
Abstract
The blood–brain barrier (BBB) constitutes the complex anatomic and physiologic interface between the intravascular compartment and the central nervous system, and its integrity is paramount for the maintenance of the very sensitive homeostasis of the central nervous system. Arterial hypertension is a leading cause of morbidity and mortality. The BBB has been shown to be disrupted in essential hypertension. BBB integrity is important for central autonomic control and this may be implicated in the pathophysiology of hypertension. On the other hand, evidence from experimental studies indicates that BBB disruption can be present in both hypertensive disease and dementia syndromes, suggesting a possibly key position of loss of BBB integrity in the pathophysiological pathways linking arterial hypertension with cognitive decline. Although much still remains to be elucidated with respect to the exact underlying mechanisms, the discovery of novel pathological pathways has changed our understanding of adult dementia and central nervous system disease overall, pointing out—in parallel—new potential therapeutic targets. The aim of this review is to summarize current scientific knowledge relevant to the pathophysiologic pathways that are involved in the disruption of the BBB function and potentially mediate hypertension-induced cognitive impairment. In parallel, we underline the differential cognition-preserving effect of several antihypertensive agents of similar blood pressure-lowering capacity, highlighting the presence of previously under-recognized BBB-protective actions of these drugs.
Similar content being viewed by others


Introduction
Blood–brain barrier (BBB) is a complex multicellular structure acting as a selective barrier between peripheral circulation and central nervous system (CNS). The integrity of this interface between intravascular compartment and brain parenchyma is essential for the maintenance of the very sensitive biological homeostasis of the CNS.
Arterial hypertension causes target organ damage (TOD) that involves vasculature and vital organs such as the heart, brain, and kidneys. Hypertension and cerebrovascular disease are strictly associated. Although the significance of stroke and ischemic disease in hypertensives is profound in literature, less interest is given in the role of BBB as a target of dysfunction in hypertensive disease. Recent evidence suggests that acute and chronic hypertension may cause BBB damage [1].
BBB disruption is a cornerstone mechanism in a variety of CNS diseases. Although the exact mechanisms underlying this pathophysiology are not fully clarified, several newly described pathophysiological pathways have been implicated. Notably, the discovery of these pathways also contributes to our understanding of adult dementia and CNS disease which is a major clinical problem in essential hypertension [2, 3]. This review primarily focuses on evidence suggesting a key position of BBB disruption in arterial hypertension and its impact on the progression to TOD mostly to cognitive decline and dementia. In addition, we briefly summarize available data regarding the superior cognition- and BBB-preserving effects of several antihypertensive agents over others with similar blood pressure-lowering capacity. We believe that the importance of BBB has not been highlighted and this review of the literature indicates its significance in clinical practice as a target of damage mediated by high blood pressure levels. However, it should be emphasized that the data at our disposal that we will cite hereafter are mostly from experimental studies in animal models and what exactly is the role of BBB in the brain of hypertensive patients, cannot be accurately described so far.
BBB physiology: evidence for its role in blood pressure regulation
The BBB is the highly selective interface between the peripheral blood circulation and the CNS, comprised of endothelial cells, the basement membrane, tight junctions (TJ), adherens junctions (AJ), pericytes, astrocytes, and neurons. The main component of BBB is the endothelial cell layer. Specific properties of the endothelial cell layer of the cerebral vasculature also contribute to the formation of the BBB; of particular importance is the relative paucity of caveolae in the endothelial cells, a feature which opposes substance transportation across the vessel wall. In addition, astrocytes, pericytes, and microglia consist an important part of the neurovascular unit [4]. Pericytes—mural cells of the microcirculation closely connected to the endothelial cells, embedded in the basement membrane—are key contributors to the formation and preservation of a functional BBB. In an age-dependent manner, pericyte number is gradually reduced, and this results in BBB weakening. Astrocytes, on the other hand, are separated from endothelial cells by the basement membrane, around which they extend cell processes termed “end-feet”. In vitro studies show that these cells are key modulators of TJ function and can also mediate distribution of transporters to the endothelium. Pericytes and astrocytes, along with endothelial cells and their TJs, as well as neurons, interneurons, and extracellular matrix, form a complex functional and anatomical structure termed the neurovascular unit, which is integral in the regulation of cerebral parenchymal homeostasis and in the process of interaction between myelinated and unmyelinated axons and brain capillaries [5].
The primary function of the BBB is to ensure a tight control of the environment of the brain, and its functionality is paramount for the maintenance of cerebral homeostasis; indeed, loss of BBB integrity constitutes a key mechanism in the pathophysiology of various neurological diseases. A healthy BBB translates into controlled traffic across the vessel wall, at a molecular and ionic level. Nondiffusible, nonlipidic molecules are transported by a highly controlled system of carriers on both sides of endothelial cells, which is both time- and concentration-dependent. AJs and TJs together form the interendothelial connections, which are cornerstone in the maintenance of a viable BBB [2].
BBB integrity is important for central autonomic control which in turn is crucial for the blood pressure regulation. A well-regulated BBB protects important anatomical areas such as the hypothalamus and medulla to respond appropriately and maintain homeostasis to the autonomic control centers [6].
On the other hand, increased angiotensin II (Ang II) availability activates intracellular pathways and increases neuronal activity within the autonomic areas as the hypothalamic paraventricular nucleus and brain stem nuclei. Consequently, an intact BBB is of great importance to protect autonomic areas from the invasion of increased Ang II levels. BBB disruption not only facilitates Ang II access, but also allows circulating inflammatory cells to enter into the brain parenchyma, contributing to further microglial activation and inflammation in autonomic areas [7].
