
Abstract
The present work highlights the versatility of a TiO2-Al2O3 mixed oxide bearing highly dispersed gold nanoparticles that was applied in the CO oxidation reaction at room temperature. The TiO2, Al2O3, and TiO2-Al2O3 supports were synthesized by the sol–gel method, while gold nanoparticles were added by the deposition–precipitation with urea method using a theoretical Au loading of 2 wt.%. A promotional effect of the TiO2-Al2O3 support on the activity of gold catalysts with respect to TiO2 and Al2O3 was observed; Au/TiO2-Al2O3 showed outstanding CO oxidation, being active from 0 °C and stable throughout a 24-h test. As for the alumina content (5, 10, and 15 wt.%) in TiO2, it improved the textural properties by retarding the crystal growth and anatase–rutile phase transformation of TiO2, suppressing the deposition of carbon on the catalyst surface and stabilizing the Au nanoparticles even at high temperatures. Gold was highly dispersed with nanoparticle sizes ranging from 1 to 2 nm when H2 was used to treat thermally the Au/TiO2-Al2O3, Au/TiO2, and Au/Al2O3 materials. In addition, the XPS technique helped elicit that Au0 and Au1+ boosted their interaction with the TiO2, Al2O3, and TiO2-Al2O3 supports by means of charge transfer, which resulted in outstanding CO oxidation activity from 0 °C. Likewise, the key factors that control the peculiar catalytic performance in the CO oxidation reaction are discussed, which represents a step forward in the versatility behavior of gold catalysts supported on mixed oxide catalysts.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Gold catalysts have been extensively studied in reactions such as low temperature CO oxidation, water gas shift (WGS), and preferential oxidation (PROX) (Romero-Sarria et al. 2008; Boaro et al. 2009; Avgouropoulos et al. 2008), where the activity of gold-based catalysts is originated by the intimate contact boundary between small gold particles and oxide supports (Haruta et al. 1987; Soares et al. 2003). As for TiO2-Al2O3 mixed oxide catalysts, they have been tested in many important industrial processes, for they have shown remarkable advantages with respect to the single metal oxides such as improved high-temperature stability, stronger surface acidity (Lewis or Brönsted), and enhanced textural properties (Tavizón-Pozos et al. 2016; Duan et al. 2009). In this context, the TiO2-Al2O3 mixed oxides have been employed in catalysis processes such as selective oxidation reactions, NOx catalytic reduction, hydrodesulfurization (Galindo and de Los Reyes 2007; Reddy et al. 2006; Camposeco et al. 2015); in photocatalysis; and in self-cleaning processes (Wu et al. 2008; Bakhshayesh et al. 2012). Regarding the modification of TiO2 by Al2O3 addition, it was found that enhanced reducibility and dispersion of noble metals, which has resulted in more active sites for heterogeneous catalysis, benefitted the catalytic properties with respect to the single Al2O3 and TiO2 supports (Huang et al. 2008). For the addition of noble metals to this mixed oxide, the sol–gel, deposition–precipitation, and hydrothermal methods have been used (Galindo and de Los Reyes 2007; Zanella and Louis 2005; Camposeco et al. 2015). From these options, the deposition–precipitation with urea method allows the synthesis of small (1–3 nm) and well-dispersed Au particles without impurities, and consequently high active catalysts for oxidation reactions, especially at low temperatures. Recently, other methods such as the biosynthesis of AuNPs by using plant extracts have offered an interesting way to obtain gold NPs, especially because they are nontoxic, simple, greener, and cost-effective synthesis methods (Hassanisaadi et al. 2021). However, the gold particle sizes obtained by biosynthesis are larger than those achieved by depositio–precipitation with urea, which when deposited on oxides would affect the catalytic activity by displacing the CO oxidation to higher reaction temperatures.
It is known that supported gold nanoparticles are active in the oxidation of carbon monoxide and generally, the activity depends on the support type, gold particle size, the synthesis method, and to lower extent on the oxide surface area (Haruta et al. 1987). Among the employed oxides, the TiO2 anatase phase has been widely used because it can stabilize the size of gold nanoparticles, providing a uniform distribution (Albonetti et al. 2008). In this way, previous reports have shown that gold catalysts supported on oxides such as TiO2 and Co3O4 exhibit higher activity than those supported on oxides like Al2O3 and SiO2 in the oxidation of carbon monoxide (Date et al. 2004; Okumura et al. 1998). Therefore, improvements in the textural, structural, and catalytic properties of TiO2 have been reported for mixed oxides prepared by the sol–gel method such as TiO2-MgO, TiO2-Al2O3, TiO2-SiO2, and TiO2-In2O3, showing strong acidities, high specific surface areas, and enhanced catalytic activities (Bokhimi et al. 1999; Morán-Pineda et al. 2002; Glez et al. 2009; Lopez et al. 2000). Moreover, it has been stated in previous works that the Au/γ-Al2O3 system is a poor catalyst for CO oxidation (Gavrila et al. 2006). In contrast, earlier works have reported that the deposition of gold on Al2O3/TiO2 produced highly stable catalysts, which preserved high activity in the CO oxidation (Wenfu et al. 2008).
CO oxidation is one of the most extensively studied reactions, because it is considered an interesting probe reaction and it is a pivotal reaction for cleaning air and lowering automotive emissions, issues that have grown exponentially during the last years (Valange and Védrine 2018). Therefore, the high activity of gold nanoparticles, with sizes lower than 5 nm supported on reducible metal oxides (as TiO2 or TiO2-Al2O3 mixed oxide), in CO oxidation is due to their ability to dissociate molecular oxygen at low temperatures. Therefore, in this work, Au/TiO2, Au/γ-Al2O3, and Au/TiO2-Al2O3 were prepared with similar gold loadings by the deposition–precipitation with urea method. Al2O3 loadings of 5, 10, and 15 wt.% were chosen to improve the textural properties of TiO2 in order to increase the specific surface and dope, at some extent, TiO2.
The aim of this work was to compare the catalytic behavior pattern of Au/TiO2-Al2O3 with those displayed by Au supported on TiO2 and γ-Al2O3 single oxides in order to understand the key factors that control the peculiar catalytic performance in the CO oxidation reaction of gold catalysts supported on a mixed oxide. The characterization of the catalysts indicated that the interaction between gold nanoparticles and the support, the dispersion of gold, and the presence of Au1+ and Au0 species improved remarkably the CO oxidation performance.
