
Abstract
Seven phthalate by-products were investigated for the first time, in target tissues of roach from a contaminated river of the Ile-de-France district. All parent phthalates were bioaccumulated in liver and muscle and liver contents were correlated with river concentrations (p < 0.01). All metabolites were found in liver, plasma and bile. The mono-iso-butyl phthalate (MiBP; 1.6 μg g−1 dw of liver), followed by mono-n-butyl phthalate (MnBP; 1.5 μg g−1 dw of liver) were the most abundant ones. Among the three metabolites of di-ethylhexyl phthalate (DEHP), mono (2-ethylhexyl) phthalate (MEHP) predominated in bile (15.5 ng ml−1) and liver (0.237 μg g−1 dw), whereas in plasma, it was mono (2-ethyl-5-hydroxyhexyl) phthalate – MEHHP (214 ng ml−1). In liver, MEHP/DEHP ratios ranged from 0.04 to 0.2. Among the oxidized metabolites, only mono (2-ethyl-5-oxohexyl) phthalate (MEOHP) was correlated (p < 0.05) with parent DEHP and appeared to be a more reliable marker of DEHP impact than the monoester.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The impact of micropollutants upon the biota was classically assessed through their accumulation in tissues. However, according to the chemical characteristics, namely lipophilic or amphiphilic properties, they may also undergo biodegradation. Phthalates are mainly used as plasticizers, but also as solubilizing or stabilizing agents in other applications such as detergents, building products, lubricating oils, carriers in pesticide formulations, solvents, personal care products, cosmetics, toys and some medical devices. Their worldwide production amounted 5.2 million tons in 2008 (German Federal Environmental Agency 2011). Because of their endocrine disrupting properties, they were investigated in mammals, namely, rodents (Kessler et al. 2004) and humans (Mortensen et al. 2005).
Phthalates appear to be easily degraded (Staples et al. 1997). A first step was hydrolysis in the digestive lumen leading to a monoester. As a matter of fact, Erkekoglu et al. (2010) reported that the monoesters might display the same toxic potencies as the parent compounds. Next, comes phase I transformation by oxidization through the cytochrome P-450 system in specific organs (liver and kidney) (Barron et al. 1995). Then, phase II by conjugation (generally glucuronidation) of the oxidized by-products occurs which facilitates their urinary excretion. Sometimes, the monoester might be directly conjugated (Silva et al. 2003).
As a matter of fact, phthalate fate in fish living in close contact with surface water regularly impacted by contamination from discharges of wastewater treatment plant, storm overflow or runoff, were scarcely investigated up to now. However, the incidence of intersex was examined in roach from three French rivers, including the Seine river, above and below major sewage treatment plants. Indeed, roach displaying primary oocytes in their testes were found in all the three rivers with the incidence ranging from 9 % to 21 % of the males (Minier et al. 2000). A set of bio-markers of endocrine disruption including gonado-somatic index, plasmatic vitellogenin, gonadal aromatase activity and histological parameters (oocyte diameter and gonad maturation) were studied in female roach from the Seine river by Gerbron et al. (2014). Female fish from polluted sites showed a number of reproductive alterations, including inhibited gonad maturation, reduced oocyte growth, reduced levels of plasmatic vitellogenin and low gonadal aromatase activity, highlighting the presence of endocrine disruption. Also, exposure of marine medaka (Oryzias latipes) to di-ethylhexyl phthalate (DEHP) and mono (2-ethylhexyl) phthalate (MEHP) from hatching to adulthood causes reproductive dysfunction and endocrine disruption (Ye et al. 2014)
Thus, our view was to explore the metabolism of seven widespread phthalates (dimethyl phthalate [DMP], diethyl phthalate [DEP], di-n-butyl phthalate [DnBP], di-iso-butyl phthalate [DiBP], butylbenzyl phthalate [BBP], DEHP and di-n-octyl phthalate [DnOP]) that were already quantified in some tissues of a ubiquitous freshwater fish species, the roach (Rutilus rutilus) of a highly polluted tributary of the Seine river in a previous work (Teil et al. 2012) and also frequently reported by a number of other authors. Particular attention was given to relationships between river water concentrations of parent phthalates and tissue contents of parent phthalates and seven monoesters (monomethyl phthalate [MMP], monoethyl phthalate [MEP], mono-iso-butyl phthalate [MiBP], mono-n-butyl phthalate [MnBP], monobenzyl phthalate [MBzP], mono-n-octyl phthalate [MnOP], MEHP) and two oxidized by-products from phase I biotransformation of DEHP, mono (2-ethyl-5-oxohexyl) phthalate [MEOHP] and mono (2-ethyl-5-hydroxyhexyl) phthalate [MEHHP]) in order to assess the respective parts played by accumulation and degradation through hydrolysis and oxidization.