Effects of oxidative stress and inflammation on BBB
Oxidative stress has been shown to exert deleterious effects on the BBB and its ultrastructure and to contribute to the process of neurodegeneration. Specifically, TJ complexes and endothelial cells seem to be particularly vulnerable, with oxidative stress inducing an alteration of their protein expression profile [8]. Similarly, an oxidative stress-induced alteration, increased transport across the BBB and disruption of its protective function, has been demonstrated in hypertensive subjects [9].
Proposed mechanisms underlying the TJ dysfunction observed under oxidative stress include an increased exposure to free radicals and cytokines, activation of MMP, as well as downregulation of TJ-proteins claudins and zonulin-1 and metalloproteinase tissue inhibitors—ultimately resulting in increased paracellular leakage [10]. Reactive oxygen species (ROS) constitute the primary oxidative stress mediators damaging the structure and function of proteins, DNA and lipids, thus increasing permeability of the BBB in a time- and concentration-dependent manner [11]. Importantly, a significant source of are beta-amyloid (Aβ) peptides, which primarily affect the cerebral microvasculature and are typically implicated in the pathogenesis of cerebral amyloid angiopathy [12]. This exaggerated generation of free radicals results in a decrease in the expression of specific TJ proteins, thus adversely affecting BBB integrity [13].
Neuroinflammation—a process that is linked with oxidative stress—seems to play a key role in both aging and degenerative processes and has also been associated with BBB dysfunction. Indeed, inflammatory cytokine levels are increased in acute ischemic stroke patients and have been associated with worse prognosis and increased risk of recurrence. Vascular dementia—apart from increased age—is also associated with increased levels of interleukins-1a, -6, and -8 (IL-1a, IL-6, IL-8, respectively), as well as tumor necrosis factor. Overexpression of interferon-γ and several chemokines such as CCL-2 and CXCL-8 also results in increased BBB permeability [14].
Nitric oxide (NO), on the other hand, appears to exert a protective effect on the BBB, by inhibiting amyloid plaque formation, tau phosphorylation, and neuronal loss. However, during oxidative stress, its metabolite peroxynitrite has neurotoxic effects similar to ROS [15].
Lipid peroxidation products, produced by the oxidative damage of polyunsaturated fatty acids, modify the function of cell membrane phospholipids, disrupt mitochondrial enzymes, and energetic and increase free radicals release. The result is perpetuation of oxidative stress BBB destabilization. This effect can be accentuated by the activation of matrix metalloproteinases (MMP), which are produced by activated microglia, negatively affecting the integrity of the endothelial basal lamina and the basement membrane of the BBB-forming neurovascular unit [16].
Renin–angiotensin aldosterone system (RAAS) system and BBB integrity
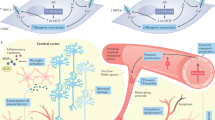
RAAS system is a highly regulated system in essential hypertension. RAAS is believed to have a pivotal role in organ damage. Especially, Ang II is a peptide with a critical role, implicated in the regulation of blood pressure. Due to its hydrophilic behavior circulating Ang II does not cross the BBB. However, elevated circulating levels of Ang II during hypertension results in disruption of the BBB integrity, allowing access of circulating Ang II (Fig. 1). There are several findings suggesting that peripheral and central RAAS, via disruption of the BBB may promote exacerbated sympathoexcitatory activity and in turn neurogenic hypertension [17]. Angiotensin 1 receptor (AT1R) activation has been shown to affect BBB permeability in cultured microvessels [18]. Chronic infusion of Ang II has been implicated in increased BBB permeability mostly due the downregulation of endothelial tight junction proteins [19]. Interestingly, local RAAS activation is also important in organ homeostasis. Components of the RAAS have been detected in various organs, such as the heart, kidney, and brain. Indeed, the brain expresses genes that encode all components of the RAAS. The local RAAS can be activated independently of the systemic RAAS, indicating that organ damage may occur even though the systemic RAAS is attenuated. Disruption of the BBB allowed the entry of peripherally injected Ang II into the rostral ventrolateral medulla which activated tyrosine hydroxylase‐expressing neurons involved in central cardiovascular regulation [20]. In addition, peripherally infused Ang II can enter a leaky BBB and influence CNS signaling [17].

Ang II angiotensin II, RAAS renin–angiotensin aldosterone system, SNS sympathetic nervous system.
A well-known risk factor for amyloid b (Ab) deposition and cognitive impairment in AD is a history of arterial hypertension several years prior to the onset of the disease. Experimental studies in mice have shown that Ang II may also be involved in Ab clearance by increasing BBB permeability through AT1R [21]. Arterial hypertension has a well-established impact on the function of cerebral vasculature and can thus affect the receptor-mediated transport across the BBB, as well as passive passage of substances across vascular basement membranes [22]. The resultant impediment of Ab elimination from the brain translates into deposition of increased amounts of Ab in various forms such as monomer, oligomer, insoluble fibrils, and plaques in the CNS. Pronounced accumulation of Ab, in turn, triggers a cascade of events culminating in neuronal damage and death, manifesting as progressive clinical dementia of the Alzheimer type [22]. It is hypothesized that Ab deposition due to a disorder of cholinergic neurons and vascular injury, respectively is a key player in dementia which is associated with both deterioration in the quality of life and poor prognosis.