Experimental
Preparation of the nanostructured catalysts
TiO2, γ-Al2O3, and TiO2-Al2O3 support nanoparticles were synthesized by the sol–gel method. The sol–gel titania and titania–alumina oxides were prepared as follows: the required quantities of aluminum isopropoxide (Aldrich 99.99%) and 18 mL of titanium isopropoxide (Aldrich 97%) were simultaneously added to a solution containing 36 mL of distilled water and 100 mL of 2-propanol (Baker 99%). Then, the gelling solution was refluxed at 80 °C under constant stirring. The quantities of aluminum isopropoxide per titanium isopropoxide were calculated to provide 5, 10, and 15 wt.% loadings of alumina in the final TiO2-Al2O3 mixed-oxide. In addition, by using the same procedure, alumina samples with TiO2 contents of 5 and 15 wt.% in the final mixed oxide, labeled as Al2O3-5TiO2 and Al2O3-15TiO2, were prepared by using titanium isopropoxide over aluminum isopropoxide as Ti and Al precursors for comparison purposes during the catalytic test. Additionally, bare sol–gel TiO2 was prepared in the same way. The formation of the gel was obtained with controlled hydrolysis-condensation reactions of titanium isopropoxide at 70 °C for 24 h at pH 3 with HNO3 and 18 mL of deionized distilled H2O. Afterwards, the samples were evaporated in a rotary evaporator at 80 °C and then dried in an oven for 16 h at 80 °C. Finally, the excess of solvent and water was removed by annealing the materials in static air at 500 °C for 4 h with a heating program rate of 3 °C/min. As for γ-Al2O3, it was prepared by the sol–gel method using aluminum isopropoxide (Aldrich 99.99%) in the presence of 2-propanol (Baker 99%); the mixture was heated to 70 °C and the pH adjusted to 3 with HNO3; then, 18 mL of deionized distilled H2O was added dropwise to the solution to perform a hydrolysis reaction; after vigorous stirring for 1 h, the gel was formed. Then, the solvents were evaporated in a rotary evaporator at 80 °C and the sample was dried in an oven for 16 h at 80 °C. To obtain the γ-Al2O3 sample, the dried sample was calcined in static air at 500 °C.
Preparation of Au nanoparticles on the TiO2, γ-Al2O3, and TiO2–Al2O3 supports
Gold nanoparticles were obtained by the deposition–precipitation with urea (DPU) method. The gold precursor, HAuCl4 and urea, were dissolved in distilled water. The gold loading was 2 wt.% and the samples were labeled as Au/TiO2, Au/Al2O3, and Au/TiO2–Al2O3. For the preparation of the Au samples, 250 mg of TiO2, γ-Al2O3, or TiO2-Al2O3 (dried at 80 °C) was added to the HAuCl4 and urea solution under constant stirring; thereafter, the suspension temperature was increased up to 80 °C and stirring was kept constant for 16 h. After the DPU procedure, all the samples were centrifuged, washed four times with water at 50 °C and centrifuged and dried under vacuum for 2.5 h at 80 °C; finally, the materials were annealed at 500 °C for 4 h under either air or hydrogen flow.
Carbon monoxide catalytic oxidation test
In a flow reactor, the CO oxidation reaction was studied at atmospheric pressure with a light-off test from 20 to 300 °C, using 40 mg of sample that was first activated in situ with 40 mL•min−1 of either air or hydrogen with a heating rate of 2 °C min−1, between 300 and 500 °C in a tubular reactor (ID = 0.001 m; L = 0.035 cm) with a porous quartz frit disk placed in the middle of the tube to support the catalyst, using a RIG-150 micro reactor system by in situ research instruments. The feed gas mixture (1 vol. % of CO and 1 vol. % of O2 balanced with N2) was introduced with a total flow rate of 100 mL•min−1 and a heating rate of 2 °C min−1. The gases were analyzed with an online gas chromatograph (7820A, Agilent Technologies) equipped with a FID detector, HP Plot Q column, and a methanizer; the GC method conditions used for the analysis were the following: in: Front inlet N2, out: back detector FID, 35 °C, 9.5636 psi, flow of 1.4 mL•min−1, average velocity of 28.96 cm•min−1, holdup time of 1.726 min, and run time of 5 min.
In situ characterization techniques
CO adsorption was tracked down by FTIR spectroscopy in a Nicolet 670-FT-IR spectrophotometer equipped with a Praying Mantis for DRIFT spectroscopy and a temperature reaction chamber by Harrick. In each experiment, approximately 25 mg of sample was packed in the sample holder and pretreated in situ under H2 flow (30 mL•min−1, heating rate of 2 °C•min−1) up to the chosen temperature. After the thermal treatment, the sample was cooled down to room temperature under the same gas flow and then purged with N2 before the introduction of 5% of CO in N2 (30 mL•min−1). A spectrum registered under N2 was used as reference; then, several spectra were recorded under CO flow until the band intensity was stable; afterwards, the temperature was increased under CO; spectra were obtained at different increasing temperatures.
Ex situ characterization techniques
X-ray diffraction patterns were acquired at room temperature with Cu Kα radiation in a Bruker Advance D-8 diffractometer having a theta–theta configuration and a graphite secondary-beam monochromator. The data were collected for scattering angles (2θ) ranging from 4 to 80° with a step size of 0.01° for 2 s per point.
Images of the samples were obtained by HR-TEM analyses, which were performed with a JEOL 2200FS microscope operating at 200 kV and equipped with a Schottky-type field emission gun and an ultrahigh resolution pole piece (Cs = 0.5 mm, point-to-point resolution, 0.190 nm). The textural properties were obtained by means of an ASAP-2000 analyzer from micromeritics. The specific surface area (SBET) was calculated from the Brunauer–Emmett–Teller (BET) equation from N2 physisorption at 77 K.
H2-TPR profiles were performed on a RIG-150 unit under flow of a 10% H2/Ar gas mixture (25 mL•min−1) with a heating rate of 10 °C min−1 from room temperature to 800 °C.
XPS analyses were carried out with a Thermo VG Scientific Escalab 250 spectrometer equipped with a hemispherical electron analyzer and an Al Kα radiation source (1486.6 eV) powered at 20 kV and 30 mA, respectively. The binding energy was determined by using carbon C (1 s) as reference line (284.6 eV). The spectrometer was operated at pass energy of 23.5 eV, and the base pressure in the analysis chamber was maintained in the order of 3 × 10−8 mbar. Deconvolution peak fitting was done by using XPSPEAK 41 with Shirley background.
