 12:723–738
Zlokovic BV (2013) Cerebrovascular effects of apolipoprotein E: implications for Alzheimer disease. JAMA Neurol 70:440–444
Acknowledgements
This work was supported by grants from the American Heart Association (to ST, MNVA, AC, and ZU), the National Center for Complementary and Alternative Medicine (R01-AT006526 to ZU), the National Institute on Aging (R01-AG047879 to AC; R01-AG038747), the NIA-supported Geroscience Training Program in Oklahoma (T32AG052363), the NIA-supported Oklahoma Nathan Shock Center (3P30AG050911-02S1), the National Institute of Neurological Disorders and Stroke (NINDS; R01-NS056218 to AC), the Oklahoma Shared Clinical and Translational Resources (to AY; NIGMS U54GM104938), the Oklahoma Center for the Advancement of Science and Technology (to AC, ZU, and AY), the Reynolds Foundation (to ZU, AC and AY), and the Presbyterian Health Foundation (to AC, ZU, and AY). We also acknowledge support from the Merit Review Award I01 BX002211-01A2 from the US Department of Veterans Affairs (to VG), the William & Ella Owens Medical Research Foundation (VG), the San Antonio Nathan Shock Center of Excellence in the Biology of Aging (2 P30 AG013319-21) (VG), and the Robert L. Bailey and daughter Lisa K. Bailey Alzheimer’s Fund in memory of Jo Nell Bailey (VG).
Author information
Authors and Affiliations
Corresponding author
About this article

Cite this article
Csiszar, A., Tarantini, S., Fülöp, G.A. et al. Hypertension impairs neurovascular coupling and promotes microvascular injury: role in exacerbation of Alzheimer’s disease. GeroScience 39, 359–372 (2017). https://doi.org/10.1007/s11357-017-9991-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11357-017-9991-9