Linking evidence between BBB dysfunction and hypertension: a focus in experimental data
Hypertension—a leading cause of cardiovascular disease—is characterized by autonomic dysregulation, sympathetic overactivity, and activation of plasma and tissue RAAS and of circulating catecholamines [23]. In addition, innate and adaptive immunity play a significant role in the pathogenesis of hypertension [24, 25]. Immune cells located in BBB may also have an impact in both pathogenesis of hypertension and BBB disruption. Such cells are the microglia which role in hypertension has increasingly been recognized. Studies have shown that microglial inhibition in hypertensive animal models leads to a decrease in blood pressure [26, 27]. On the other hand, astrocytes are also involved in the pathogenesis of hypertension through the signaling cascade of the sympathoexcitatory effects of Ang II in the PVN [28]. Experimental studies have shown that pharmacological inhibition of astrocytes using sodium fluoroacetate causes an increase in blood pressure and a decrease in BBB permeability [29].
The cerebrospinal fluid (CSF) is a clear colorless fluid that is produced by the choroid plexus and provides the brain with mechanical and immunological protection. In parallel, the CSF plays an active role in cerebral metabolism. Loss of BBB integrity results in a leak of substances—including proteins—into the CSF, which, when detected, can be regarded as an indicator of BBB insufficiency [30]. In this context, in a recent study, the CSF was analyzed in spontaneously hypertensive rats (SHR) and control rats and significant differences in protein concentration were detected between the two groups. The BBB disruption observed in SHRs could be explained by the susceptibility of arteries with profoundly reduced perfusion to endothelial damage. In the SHRs, upregulation of α-1-antitrypsin, apolipoprotein A1, albumin, immunoglobulin G, vitamin D binding protein, haptoglobin, and α-macroglobulin was observed [30].
In normotensive rats, acute increases in arterial blood pressure induced a marked increase in CSF pressure, whereas in SHRs the effects of phenylephrine administration were milder, suggesting the development of a cerebroprotective mechanism with chronic hypertension. However, an increased permeability for sucrose and lower brain uptake rate was identified in chronic hypertensive subjects, suggesting a decreased brain capillary density a reduction in cerebral blood flow (CBF) [29].
Impairment of endothelial homeostasis in SHRs may also contribute to the selective BBB disruption. In chronic ischemia, long-standing hypertension may impair of BBB integrity and induce white matter dysfunction, leading to worsening of spatial cognitive performance. Leaked plasma components, known to exert toxic effects on perivascular neurons, may access periaxonal environment through disrupted paranodal axon-glia junctions, thereby impairing axon conduction [31].
In parallel, research is focusing on the elucidation of pathways by which the BBB may be involved in the neuronal dysfunction caused by accumulating ß-amyloid in the brain [32]. Therapies aiming to increase low-density lipoprotein receptor related protein-1 expression or reduce RAGE activity at the BBB and/or restore the peripheral β-amyloid “sink” action, have been shown to inhibit brain β-amyloid accumulation and inflammation, and improve CBF and functional recovery in AD models. Thus, such therapies have emerged as promising future treatment modalities for AD patients [33].
Prolonged Ang II-induced hypertension is associated with large BBB leaks, microglial activation, myelin loss, and cognitive impairment in the absence of stroke. Finally, cerebral autoregulatory dysfunction ultimately results in BBB leakage, allowing the circulating inflammatory factors to infiltrate the brain to activate glia, especially in response to elevations in cerebral arterial pressure as it has been shown in hypertensive animal models [34]. Finally, hypertension promotes highly distinctive alterations in small arteries which in turn has been shown in animal models to cause early dysfunction in ΒΒΒ [35].
BBB dysfunction as a link between hypertension and cognitive decline in human studies
Accumulating evidence has shown that chronic hypertension is accompanied by BBB dysfunction. The brain, an organ accounting for 20–25% of the body’s total oxygen consumption, is critically dependent on the cardiovascular system. Hypertension is a risk factor for the aggravation of cognitive impairment [34]. Importantly, recent evidence suggests the existence of a key vascular component in the pathophysiology of Alzheimer’s disease (AD), which has been for long considered as a primarily neurodegenerative entity [36, 37]. Although much remains to be clarified with respect to the exact mechanisms, BBB dysfunction appears to be involved in the association between arterial hypertension and dementia syndromes, as it can be present in both conditions. However, the impact of blood pressure perturbations on long-term alterations in cognitive function appears to be multifactorial and much remains to be elucidated. Indeed, although several studies report that a history of hypertension decades before the onset of the disease is correlated with the later occurrence of AD, the time evolution of the blood pressure profile of future AD patients may demonstrate a significant drop in blood pressure levels close to the onset of the disease as well as a consistent decrease throughout the dementia phase [38].
Dementia syndromes, primarily AD, are characterized by hippocampal atrophy which typically develop early in the disease course [39]. Similarly, the entorhinal cortex, the cornu ammonis 1 (CA1) sector and subicular areas are also affected early, followed by CA2, 3, and 4, whereas the neocortex is usually involved later in the course of the disease. The involvement of the CA1 pyramidal cells—and the sector as a whole—results in the loss of anterograde memory, resulting in significant impairment of cognitive functions. CA2, 3, and 4 hippocampal subfields, on the other hand, are much less affected by the disease, as suggested by evidence from postmortem AD studies [40].
Memory performance in hypertensive individuals is related to a blunted regional CBF response [41]. However, there are sufficient data based on postmortem investigations showing significant BBB damage in dementia patients [42]. On top of that, numerous studies with magnetic resonance imaging and positron emission tomography have proved alterations in regional BBB permeability and cerebral metabolism in the living human brain in dementia and AD [43].