Results and discussion
X-ray diffraction
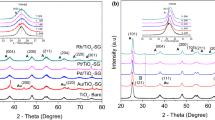
In order to investigate the structure of the Au/TiO2, Au/Al2O3, and Au/TiO2-15Al2O3 mixed-oxide catalysts, X-ray diffraction patterns of the as-prepared catalysts are presented in Fig. 1. The diffraction peaks at 2θ = 19.44° (111), 37.59° (311), 39.47° (222), 45.84° (400), and 67.00° (440) are assigned to γ-Al2O3, corresponding to the standard (JCPDS 010–0425). Likewise, for the Au/TiO2 and Au/TiO2-15Al2O3 catalysts, diffraction peaks at 2θ = 25.37° (101), 37.80° (004), 48.04° (200), 53.89° (105), 62.68° (204), 68.76° (116), 70.30° (220), and 75.02° (215), corresponding to the standards (JCPDS: 21–1272) are assigned to the TiO2 anatase phase. For the Au/TiO2 and Au/TiO2-15Al2O3 catalysts, diffraction peaks ascribed to rutile and brookite were not detected as the main phase of TiO2, suggesting that calcination at 500 °C leads only to the existence of the TiO2 anatase phase. Gold peaks were not observed according to (JPDCS 04–0784) due to the high dispersion and low Au loading (2 wt.%) used in the three different supports. In contrast, it is apparent that for the Au/TiO2-15Al2O3 catalyst annealed at 500 °C, amorphous structures are displayed with respect to Au/TiO2 catalysts, which showed higher crystallinity; so, the addition of Al2O3 suppressed the transition of anatase to rutile in agreement with a previous work (Lakshmanan et al. 2014; Morán-Pineda et al. 2002). Also, in Fig. 1, it is observed that the addition of Al2O3 decreased the TiO2 crystallite size, which was corroborated by the Scherrer equation at 2θ = 25.37° (101), obtaining for the Au/TiO2 catalyst a crystallite size of 15 nm, while for the Au/TiO2-15Al2O3 mixed-oxide catalyst, it was 7 nm. Therefore, this result confirms that the presence of alumina on TiO2 allows an improvement on the textural and structural properties.

X-ray diffraction patterns of the Au/TiO2, Au/TiO2-15Al2O3, and Au/Al2O3 catalysts treated at 500 °C in H2
H2/TPR
The Au/TiO2, Au/Al2O3, and Au/TiO2-15Al2O3 mixed-oxide catalysts were analyzed by temperature programmed reduction as shown in Fig. 2. The Au/Al2O3 catalyst showed one peak related to gold reduction (Au3+ to Au0) at 180 °C, suggesting that the reduction took place in the temperature interval starting at 110 and ending at 220 °C, in agreement with a previous work (Ma and Dai 2011). In this way, Au/TiO2 presented two peaks at 145 and 400 °C; the first intense peak is related to the reduction of Au3+ to Au0 species, in agreement with an earlier work (Bouslama et al. 2012). The broad and low intensity peak at 400 °C indicates the reduction, at least at some extent, from Ti4+ to Ti3+ of the support. The Au/TiO2-15Al2O3 mixed-oxide catalysts displayed the lowest temperature reduction peak at 95 °C, indicating the reduction of Au3+ to Au0 species, with respect to Au/TiO2 and Au/Al2O3, highlighting that the addition of gold to the TiO2-15Al2O3 mixed oxide fosters a strong interaction with regard to the Au/TiO2 and Au/Al2O3 catalysts (see Fig. 2); so, in the mixed sample, the reduction of gold is carried out at lower temperatures and the interaction between Au and the TiO2-Al2O3 support is improved. Earlier research works have found comparable effects after adding gold to mixed oxides, detecting reduced gold species at lower temperatures (Oliveira et al. 2013). The Au/TiO2-15Al2O3 mixed-oxide catalysts displayed an additional broad reduction peak with maximum at 540 °C, which may be associated with the interaction between TiO2 and Al2O3, which modifies the reduction temperature of Ti4+ to Ti3+.

H2/TPR profiles for the Au/TiO2, Au/TiO2-15Al2O3, and Au/Al2O3, catalysts
TEM
According to the TEM analysis, the Au/TiO2, Au/Al2O3, and Au/TiO2-15Al2O3 mixed-oxide catalysts had a narrow particle size distribution with mean particle sizes of 2.0, 1.8, and 1.6 nm, respectively; see frequency histograms in Fig. 3A–C. In order to obtain larger gold particles in the Au/Al2O3 catalysts to study the effect of the gold particle size on the CO oxidation reaction, for the Au/Al2O3 catalyst, an additional Au/Al2O3 sample was prepared following the procedure previously described in the experimental section, but the HAuCl4 solution was first put in contact with 0.1 M of NaBH4 then mixed with urea and Al2O3 to carry out the DPU method. This sample was labeled as Au/Al2O3-B. Subsequently, this sample was also thermally treated in H2 at 500 °C. Under these thermal treatment conditions, the average gold particle size was 5.2 nm, Fig. 3D. Due to the different particle sizes, the gold dispersion of the catalysts thermally treated in hydrogen was 60, 63, and 68% for the Au/TiO2, Au/Al2O3, and Au/TiO2-15Al2O3 catalysts, respectively. Likewise, the mean semi-spherical particle size, gold dispersion, and gold loading of the catalysts are summarized in Table 1. As for the calculated value of the crystalline interplanar distance, it was equal to 0.20 nm, which corresponds to gold (111) lattice planes (JCPDF 04–0784); see Fig. 3A and C. The calculated value of the crystalline interplanar distance was 0.2 nm, which corresponds to gold (111) and 0.19 nm that is associated with (200) lattice planes (JCPDF04-0784); see Fig. 3B. Moreover, the lattice image of the support confirmed that the TiO2 anatase crystals were oriented principally into the (101) interplanar spacing of 0.35 nm; see inset in Fig. 3A.

HAADF-STEM images of the catalysts annealed in hydrogen at 500 °C: A Au/TiO2, B Au/TiO2-15Al2O3, C Au/Al2O3 and D Au/Al2O3-B catalysts, and corresponding particle size frequency histograms
XPS
The electronic structure and chemical composition of the Au/TiO2, Au/Al2O3, and Au/TiO2-15Al2O3 mixed-oxide catalysts were elicited by XPS analysis. Figure 4 shows the core-level high resolution XPS spectra of Ti 2p, Al 2p, O 1s, and Au 4f regions for all the catalysts. For the Au/Al2O3 and Au/TiO2-15Al2O3 catalysts, the Al 2p spectrum shows well-separated oxide and metal peaks at 73.6 and 76.5 eV, respectively; in addition, a binding energy shift and an outstanding intensity increase of the Al 2p peak are observed (Fig. 4) with regard to the Au/TiO2-15Al2O3 mixed-oxide catalysts. The O 1s spectra were deconvoluted as three characteristic species at 530.1, 531.2, and 532.4 eV; the first peak (I) is related to lattice oxygen present in TiO2 and Al2O3; the second peak (II) is due to OH groups; and the third small peak (III) stemmed from both organic carbon contaminants and H2O according to previous studies (Gualteros et al. 2019); furthermore, in all the catalysts, an extra peak (IV) at 530.6 eV attributed to (Au-O) species, according to previous works (Masoud et al. 2019), was identified. In Fig. 4, a slight shift in the O 1s peaks for the Au/TiO2-Al2O3 mixed oxide occurred with regard to the Au/TiO2 and Au/Al2O3 catalysts, which may have been due to oxygen atom substitution in either Al–O–Al or Ti–O–Ti bonds.

XPS spectra of the Ti 2p, Au 4f, O 1s, and Al 2p deconvolution for the Au/TiO2, Au/TiO2-15Al2O3, and Au/Al2O3 catalysts thermally treated under H2 at 500 °C
Likewise, the Au 4f characteristic peaks are located at 85.8 eV (Au 4f 5∕2) and 82.15 eV (Au 4f 7∕2) according to the deconvolution of gold species (Fig. 4), where Au0 (81.5 eV) and Au1+ (82.1 eV) are the foremost species present in the Au/TiO2, Au/Al2O3, and Au/TiO2-15Al2O3 mixed-oxide catalysts (see Table 2); previous studies (Masoud et al. 2019; Tsai et al. 2009) have shown that the existence of Au1+ species has a great influence on the CO oxidation as observed in the Au/TiO2-15Al2O3 catalysts, which displayed the highest contribution of Au1+ species with regard to the Au/TiO2 and Au/Al2O3 catalysts; therefore, outstanding CO oxidation at 0 °C was observed in the mixed-oxide catalysts; see the catalytic activity section below.
As for the Au/TiO2 and Au/TiO2-15Al2O3 catalysts, they showed Ti 2p photoelectron peaks at 458.5 and 464.4 eV corresponding to Ti 2p3/2 and Ti 2p1/2, respectively, where the position of Ti 2p peaks was constant for Au/TiO2 and Au/TiO2-15Al2O3, indicating that mainly the Ti4+ oxidation state was found in both catalysts. Meanwhile, the peaks at 455.6 and 461.4 eV due to presence of Ti 2p3/2 and Ti 2p1/2 suggest the existence of Ti3+ species in the Au/TiO2 and Au/TiO2-Al2O3 catalysts, in agreement with previous studies (Mendialdua et al. 1995). The intensity of this peak is increased by the Al2O3 addition; see Table 2. This increase can been due to a change in coordination environment, electronic properties, and bonding geometries (Zhang and Yates 2012).
CO adsorption
The in situ DRIFTS spectra of CO adsorbed on the Au/TiO2, Au/Al2O3, and Au/TiO2-15Al2O3 mixed-oxide catalysts are shown in Fig. 5A–C. According to the literature, CO adsorption has revealed two bands at ~ 2172 and 2118 cm−1; the band at ~ 2172 was due to CO adsorbed on Ti4+-CO acid sites of the TiO2 supports (β), and the second one at ~ 2118 has been assigned to CO linearly bound to Au0-CO species present in the Au/TiO2, and Au/TiO2-15Al2O3 mixed-oxide catalysts (Saavedra et al. 2018; Tsai et al. 2009; Yang et al. 2006); see Fig. 5. For the Au/Al2O3 catalyst, the band observed at ~ 2172 is due to CO adsorbed on the Lewis acid sites of the Al2O3 surface. In the Au/TiO2 and Au/TiO2-15Al2O3 mixed-oxide catalysts, the Au0-CO band at ~ 2118 diminished in intensity as the temperature increased and a blue shifting wave number at ~ 2090 is observed. Furthermore, an additional band appeared at around ~ 2016 cm−1, which represents active sites produced during the CO adsorption. This band, assigned to Auδ+-CO species, is related to the symmetric and asymmetric vibrations of gem-dicarbonyl doublet-CO on the positively charged Au1+ atom (Guan et al. 2016), suggesting the binding formation of Au1+-CO, Auδ+-CO, or Au3+-CO (Tsai et al. 2009; Yang et al. 2006) on the Au/TiO2 and Au/TiO2-15Al2O3 mixed-oxide catalysts. In this way, Fig. 5B shows that for Au/Al2O3, two bands at ~ 2172 and 2118 cm−1 are present; the band at ~ 2172 is associated with CO adsorbed on the Al3+-CO acid sites of the Al2O3 supports, and the second one, noted as a weak peak at ~ 2118, is assigned to CO linearly bound to Au0 species. The weak band observed at ~ 2020 cm−1 is associated to Au1+-CO, Auδ+-CO, and Au3+-CO on Au/Al2O3 sites (Saavedra et al. 2018); this band is observed even at 0 °C, disappearing at 100 °C; see Fig. 5B. In fact, the in situ DRIFTS spectra of CO adsorbed on the Au/TiO2, Au/Al2O3, or Au/TiO2-15Al2O3 mixed-oxide catalysts demonstrate that there is a small fraction of oxidized gold sites remaining on the surface of the Au/TiO2, Au/Al2O3, and Au/TiO2-15Al2O3 mixed-oxide catalysts, where gold is mainly in the form of metal Au0; likewise, the Auδ+ sites were stable up to 100 °C in all the samples, which correlates well with the Au0/Au1+ species ratio, as corroborated by the XPS results presented above. The CO-DRIFTS profile evolution for the Au/TiO2-15Al2O3 mixed-oxide catalyst, as a function of the temperature, was totally different with respect to that displayed by the Au/TiO2 and Au/Al2O3 catalysts. These CO adsorption outcomes indicate that the gold particle absorption properties of Au/TiO2-15Al2O3 are different from those in the Au/TiO2 and Au/Al2O3 catalysts. Also, the Au1+ species are present mainly in the Au/TiO2-15Al2O3 catalyst (XPS analysis); these results suggest that oxygen can be activated at the Au1+ sites to a higher extent on the Au/TiO2-Al2O3 catalysts and increase the CO conversion at low temperatures with regard to the Au/TiO2 and Au/Al2O3 catalysts. It can be highlighted that the Au1+ species could work as a linkage that interacted with the TiO2-15Al2O3 mixed oxide, giving reactive oxygen for the carbon monoxide oxidation, in accordance with earlier studies (Trautmann and Baerns 1994).