Antihypertensive medication for the prevention of BBB dysfunction and dementia
Angiotensin-converting enzyme inhibitors (ACEIs) and Ang II receptor blockers
Since Ang II has emerged as a critical factor of BBB disruption in hypertension, medication targeting to RAAS may offer protective mechanisms to these structural changes. Results of experimental studies suggest that treatment with ACEIs and Ang II type 1 receptor blockers could attenuate progression of cognitive decline and oppose the development of dementia. Ang II likely participates in a feed-forward mechanism inducing neurogenic hypertension via neurohumoral activation and reaches the hypothalamus and brain stem through the disrupted BBB. Recent animal studies show that Ang II also plays a critical role in BBB integrity under hypertensive conditions. Indeed, administration of Ang II led to increased BBB permeability and downregulation of constituent BBB proteins, facilitating its extravasation to critical blood pressure-regulating brain regions and its interaction with neurons and glial cells, while treatment with losartan appears to inhibit these effects and restore, in part, blood pressure homeostasis [17, 44]. In contrast, administration of hydralazine, a direct vasodilator, failed to restore BBB disruption. This supports the evidence that Ang II–AT1R mechanisms are implicated in BBB dysregulation. Tables 1 and 2 present studies addressing the effects of antihypertensive medication on BBB function.
Brain RAS hyperactivity has been implicated in the pathogenesis of essential hypertension in many experimental studies [45,46,47]. Aminopeptidase A (APA) a membrane-bound zinc metalloprotease, is involved in the metabolism of Ang II, The inhibition of central, but not peripheral, APA by specific and selective inhibitors leads to a large and sustained decrease in BP via inhibition of AVP release and sympathetic neuron activity [48].
The molecular weight of ACE inhibitors and ARBs is small permitting them their passage from BBB depending on their lipophilicity and concentration of the drug in plasma. Especially, lisinopril, which is hydrophilic, has been shown to inhibit central ACE, however trandolapril, a lipophilic agent, caused more ready inhibition of ACE binding in cardiovascular regulatory centers inside the BBB [49]. AT1R blockade has been shown to have neuroprotective effects when used for inflammatory brain disorders that accompany BBB disruption [50]. Evidence supporting a potential dementia-protective effect of ARBs was provided by an animal study by Takeda et al. whereby pretreatment with a low dose of olmesartan was shown to prevent β-amyloid-induced vascular dysregulation and to attenuate the impairment of hippocampal synaptic plasticity [51] and decreased brain Ang II levels and restored mRNA expression of TJs and collagen-IV in Dahl salt-sensitive rats [52]. However, no effect on cerebral β-amyloid levels was observed [51]. Other ARBs have also been pointed out as neuroprotective agents in animal studies [53]. Characteristically, in rodent models of AD, administration of several angiotensin receptor blockers—namely candesartan, losartan, valsartan, and telmisartan—appeared to preserve CBF and cognition during stroke, decrease inflammation and Aβ neurotoxicity and to exert cerebroprotective effects in the context of traumatic brain injury [54]. Similarly, human cohort analyses indicate that treatment of hypertension with angiotensin receptor blockers can significantly reduce the incidence and progression of AD [54]. In patients with dementia, treatment with perindopril—besides effective control of blood pressure—was associated with an improvement of the cognitive function of the patients as represented by performance on the Mini-Mental State Examination and this result was sustained after a 12-months follow-up [55]. However, it should be emphasized that more data are needed for definite conclusions to be drawn with respect to preference of specific AT1 receptor blockers over others in the context of neuroprotection.
Several studies suggest that centrally acting ACEΙs could decrease the risk for dementia to a greater degree compared with other antihypertensives; indeed, a reduction in the incidence of a decline in cognitive scores of up to 65% per year of exposure has been reported [56,57,58,59]. Centrally acting ACE inhibitors include captopril, lisinopril, fosinopril, perindopril, ramipril, and trandolapril and their more pronounced dementia-protective effect compared with non-centrally acting representatives of the class could be primarily attributed to the fact that the brain’s intrinsic renin–angiotensin system is involved in memory and cognition. Nevertheless, the aforementioned BBB-preserving potential of these agents might also be implicated [57]. In general, antihypertensives that cross the BBB and affect the renin–angiotensin-aldosterone system (such as perindopril or losartan) may provide additional protection beyond BP control against cognitive decline.
These data clearly raise the question whether centrally acting ACEIs should be preferred over non-centrally acting agents, at least in patients at risk for the development of dementia. In this context, a recent study evaluating the effects of initiating centrally acting ACEIs in an unselected outpatient group of elderly patients showed a significant decrease in the degree of cognitive decline relative to other patients already on therapy or treated with other medications. This decrease in cognitive decline was observed as early as within 6 months from the initiation of treatment [57]. Furthermore, progression of cognitive decline seemed to be inhibited in patients with established AD once centrally acting ACEIs—particularly perindopril—were initiated. Importantly, the magnitude of the cognitive function-preserving effect did not demonstrate correlation to the degree of change in blood pressure during the 12-month follow-up period, suggesting a mechanism of action separate from the one that mediates the antihypertensive effect of these agents [57].
However, large clinical trials showed conflicting results regarding the beneficial effects of RAAS blockers in cognitive function in hypertensive populations [60].
Calcium channel blockers (CCBs)
Dihydropyridine (DHP)-type CCBs, on the other hand, also appear to slow the progression of dementia and prevent BBB damage, via increasing the relative regional CBF dose-dependently. Recent research has been especially focusing on nilvadipine, as this agent has been shown to be a much more potent effector of regional CBF functionalities compared with nicardipine [61]. In fact, in the 20-month follow-up, hypertensive patients treated with nilvadipine were less likely to develop AD. Another representative of this drug class, nimodipine, is also of special interest in this context, as appears to exert more pronounced neuroprotective effects than other CCBs, at least in specific clinical situations; indeed, the agent has been shown to improve neurological outcome in patients with subarachnoid hemorrhage from ruptured intracranial berry aneurysms (Hunt and Hess Grades I–V) and its use is indicated in this clinical scenario, regardless of their postictal neurological condition. Even though some studies suggest that nicardipine might be a better choice in terms of treating disorders associated with arterial hypertension than nimodipine, a Cochrane review of the clinical efficacy of the latter found pronounced benefit associated with the administration of the drug compared with placebo on cognitive function, when the subjects suffered from either AD or vascular dementia [62].
The lipophilic CCBs easily cross the intact BBB, and this property enables more pronounced effects in the CNS. It is hypothesized that DHP CCBs contribute to the amelioration of cerebral hypoperfusion that precedes clinical manifestations of both AD and vascular dementia. Interestingly, DHP-CCBs appear to antagonize the β-amyloid-induced vasoconstriction associated with AD [30]. However, amlodipine, another DHP-CCB, has been found to be much less potent in terms of preservation of cognitive functions. Amlodipine and nifedipine were found to be ineffective in impeding the accumulation of β-amyloid in the brain. This, coupled with the well-established fact that antihypertensive activity is a class effect of CCBs, suggests that the Aβ-lowering capacity of the DHPs is independent of their blood pressure-lowering capacity [63]. Furthermore, nilvadipine and nitrendipine were shown to inhibit Aβ production in in vitro studies, whereas other representatives of the class had either no effect or were associated with a rise in Aβ levels. In vivo, nilvadipine and nitrendipine acutely reduced brain Aβ levels in a transgenic mouse model of AD (Tg PS1/APPsw) and improved Aβ clearance across the BBB [61]. Importantly, this Aβ-lowering action could itself translate into improved BBB function and, in turn, inhibition of cognitive decline.
Alterations in cellular calcium handling could constitute another possible neuroprotective mechanism of action of CCBs, independent of their blood pressure-lowering effect. With ageing, the brain loses the capacity to efficiently regulate intracellular calcium levels. This calcium “mishandling” facilitates cell death, while also increasing the risk of AD development. In this regard, it has been postulated that, to some degree, the cognition-preserving actions of DHP-CCBs could be mediated by prevention of intracellular calcium overload [62, 63].
Although data are contradictory, it has been suggested a beneficial role of CCBs in BBB disruption of hypertensive animal models [64]. More specifically, nifedipine can modify the permeability disruptions observed in acutely hypertensive rats [65].
Nevertheless, much still remains to be clarified and further research in humans is required for any CCBs to be recommended specifically for the prevention of dementia in hypertensive patients.
Other antihypertensive drugs
A recent experimental study examining different beta blockers has shown that propranolol is able to cross the BBB and is found in brain tissue in higher amounts than atenolol and nadolol but were able to induce the secretion of signaling molecules in the hypothalamus, independently of their ability to cross the BBB [66].
The Honolulu-Asia Aging Study determined the associations between classes of antihypertensive medication use and the risk of cognitive impairment among elderly hypertensive men [67]. The authors found that β-blocker use as the sole antihypertensive drug at baseline was consistently associated with a lower risk of cognitive impairment as compared with men not taking any antihypertensive medications. Diuretics, CCBs, ACEIs, or vasodilators alone were not significantly associated with cognitive impairment. The authors hypothesized that the neuroprotective effects of b-blockers may be attributed to the improvement of microvascular integrity, amyloid deposition, widening of periarteriolar spaces, and microinfarct reduction. However, their protective effect has not been confirmed by solid data.
The Cache County Study examined the relationship of antihypertensive treatment with the incidence of Alzheimer disease [68]. The study showed that potassium-sparing diuretics, is associated with reduced incidence of Alzheimer disease.
Similarly, there are scarred data with the other antihypertensive categories, mostly experimental. In an experimental study with rats, it has been shown that diazoxide significantly inhibited the extravasation, depolarized the mitochondrial membrane, suggesting a direct diazoxide effect on the endothelial mitochondria, and protects the BBB against ischemic stress [69]. In another study, indapamide may also have a protective role against ischemia-induced injury and dysfunction of the BBB [70].
Future perspectives
Future studies will advance the current limited knowledge of the pathophysiology of BBB disorders that occur in various cardiovascular diseases. Drug delivery to the brain remains a major obstacle for the treatment of BBB disruption. There is however emerging evidence that exercise may prove beneficial in maintaining BBB integrity by reducing local Ang II [71].
The future emphasis will be on developing drug delivery systems for the brain that can deliver adequate amounts of neuroprotective agents in a controlled manner in order to treat neurological complications of arterial hypertension. This may include nanobiotechnology-based delivery of therapeutics such as gene therapy and siRNAs.
Conclusions
Taken together, the aforementioned data indicate a central position of BBB disruption in the pathophysiological pathways of arterial hypertension that are also linked with the development of cognitive decline and dementia. As the correlation between hypertension, RAAS activation, BBB dysfunction, and neurodegeneration becomes clearer, early and tailored antihypertensive treatment—with meticulous attention paid to the avoidance of hypotensive episodes—could probably exert cognition-preserving effects. Antihypertensive agents with independent BBB-sparing and neuroprotective effects—such as centrally acting ACEIs—would be theoretically be ideal choices in this context. It should be emphasized, however, that much more evidence from clinical trials is needed until a specific drug class can be strongly recommended over another for the sole purpose of dementia prevention.
References
Mohammadi MT, Dehghani GA. Acute hypertension induces brain injury and blood–brain barrier disruption through reduction of claudins mRNA expression in rat. Pathol Res Pr. 2014;210:985–89.
Kalaria RN. Vascular basis for brain degeneration: faltering controls and risk factors for dementia. Nutr Rev. 2010;6:S74–S87.