DRIFTS of the CO desorption as a function of the temperature of the A Au/TiO2, B Au/Al2O3 and C Au/TiO2-15Al2O3 catalysts
Catalytic activity
Figure 6A shows the light-off CO oxidation curves for the Au/TiO2, Au/Al2O3, and Au/TiO2-15Al2O3 mixed-oxide catalysts treated at 500 °C under H2 atmosphere. The Au/TiO2-15Al2O3 mixed-oxide catalyst exhibited superior catalytic activity with regard to the Au/TiO2 and Au/Al2O3 catalysts (Fig. 6 and Table 1); the addition of gold nanoparticles to TiO2-15Al2O3 promoted the catalytic oxidation at 0 °C (86% of conversion), reaching 100% of conversion at 160 °C, while for Au/TiO2, 46% of conversion was achieved at 0 °C and 100% of conversion was reached at 170 °C; in the case of Au/Al2O3, 20% of conversion was achieved at 0 °C and 100% of conversion was possible at 200 °C; in this context, it is worth mentioning the peculiar behavior displayed by the Au/Al2O3 catalyst as a function of the reaction temperature, which has been related mostly to small gold nanoparticle sizes, local overheating of catalyst surfaces, local heating, and heat exchange by the presence of some steady states in the catalytic system (Grunwaldt et al. 1999; Gómez-Cortés et al. 2009; Engel and Ertl 1979), as discussed below.

CO oxidation reaction on the Au/TiO2, Au/TiO2-15Al2O3, and Au/Al2O3 catalysts for A thermally treated at 500 °C under H2 atmosphere, B thermally treated at 300 °C under H2 atmosphere
Figure 6B shows the light-off CO oxidation curves for the same samples shown in Fig. 6A (Au/TiO2, Au/Al2O3, and Au/TiO2-15Al2O3) but now thermally treated under H2 atmosphere at 300 °C instead of 500 °C. All the catalysts thermally treated at 300 °C showed lower activity than the ones thermally treated at 500 °C. The CO conversion of the Au/TiO2 catalysts at 300 °C was 18% at 0 °C, reaching 100% of CO conversion at 250 °C; for the Au/Al2O3 catalyst, the CO conversion at 0 °C was 19%, reaching 100% of CO conversion at 200 °C, while for the Au/TiO2-15Al2O3 catalyst, the CO conversion at 0 °C was 80%, reaching 100% of CO conversion at 150 °C; it is worth noting that the thermal treatment temperature had a significant impact on the Au/TiO2 catalyst, while for the Au/Al2O3 and Au/TiO2-15Al2O3 catalysts, the CO conversion also decreased, but only by 5% at 0 °C, when they were treated at 300 °C compared to those treated at 500 °C. Therefore, some works have reported that the catalytic activity in the CO oxidation displayed by supported gold particles depends on the calcination temperature and atmosphere used during the thermal treatment, showing that gold particles deposited on the TiO2 anatase support synthesized by the deposition–precipitation method grow when the annealing temperature is increased, thus making the catalysts less active (Salanov and Savchenko 1985). However, in the here studied catalysts, the highest activity at 500 °C under hydrogen thermal treatment may be related to the high dispersion of gold nanoparticles mainly in the TiO2-15Al2O3 support (Table 1). The addition of Al2O3 to TiO2 allows to stabilize the TiO2 anatase phase at 500 °C and does not allow the sintering of gold nanoparticles. So, the activation temperature is fundamental in the CO oxidation, evidencing that the CO oxidation increases when the activation temperature increases as follows: 500 > 300 °C, (Camposeco and Zanella 2022); see Fig. 6.
Impact of the TiO2-Al2O3 composition
A positive catalytic promotion when TiO2 is in a higher proportion than alumina in the Au/TiO2-Al2O3 catalysts was observed; see Fig. 7A. The highest CO conversion was obtained for the Au/TiO2-15Al2O3 catalysts, reaching a conversion of 86% at 0 °C, followed by the Au/TiO2-10Al2O3 and Au/TiO2-5Al2O3 catalysts with CO conversions of 76 and 73% at 0 °C, respectively. As expected, in the case of the Au/Al2O3-15TiO2 and Au/Al2O3-5TiO2 catalysts with higher Al2O3 content than TiO2 (15% and 5% wt.% of TiO2 in the final Al2O3-TiO2 mixed oxide, respectively), lower activity than that of the samples with higher TiO2 content was observed. The CO conversions at 0 °C reached 22 and 13% by the Au/Al2O3-15TiO2 and Au/Al2O3-5TiO2 catalysts, respectively; according to these results, the addition of small amounts of alumina (5, 10, and 15 wt.%) to TiO2 allows to delay the anatase–rutile transition, stabilizes the gold nanoparticle size, and reaches higher CO conversion at 0 °C than that displayed by catalysts with higher Al2O3 amounts. Previous studies have reported that the phase composition of the mixed oxide significantly affects the size of the gold crystallites and stabilizes their growth, thus positively affecting the catalytic activity (Zanella and Louis 2005). The combination of Al2O3 and TiO2 proved effective in the enhancement of the activity of the gold catalysts and a higher thermal treatment temperature (500 °C) could produce even more active catalysts in the CO oxidation for specific compositions.

CO conversions for the A Au/TiO2-Al2O3 catalyst with different compositions thermally treated at 500 °C under hydrogen; B Au/TiO2-15Al2O3 thermally treated at 500 °C under air or hydrogen; C Au/Al2O3 thermally treated at 500 °C under air, hydrogen, or nitrogen; D) Au/TiO2 thermally treated at 500 °C under air or hydrogen, and E stability tests for Au/TiO2-15Al2O3 thermally treated at 500 under H2 atmosphere
Effect of pretreatment gas
To observe the influence of the nature of the gas used in the pretreatment of the catalyst, a thermal treatment with either air or hydrogen was performed for the Au/TiO2-15Al2O3, Au/Al2O3, and Au/TiO2 catalysts at 500 °C; see Fig. 7B, C, and D. Earliest studies have reported that materials synthesized by the deposition–precipitation with urea method produce Au3+ species that can be reduced in air or hydrogen to Au0 in different steps (Brijaldo et al., 2014). It is shown in Fig. 7B, C, and D that the activity of the Au/TiO2, Au/Al2O3, and Au/TiO2-15Al2O3 catalysts was higher when the samples were thermally treated in hydrogen instead of in air. These results confirm that the gas used in the thermal treatment (hydrogen or air) significantly affects the gold particle size and therefore, the metal dispersion. As for the Au/Al2O3 catalyst, it was also treated with N2 (Fig. 7C); in this case, the peculiar CO conversion behavior as a function of the temperature was less important. It is worth mentioning that even if N2 is an inert gas, the reduction of gold is expected when the temperature of the sample is increased (Giorgio et al., 2004); however, the thermal treatment at high temperature can affect the physiochemical properties of the Au/Al2O3 catalyst. The CO conversion behavior as a function of the temperature when the Au/Al2O3 sample was thermally treated in hydrogen or air could be associated with the existence of some steady states in the catalytic system related to a slow transition from an oxygen enriched surface on Al2O3 increasing the presence of Au-OH groups that together with Al–OH− modify the oxidation pathway and also to Au0-Au1+ transitions during the heating of the sample when H2 or air treatment was used, while in the case of a thermal treatment in N2, almost a typical light-off behavior is observed, resulting in the narrowing of the hysteresis curve compared to the samples thermally treated in H2 or air. Figure 7E shows that Au/TiO2-15Al2O3 thermally treated under H2 atmosphere at 500 °C displayed an initial decrease in the CO conversion for the first 2 h of reaction at constant reaction temperature (15 °C) and then remained stable, showing CO conversion of 80% after 24 h under reaction conditions.
Hysteresis effect
In heterogeneous catalysis, hysteresis loops are frequently observed in heating–cooling reaction cycles, which mean that conversion does not match the heating and cooling processes. This interesting behavior is observed in the Au/Al2O3 and Au/TiO2-15Al2O3 catalysts, as it can be seen in Fig. 8, while it is not observed for Au/TiO2 sample. There are many factors that can induce a hysteresis loop such as changes in particle size and gas composition and impurities in the catalysts, among others (Yablonskii et al., 1996). In our case, it seems to be related to the presence of Al2O3 and gold nanoparticle size.

CO oxidation profiles for the Au/TiO2-15Al2O3 mixed-oxide A; Au/TiO2-10Al2O3 mixed oxide B; and Au/Al2O3 catalyst thermally treated under hydrogen at 300 °C C and at 500 °C D
The results obtained for the Au/Al2O3 and Au/TiO2-15Al2O3 catalysts clearly point toward the impact of the gold nanoparticle size and Al2O3 content on the light-off temperature and particularly on the hysteresis loops. As observed in Fig. 8A–D, an inverse hysteresis loop appears when the gold nanoparticles are smaller than 2 nm as is the case of the Au/Al2O3 and Au/TiO2-15Al2O3 catalysts; likewise, with the increase in particle size and decrease in alumina content in the catalyst (Au/TiO2-5Al2O3), the catalytic activity increases and the hysteresis effect is almost not observed; see Fig. 8A. The current study shows that the nanoparticle size is found between 1.8 and 2 nm when Al2O3 (between 5 and 15 wt.%) is added to TiO2. As previously mentioned, Au/TiO2-Al2O3 is more active at 0 °C than the Au/TiO2 catalyst. Therefore, the results obtained in these binary and ternary catalysts are important for understanding the hysteresis phenomenon because it is not well developed in the literature. In the present investigation, meaningful experiments were performed to try to understand the effect of parameters such as CO/O2 ratio, particle size, structure, and nature of the catalyst on the hysteresis effects. It is observed that the hysteresis effect on the CO oxidation employing the Au/TiO2-Al2O3 catalyst is highly dependent on the gold particle size; for the catalysts with average particle size of 1.8 nm, a hysteresis effect is observed, while for a catalyst with larger particle size (5.2 nm), almost no hysteresis effect was observed; see Fig. 9A, which is in good agreement with observations reported in previous works (Casapu et al., 2016). Besides, another studied parameter was the inlet gas stoichiometric ratio (CO to O2), observing that for higher O2 concentration, a very slight hysteresis loop was obtained, while it was larger and more pronounced for lower oxygen concentrations as shown in Fig. 9B. The peculiar behavior observed at low reaction temperatures for the Au/Al2O3 catalyst may be due to the fact that at low temperatures and low O2 to CO ratios, carbon monoxide is heavily adsorbed on the catalyst surface and prevents the O2 adsorption. This means that two states are predominant, the CO and O2 coverage on the surface of the Au/Al2O3 catalysts, while for higher temperatures or high O2 to CO ratios, the Au/Al2O3 catalyst surface is saturated with oxygen atoms, and therefore, the CO reaction proceeds more rapidly; this result is in agreement with Newton et al. (2017), concluding that both the particle size and O2 to CO ratio are factors that influence the behavior of the hysteresis effect in Au/TiO2-Al2O3 and above all in the Au/Al2O3 catalyst.

A CO oxidation profiles with different gold sizes for Au/Al2O3 and Au/Al2O3-B catalysts; and B CO oxidation at different CO/O2 ratios for Au/Al2O3 catalyst. The samples were thermally treated at 500 °C under H2 flow
Regarding the reaction mechanism, various pathways for CO oxidation have been reported (Salomons et al., 2006; Carlsson et al., 2004); however, one of the most accepted for gold on reducible supports, that can govern the CO oxidation reaction in the catalysts here studied within the low temperature region, is the one in which the support acts as the oxygen supplier (Salomons et al., 2006). Figure 10 displays possible mechanisms on the surface of the catalysts thermally treated at 500 °C under H2 atmosphere, consisting in the adsorption of CO on the surface of gold, and the participation of a support lattice oxygen atom, resulting in the formation and desorption of CO2 and an oxygen vacancy. Then, the oxygen vacancy site is wrapped by O2 from the gas phase, which re-oxidizes the surface of the oxide. With respect to the reaction mechanism on the Au/Al2O3 catalyst, it is still a topic under debate; the most accepted mechanism for the CO oxidation reaction by Au catalysts is the one involving OH coming from the Al2O3 surface (Costello et al., 2002). The OH content on the surface of Al2O3 may also explain the hysteresis effect observed in Au/Al2O3 catalysts as a function of the reaction temperature.

Schematic representation of the possible mechanism for CO oxidation over reducible oxide supports
Conclusions
Au/TiO2–Al2O3, Au/TiO2, and Au/Al2O3 catalysts with highly dispersed gold species were synthesized. The Au/TiO2-15Al2O3 mixed oxide showed both the best performance in the oxidation of CO and resistance to deactivation for 24 h at 15 °C, achieving full CO conversion at 160 °C. Characterization of the catalysts by TEM and XPS revealed steady and well-dispersed gold particles whose dimensions oscillated between 1.6 and 2.0 nm, with higher contents of Au0 and Au1+ species that seemed to be the main factors for achieving outstanding CO oxidation activity, allowing the occurrence of a strong metal-support interaction and featuring scattered gold on TiO2-Al2O3. Likewise, the catalytic activity displayed by Au/Al2O3 was high due to the presence of remarkably small gold particles (1.8 nm) interacting with the Al2O3 support. The peculiar CO conversion behavior shown by the catalysts as a function of the reaction temperature became more evident with the samples having smaller gold nanoparticles and higher Al2O3 content in the mixed catalysts. As for the oxygen vacancies present in the Au/TiO2-15Al2O3 mixed oxide, it seems that they contribute to upgrade the oxygen mobility and improve the catalytic activity in CO oxidation reactions. In contrast, gold particle sizes between 4 and 7 nm in the Au/Al2O3 sample confirmed the assumption that gold on a not reducible oxide presents weak-metal-support-interaction, for it did not show hysteresis behavior. In addition, by comparing the three gold catalytic systems here studied (Au/TiO2–Al2O3, Au/TiO2, and Au/Al2O3) in the CO oxidation, it is concluded that the interaction between gold nanoparticles and the support is pivotal to enhance both the catalytic performance and gold dispersion.