Keller A. Breaking and building the wall: the biology of the blood–brain barrier in health and disease. Swiss Med Wkly. 2013;143:w13892.
Obermeier B, Daneman R, Ransohoff RM. Development, maintenance and disruption of the blood–brain barrier. Nat Med. 2013;19:1584–96.
Serlin Y, Shelef I, Knyazer B, Friedman A. Anatomy and physiology of the blood–brain barrier. Semin Cell Dev Biol. 2015;38:2–6.
Smith PM, Ferguson AV. Circulating signals as critical regulators of autonomic state–central roles for the subfornical organ. Am J Physiol Regul Integr Comp Physiol. 2010;299:R405–15.
Waki H, Gouraud SS, Maeda M, Raizada MK, Paton JF. Contributions of vascular inflammation in the brainstem for neurogenic hypertension. Respir Physiol Neurobiol. 2011;178:422–8.
Ronaldson PT, Davis TP. Targeting transporters: promoting blood–brain barrier repair in response to oxidative stress injury. Brain Res. 2015;1623:39–52.
Pires PW, Dams Ramos CM, Matin N, Dorrance AM. The effects of hypertension on the cerebral circulation. Am J Physiol Heart Circ Physiol. 2013;304:H1598–H1614.
Lippoldt A, Kniesel U, Liebner S, Kalbacher H, Kirsch T, Wolburg H, et al. Structural alterations of tight junctions are associated with loss of polarity instroke-prone spontaneously hypertensive rat blood–brain barrier endothelial cells. Brain Res. 2000;885:251–61.
Tan KH, Harrington S, Purcell WM, Hurst RD. Peroxynitrite mediatesnitric oxide-induced blood–brain barrier damage. Neurochem Res. 2004;29:579–87.
Sparks DL, Scheff SW, Liu H, Landers TM, Coyne CM, Hunsaker JC 3rd. Increased incidence of neurofibrillary tangles (NFT) in non-demented individuals with hypertension. J Neurol Sci. 1995;131:162–9.
Witt KA, Mark KS, Hom S, Davis TP. Effects of hypoxia-reoxygenation on rat blood–brain barrier permeability and tight junctional protein expression. Am J Physiol Heart Circ Physiol. 2003;285:2820–31.
Szarka N, Toth L, Czigler A, Kellermayer Z, Ungvari Z, Amrein K, et al. Single mild traumatic brain injury induces persistent disruption of the blood–brain barrier, neuroinflammation and cognitive decline in hypertensive rats. Int J Mol Sci. 2019;20:3223.
Parathath SR, Parathath S, Tsirka SE. Nitric oxide mediates neurodegeneration and breakdown of the blood–brain barrier in tPA-dependent excitotoxic injury in mice. J Cell Sci. 2006;119:339–49.
Enciu AM, Gherghiceanu M, Popescu BO. Triggers and effectors of oxidative stress at blood–brain barrier level: relevance for brain ageing and neurodegeneration. Oxid Med Cell Longev. 2013;2013:297512.
Biancardi VC, Son SJ, Ahmadi S, Filosa JA, Stern JE. Circulating angiotensin II gains access to the hypothalamus and brain stem during hypertension via breakdown of the blood–brain barrier. Hypertension. 2014;63:572–9.
Fleegal‐DeMotta MA, Doghu S, Banks WA. Angiotensin II modulates BBB permeability via activation of the AT1 receptor in brain endothelial cells. J Cereb Blood Flow Metab. 2009;29:640–7.
Vital SA, Terao S, Nagai M, Granger DN. Mechanisms underlying the cerebral microvascular responses to angiotensin II‐induced hypertension. Microcirculation. 2010;17:641–9.
Yao ST, May CN. Intra‐carotid angiotensin II activates tyrosine hydroxylase‐expressing rostral ventrolateral medulla neurons following blood–brain barrier disruption in rats. Neuroscience. 2013;245:148–56.
Zhang M, Mao Y, Ramirez SH, Tuma RF, Chabrashvili T. Angiotensin II induced cerebral microvascular inflammation and increased blood–brain barrier permeability via oxidative stress. Neuroscience. 2010;171:852–8.
Carnevale D, Mascio G, D’Andrea I, Fardella V, Bell RD, Branchi I, et al. Hypertension induces brain β-amyloid accumulation, cognitive impairment, and memory deterioration through activation of receptor for advanced glycation end products in brain vasculature. Hypertension. 2012;60:188–97.
Oparil S, Zaman MA, Calhoun DA. Pathogenesis of hypertension. Ann Intern Med. 2003;139:761–76.
Rodriguez-Iturbe B, Pons H, Johnson RJ. Role of the immune system in hypertension. Physiol Rev. 2017;97:1127–64.
González-Marrero I, Castañeyra-Ruiz L, González-Toledo JM, Castañeyra-Ruiz A, de Paz-Carmona H, Castro R, et al. High blood pressure effects on the blood to cerebrospinal fluid barrier and cerebrospinal fluid protein composition: a two-dimensional electrophoresis study in spontaneously hypertensive rats. Int J Hypertens. 2013;2013:164653.
Shen XZ, Li Y, Li L, Shah KH, Bernstein KE, Lyden P, et al. Microglia participate in neurogenic regulation of hypertension. Hypertension. 2015;66:309–16.
Shi P, Raizada MK, Sumners C. Brain cytokines as neuromodulators in cardiovascular control. Clin Exp Pharm Physiol. 2010;37:e52–7.
Stern JE, Son S, Biancardi VC, Zheng H, Sharma N, Patel KP. Astrocytes contribute to angiotensin II stimulation of hypothalamic neuronal activity and sympathetic outflow. Hypertension. 2016;68:1483–93.