Data availability
Not applicable.
References
Albonetti S, Bonelli R, EpoupaMengou J, Femoni C, Tiozzo C, Zacchini S, Trifiro F (2008) Gold/iron carbonyl clusters as precursors for TiO2 supported catalysts. Catal Today 137:483–488
Avgouropoulos G, Papavasiliou J, Ioannides T (2008) PROX reaction over CuO–CeO2 catalyst with reformate gas containing methanol. Catal Commun 9:1656–1660
Bakhshayesh AM, Mohammadi MR, Fray DJ (2012) Controlling electron transport rate and recombination process of TiO2 dye-sensitized solar cells by design of double-layer films with different arrangement modes. Electrochim Acta 78:384–391
Boaro M, Vicario M, Llorca J, de Leitenburg C, Dolcetti G, Trovarelli A (2009) A comparative study of water gas shift reaction over gold and platinum supported on ZrO2 and CeO2–ZrO2. App Catal B 88:272–282
Bokhimi X, Boldu JL, Muñoz E, Novaro O, Lopez T, Hernandez J, Gomez R, Garcia-Ruiz A (1999) Structure and composition of the nanocrystalline phases in a MgO-TiO2 system prepared via sol−gel technique. Chem Mater 11:2716–2721
Bouslama M, Amamra MC, Jia Z, Ben Amar MK, Chhor O, Brinza M, AbderrabbaVignes JL, Kanaev A (2012) Nanoparticulate TiO2–Al2O3 photocatalytic media: effect of particle size and polymorphism on photocatalytic activity. ACS Catal 2(9):1884–1892
Brijaldo MH, Passos FB, Rojas HA et al (2014) Hydrogenation of m-dinitrobenzene over Pt supported catalysts on TiO2–Al2O3 binary oxides. Catal Lett 144:860–866
Camposeco R, Zanella R (2022) Activity boosting of gold nanoparticles supported on V2O5/TiO2 nanostructures for CO oxidation at low temperature. Catal Today 392–393:49–59
Camposeco R, Castillo S, Mejía-Centeno I, Navarrete J, Nava N (2015) Boosted surface acidity in TiO2 and Al2O3-TiO2 nanotubes as catalytic supports. Appl Surf Sci 356:115–123
Carlsson P, Osterlund L, Thormahlen P, Palmqvist A, Fridell E, Jansson J, Skoglundh MA (2004) Transient in situ FTIR and XANES study of CO oxidation over Pt/AlO catalysts. J Catal 226:422–434
Costello CK, Kung MC, Oh HS, Wang Y, Kung HH (2002) Nature of the active site for CO oxidation on highly active Au/γ-Al2O3. Appl Catal A 232(1–2):159–168
Casapu M, Fischer A, Gänzler AM, Popescu R, Crone M, Gerthsen D, Türk M, Grunwaldt JD (2016) Origin of the normal and inverse hysteresis behavior during CO oxidation over Pt/Al2O3. ACS Catal 7:343–355
Date M, Okumura M, Tsubota S, Haruta M (2004) Vital role of moisture in the catalytic activity of supported gold nanoparticles. Angew Chem Int Ed 43(16):2129–2132
Duan A, Li R, Jiang G, Gao J, Zhao Z, Wan G, Zhang D, Huang W, Chung KH (2009) Hydrodesulphurization performance of NiW/TiO2-Al2O3 catalyst for ultra clean diesel. Catal Today 140(3–4):187–191
Engel T, Ertl G (1979) Elementary steps in the catalytic oxidation of carbon monoxide on platinum metals. In: Advances in catalysis, vol 28. Academic Press, Cambridge, pp 1–78
Galindo I, de Los Reyes J (2007) Effect of alumina–titania supports on the activity of Pd, Pt and bimetallic Pd–Pt catalysts for hydrorefining applications. Fuel Process Technol 88(9):859–863
Gavrila D, Georgakab A, Loukopoulosb V et al (2006) On the mechanism of selective CO oxidation on nanosized Au/γ-Al2O3 catalysts. Gold Bull 39:192–199
Glez V, Castillo S, Morán-Pineda M, Zanella R, Gomez R (2009) Effect of TiO2 In2O3 and TiO2 Al2O3 sol-gel supports on the morphology of gold nanoparticles. J Nano Res 5:1–12
Gómez-Cortés A, Díaz G, Zanella R, Ramírez H, Santiago P, Saniger JM (2009) Au-Ir/TiO2 prepared by deposition precipitation with urea: improved activity and stability in CO oxidation. J Phys Chem C 113:9710–9720
Gualteros JAD, Garcia MAS, da Silva AGM et al (2019) Synthesis of highly dispersed gold nanoparticles on Al2O3, SiO2, and TiO2 for the solvent-free oxidation of benzyl alcohol under low metal loadings. J Mater Sci 54:238–251
Guan H, Lin J, Qiao B, Yang X, Li L, Miao S, Liu J, Wang A, Wang X, Zhang T (2016) Catalytically active Rh sub-nanoclusters on TiO2 for CO oxidation at cryogenic temperatures. Angew Chem Int 55:2820–2824
Grunwaldt JD, Maciejewski M, Becker OS, Fabrizioli P, Baiker A (1999) Comparative study of Au/TiO2 and Au/ZrO2 catalysts for low-temperature CO oxidation. J Catal 186:458–469
Haruta M, Kobayashi T, Sano H, Yamada N (1987) Novel gold catalysts for the oxidation of carbon-monoxide at a temperature far below 0 °C. Chem Lett 16:405–408
Hassanisaadi M, Bonjar GHS, Rahdar A, Pandey S, Hosseinipour A, Abdolshahi R (2021) Environmentally safe biosynthesis of gold nanoparticles using plant water extracts. Nanomaterials 11(8):2033
Huang W, Duan A, Zhao Z, Wan G, Jiang G, Dou T (2008) Ti-modified alumina supports prepared by sol–gel method used for deep HDS catalysts. Catal Today 131:314–321
Lakshmanan P, Park J, Park E (2014) Recent advances in preferential oxidation of CO in H2 over gold catalysts. Catal Surv Asia 18(2–3):75–88
Lopez T, Bosch P, Tzompantzi F, Gomez R, Navarrete J, Lopez-Salinas E, Llanos ME (2000) Effect of sulfation methods on TiO2–SiO2 sol–gel catalyst acidity. Appl Catal A 197:107–117
Ma Z, Dai S (2011) Development of novel supported gold catalysts: a materials perspective. Nano Res 4:3–32
Masoud N, Partsch T, de Jong KP (2019) Thermal stability of oxide-supported gold nanoparticles. Gold Bull 52:105–114
Mendialdua J, Casanova R, Barbaux Y (1995) XPS studies of V2O5, V6O13, VO2 and V2O3. J Electron Spectrosc Relat Phenom 71:249–261
Morán-Pineda M, Castillo S, Asomoza M, Gomez R (2002) Al2O3-TiO2 sol-gel mixed oxides as suitable supports for the reduction of NO by CO. React Kinet Catal Lett 76:75–81
Newton M (2017) Time resolved operando X-ray techniques in catalysis, a case study: CO oxidation by O2 over Pt surfaces and alumina supported Pt catalysts. Catalysts 7(2):58
Okumura M, Nakamura S, Tsubota S, Nakamura T, Azuma M, Haruta M (1998) Chemical vapor deposition of gold on Al2O3, SiO2, and TiO2 for the oxidation of CO and of H2. Catal Lett 51:53–58
Oliveira RL, Bitencourt IG, Passos FB (2013) Partial oxidation of methane to syngas on Rh/Al2O3 and Rh/Ce-ZrO2 catalysts. J Braz Chem Soc 24:68–75
Reddy BM, Rao KN, Reddy GK, Bharali PJ (2006) Characterization and catalytic activity of V2O5/Al2O3-TiO2 for selective oxidation of 4-methylanisole. Mol Catal A 253(1):44–51
Romero-Sarria F, Penkova A, Martinez LM, Centeno MA, Hadjiivanov K, Odriozola JA (2008) Role of water in the CO oxidation reaction on Au/CeO2: modification of the surface properties. Appl Catal B 84:119–124
Saavedra J, Pursell CJ, Chandler BD (2018) CO oxidation kinetics over Au/TiO2 and Au/Al2O3 catalysts: evidence for a common water-assisted mechanism. J Am Chem Soc 140(10):3712–3723
Salanov AN, Savchenko VI (1985) TD and AES studies of the interaction of oxygen with Rh(100). React Kinet Catal Lett 29:101–109
Salomons S, Votsmeier M, Hayes RE, Drochner A, Vogel H, Gieshof J (2006) CO and H2 oxidation on a platinum monolith diesel oxidation catalyst. Catal Today 117:491–497
Soares JMC, Morrall P, Crossley A, Harris P, Bowker M (2003) Catalytic and noncatalytic CO oxidation on Au/TiO2 catalysts. J Catal 219:17–24
Tavizón-Pozos, JA, Suárez-Toriello VA, de los Reyes JA, Guevara-Lara A, Pawelec B, Fierro JLG, Vrinat M, Geantet C (2016) Deep hydrodesulfurization of dibenzothiophenes over niw sulfide catalysts supported on sol-gel titania-alumina. Top Catal 59:241–251
Trautmann S, Baerns M (1994) Infrared spectroscopic studies of CO adsorption on rhodium supported by SiO2, Al2O3, and TiO2. J Catal 150:335–344
Tsai Y, Chao H, Lin H (2009) Low temperature carbon monoxide oxidation over gold nanoparticles supported on sodium titanate nanotubes. J of Mol Catal A Chem 298:115–124
Valange S, Védrine J (2018) General and prospective views on oxidation reactions in heterogeneous catalysis. Catalysts 8:483
Wenfu Y, Zhen M, Shannon M, Jian J, Edward H, Steven O, Sheng D (2008) Novel Au/TiO2/Al2O3·xH2O catalysts for CO oxidation. Catal Lett 21(3–4):209–218
Wu S, Han H, Tai Q, Zhang J, Xu S, Zhou C, Yang Y, Hu H, Chen B, Zhao X (2008) Improvement in dye-sensitized solar cells employing TiO2 electrodes coated with Al2O3 by reactive direct current magnetron sputtering. J Power Source 182(1):119–123
Yablonskii GS, Lazman MZ (1996) New correlations to analyze isothermal critical phenomena in heterogeneous catalysis reactions (“Critical simplification”, “hysteresis thermodynamics”). React Kinet Catal Lett 59:145–150
Yang J, Bai H, Tan X, Lian J (2006) IR and XPS investigation of visible-light photocatalysis-nitrogen-carbon-doped TiO2 film. App Surf Sci 253:1988–1994
Zanella R, Louis C (2005) Influence of the conditions of thermal treatments and of storage on the size of the gold particles in Au/TiO2 samples. Catal Today 107–108:768–777
Zanella R, Giorgio S, Shin CH, Henry CR, Louis C (2004) Characterization and reactivity in CO oxidation of gold nanoparticles supported on TiO2 prepared by deposition-precipitation with NaOH and urea. J Catal 222:357–367
Zhang ZJ, Yates T (2012) Band bending in semiconductors: chemical and physical consequences at surfaces and interfaces. Chem Rev 112:5520–5551
Acknowledgements
The authors want to thank the financial support provided by the Consejo Nacional de Ciencia y Tecnología (CONACYT) through the CB A1-S-18269 grant, Dirección General de Asuntos del Personal Académico-UNAM through the PAPIIT IN104022 grant.
Funding
Consejo Nacional de Ciencia y Tecnología (CONACYT) through the CB A1-S-18269 grant, Dirección General de Asuntos del Personal Académico-UNAM through the PAPIIT IN104022 grant.
Author information
Authors and Affiliations
Contributions
Roberto Camposeco: formal analysis, writing—original draft preparation, conceptualization, and validation; investigation; Rodolfo Zanella: formal analysis, investigation, writing—original draft preparation, supervision, and project administration.
Corresponding author
Ethics declarations
Ethics approval
Not applicable.
Consent for publication
Not applicable.
Consent to participate
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Responsible Editor: George Z. Kyzas
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Camposeco, R., Zanella, R. Catalytic behavior of gold nanoparticles supported on a TiO2-Al2O3 mixed oxide for CO oxidation at low temperature. Environ Sci Pollut Res 29, 76992–77006 (2022). https://doi.org/10.1007/s11356-022-21076-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-022-21076-2