Setiadi A, May CN, Yao ST. Ablation of astrocytes in the paraventricular nucleus disrupts the blood‐brain barrier and increases blood pressure in rats. FASEB J. 2017;31:1010.
Michalak Z, Lebrun A, Di Miceli M, Rousset MC, Crespel A, Coubes P, et al. IgG leakage may contribute to neuronal dysfunction in drug-refractory epilepsies with blood–brain barrier disruption. J Neuropathol Exp Neurol. 2012;71:826–38.
Meissner A, Minnerup J, Soria G, Planas AM. Structural and functional brain alterations in a murine model of Angiotensin II-induced hypertension. J Neurochem. 2017;140:509–21.
Yamazaki Y, Kanekiyo T. Blood–brain barrier dysfunction and the pathogenesis of Alzheimer’s disease. Int J Mol Sci. 2017;18:1965.
Sagare AP, Deane R, Zlokovic BV. Low-density lipoprotein receptor-related protein 1: a physiological Aβ homeostatic mechanism with multiple therapeutic opportunities. Pharm Ther. 2012;136:94–105.
Foulquier S, Namsolleck P, Van Hagen BT, Milanova I, Post MJ, Blankesteijn WM, et al. Hypertension-induced cognitive impairment: insights from prolonged angiotensin II infusion in mice. Hypertens Res. 2018;41:827.
Toth P, Tucsek Z, Sosnowska D, Gautam T, Mitschelen M, Tarantini S, et al. Age-related autoregulatory dysfunction and cerebromicrovascular injury in mice with angiotensin II-induced hypertension. J Cereb Blood Flow Metab. 2013;33:1732–42.
de la Torre JC, Stefano GB. Evidence that Alzheimer’s disease is a microvascular disorder: the role of constitutive nitric oxide. Brain Res Brain Res Rev. 2000;34:119–36.
Bueche CZ, Hawkes C, Garz C, Vielhaber S, Attems J, Knight RT, et al. Hypertension drives parenchymal β-amyloid accumulation in the brain parenchyma. Annals of Clinical and Translational. Neurology. 2014;1:124–9.
Faraco G, Iadecola C. Hypertension: a harbinger of stroke and dementia. Hypertension. 2013;62:810–7.
Apostolova LG, Mosconi L, Thompson PM, Green AE, Hwang KS, Ramirez, et al. Subregional hippocampal atrophy predicts Alzheimer’s dementia in the cognitively normal. Neurobiol Aging. 2010;31:1077–88.
Bobinski M, Wegiel J, Wisniewski HM, Tarnawski M, Reisberg B, Mlodzik B, et al. Atrophy of hippocampal formation subdivisions correlates with stage and duration of Alzheimer disease. Dementia. 1995;6:205–10.
Jennings JR, Muldoon MF, Ryan C, Price JC, Greer P, Sutton-Tyrrell K, et al. Reduced cerebral blood flow response and compensation among patients with untreated hypertension. Neurology. 2005;64:1358–65.
Tomimoto H, Akiguchi I, Suenaga T, Nishimura M, Wakita H, Nakamura S, et al. Alterations of the blood–brain barrier and glial cells in white-matter lesions in cerebrovascular and Alzheimer’s disease patients. Stroke. 1996;27:2069–74.
Montagne A, Nation DA, Pa J, Sweeney MD, Toga AW, Zlokovic BV. Brain imaging of neurovascular dysfunction in Alzheimer’s disease. Acta Neuropathol. 2016;131:687–707.
Kaya M, Kalayci R, Küçük M, Arican N, Elmas I, Kudat H, et al. Effect of losartan on the blood–brain barrier permeability in diabetic hypertensive rats. Life Sci. 2003;73:3235–44.
Ganten D, Hermann K, Bayer C, Unger T, Lang RE. Angiotensin synthesis in the brain and increased turnover in hypertensive rats. Science. 1983;221:869–71.
Guyenet P. The sympathetic control of blood pressure. Nat Rev Neurosci. 2006;7:335–46.
Reaux A, Fournie-Zaluski MC, David C, Zini S, Roques BP, Corvol P, et al. Aminopeptidase A inhibitors as potential central antihypertensive agents. Proc Natl Acad Sci USA. 1999;96:13415–20.
Gao J, Marc Y, Iturrioz X, Leroux V, Balavoine F, Llorens-Cortes C. A new strategy for treating hypertension by blocking the activity of the brain renin-angiotensin system with aminopeptidase A inhibitors. Clin Sci. 2014;127:135–48.
Tan J, Wang JM, Leenen FH. Inhibition of brain angiotensin converting enzyme by peripheral administration of trandolapril versus lisinopril in Wistar rats. Am J Hypertens. 2005;18:158–64.
Perret‐Guillaume C, Joly L, Jankowski P, Benetos A. Benefits of the RAS blockade: clinical evidence before the ONTARGET study. J Hypertens Suppl. 2009;27:S3–7.
Takeda S, Sato N, Takeuchi D, Kurinami H, Shinohara M, Niisato M, et al. Angiotensin receptor blocker prevented β-amyloid-induced cognitive impairment associated with recovery of neurovascular coupling. Hypertension. 2009;54:6.
Pelisch N, Hosomi N, Ueno M, Nakano D, Hitomi H, Mogi M, et al. Blockade of AT1 receptors protects the blood–brain barrier and improves cognition in Dahl salt-sensitive hypertensive rats. Am J Hypertens. 2011;24:362–8.s.
Elkahloun AG, Hafko R, Saavedra JM. An integrative genome-wide transcriptome reveals that candesartan is neuroprotective and a candidate therapeutic for Alzheimer’s disease. Alzheimers Res Ther. 2016;28:5.
Davies NM, Kehoe PG, Ben-Shlomo Y, Martin RM. Associations of antihypertensive treatments with Alzheimer’s disease, vascular dementia, and other dementias. J Alzheimers Dis. 2011;26:699–708.
O’Caoimh R, Healy L, Gao Y, Svendrovski A, Kerins DM, Eustace J, et al. Effects of centrally acting angiotensin converting enzyme inhibitors on functional decline in patients with Alzheimer’s disease. J Alzheimers Dis. 2014;40:595–603.
Sink KM, Leng X, Williamson J, Kritchevsky SB, Yaffe K, Kuller L, et al. Angiotensin-converting enzyme inhibitors and cognitive decline in older adults with hypertension: results from the Cardiovascular Health Study. Arch Intern Med. 2009;169:1195–202.
Gao Y, O’Caoimh R, Healy L, Kerins DM, Eustace J, Guyatt G, et al. Effects of centrally acting ACE inhibitors on the rate of cognitive decline in dementia. BMJ Open. 2013;3:e002881.
Tzourio C, Anderson C, Chapman N, Woodward M, Neal B, MacMahon S, et al. PROGRESS Collaborative Group. Effects of blood pressure lowering with perindopril and indapamide therapy on dementia and cognitive decline in patients with cerebrovascular disease. Arch Intern Med. 2003;163:1069–75.
Takakura S, Satoh Y, Satoh H, Mori J, Kohsaka M. Effects of nilvadipine on regional cerebral blood flow and skin blood flow in anesthetized cats. Arch Int Pharmacodyn Ther. 1992;319:38–48.
Anderson C, Teo K, Gao P, Arima H, Dans A, Unger T, et al. ONTARGET and TRANSCEND Investigators. Renin-angiotensin system blockade and cognitive function in patients at high risk of cardiovascular disease: analysis of data from the ONTARGET and TRANSCEND studies. Lancet Neurol. 2011;10:43–53.
Hanyu H, Hirao K, Shimizu S, Sato T, Kiuchi A, Iwamoto T. Nilvadipine prevents cognitive decline of patients with mild cognitive impairment. Int J Geriatr Psychiatry. 2007;22:1264–6.
Amenta F, Lanari A, Mignini F, Silvestrelli G, Traini E, Tomassoni D. Nicardipine use in cerebrovascular disease: a review of controlled clinical studies. J Neurological Sci. 2009;283:219–23.
Paris D, Quadros A, Humphrey J, Patel N, Crescentini R, Crawford F, et al. Nilvadipine antagonizes both A beta vasoactivity in isolated arteries, and the reduced cerebral blood flow in APPSW transgenic mice. Brain Res. 2004;999:53–61.
Edvinsson L, Johansson BB, Larsson B, MacKenzie ET, Skärby T, Young AR. Calcium antagonists: effects on cerebral blood flow and blood–brain barrier permeability in the rat. Br J Pharm. 1983;79:141–8.
Nukhet Turkel A, Ziya Ziylan Y. Protection of blood–brain barrier breakdown by nifedipine in adrenaline-induced acute hypertension. Int J Neurosci. 2004;114:517–28.
Laurens C, Abot A, Delarue A, Knauf C. Central effects of beta-blockers may be due to nitric oxide and hydrogen peroxide release independently of their ability to cross the blood–brain barrier. Front Neurosci. 2019;13:33.
Gelber RP, Ross GW, Petrovitch H, Masaki KH, Launer LJ, White LR. Antihypertensive medication use and risk of cognitive impairment: the Honolulu-Asia Aging Study. Neurology. 2013;81:888–95.
Khachaturian AS, Zandi PP, Lyketsos CG, Hayden KM, Skoog I, Norton MC, et al. Antihypertensive medication use and incident Alzheimer disease: the Cache County Study. Arch Neurol. 2006;63:686–92.
Lenzsér G, Kis B, Bari F, Busija DW. Diazoxide preconditioning attenuates global cerebral ischemia-induced BBB permeability. Brain Res. 2005;1051:72–80.
Nishioku T, Takata F, Yamauchi A, Sumi N, Yamamoto I, Fujino A, et al. Protective action of indapamide, a thiazide-like diuretic, on ischemia-induced injury and barrier dysfunction in mouse brain microvascular endothelial cells. J Pharm Sci. 2007;103:323–7.
Buttler L, Jordão MT, Fragas MG, Ruggeri A, Ceroni A, Michelini LC. Maintenance of blood–brain barrier integrity in hypertension: a novel benefit of exercise training for autonomic control. Front Physiol. 2017;8:1048.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Katsi, V., Marketou, M., Maragkoudakis, S. et al. Blood–brain barrier dysfunction: the undervalued frontier of hypertension. J Hum Hypertens 34, 682–691 (2020). https://doi.org/10.1038/s41371-020-0352-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41371-020-0352-2
- Springer Nature Limited
This article is cited by
-
Arterial hypertension in the chronic evolution of migraine: bystander or risk factor? An overview
The Journal of Headache and Pain (2024)
-
Methylglyoxal, a highly reactive dicarbonyl compound, as a threat for blood brain barrier integrity
Fluids and Barriers of the CNS (2023)
-
Predisposition to cortical neurodegenerative changes in brains of hypertension prone rats
Journal of Translational Medicine (2023)
-
Contributions of neuroimaging to the knowledge of the relationship between arterial hypertension and cognitive decline
Hypertension Research (2023)
-
Blood–Brain Barrier Dysfunction in Hypertensive Disorders of Pregnancy
Current Hypertension Reports (2023)