We focused upon target organs: (1) the liver where phthalates from digestive intake and brought through the portal vein undergo biotransformation before they could reach the general blood flow; (2) the bile fluid, as an excretion medium; (3) the blood plasma, integrating the systemic contamination; (4) the muscle fillet as a current tissue for environmental contamination monitoring.
Material and methods
Chemicals and reagents
Solvents for cleaning and extraction purposes of ultrapure quality and free from phthalate residues were supplied by Merck-Chimie (Fontenay-sous-Bois, France). Sodium sulphate under granular form (Merck-Chimie) was burned at 400 °C, for 10 h. Superclean LC-Florisil (6 ml/1 g) cartridges for solid–liquid extraction (SPE) clean-up, were obtained from Sigma-Aldrich (Saint-Quentin Fallavier, France). Oasis HLB cartridges (3 ml/60 mg and 6 ml/200 mg) were provided by Waters (Guyancourt, France). Mobile phase for GC/MS, helium and nitrogen (99.999 %), were purchased from Air-Liquide (Paris, France). Mobile phase for LC/MS/MS was prepared with acetonitrile (can) (liquid chromatography quality solvent; Merck), formic acid (Carlo-Erba for analysis) and ultrapure water from a Milli-Q system. A standard solution of six phthalates in isooctane (DMP, DEP, DnBP, BBP, DEHP, DnOP), was purchased from Supelco (via Sigma-Aldrich) and DiBP (Supelco) was added to the standard mixture at a final concentration of 8 μg ml−1 for each compound. A solution of 0.4 μg μl−1 of di-pentyl phthalate (DPP; Supelco) in isooctane was used as internal standard (IS) as 4 μg by sample and a solution of 0.1 μg μl−1 of benzyl benzoate (Supelco) as surrogate standard (SrS) as 1 μg per sample. Seven phthalate monoesters (MMP, MEP, MiBP, MnBP, MBzP, MnOP, MEHP) and two oxidized by-products (MEOHP and MEHHP) were purchased from Cambridge Isotope Laboratories, Inc. (UK). A mixture of four internal standards (from Cambridge Isotope Laboratories), 4-methyl umbelliferone 13C12 (4-MU-13C12), MnBP-13C12, MEHP-13C12 and MiNP-13C12, each one at 25 ng μl−1 in ACN was prepared and used as 10 μl per sample. β-Glucuronidase from Escherichia coli K12, 200 units ml−1 was provided by Roche Biomedical (France).
Sample collection
Adult roach were caught by electro-fishing operation at the outlet of the Orge river basin characterized by a strong man induced impact both upstream with intensive agriculture activities and downstream with highly urbanized and industrialized areas (Fig. 1). They were then, anesthetized with tricaine methane sulphonate (MS222; 1 g l−1 in river water). Fish length and weight averaged 230 cm and 150 g, respectively. Blood plasma were sampled by puncture into the tail vein and stored in heparinized vials (n = 4). The whole biliary bladders were dissected and the bile was collected. Whole livers (n = 4) and muscles as fillets (n = 4) were sampled.

Sampling location
Simultaneously, water samples were collected with stainless steel bottles and stored in amber glass flasks with Teflon® stoppers.
Sample treatment and extraction
In order to avoid sample contamination by phthalates, the equipment used (flask, pipet) was glassware exclusively. After washing twice with acetone and with hexane, the glassware (except for volumetric material) was baked at 400 °C in a furnace, for 4 h and stored with glass stoppers in aluminum foils. For micro-volumes, automated analytical glass syringes (Thermo Fisher Scientific, UK) were used. Moreover, laboratory blanks were performed, following the same treatment steps than the samples.
The internal standards were added to the different types of samples, prior to their extraction.
Concentrations of seven parent phthalates: DMP, DEP, DnBP, DiBP, BBP, DEHP and DnOP were quantified in surface water, fish liver and fish muscle after liquid extraction and purification by solid phase extraction (SPE) upon Florisil cartridges as previously described (Teil et al. 2012).
First, 3.5 l raw water samples were treated with 350 ml of a solvent mixture (75 % hexane and 25 % methylene chloride) by shaking for 20 min, three times in succession. The extracts were then dried with anhydrous sodium sulphate then, concentrated to a volume of 10 ml by rotary evaporation under vacuum and transferred into a glass tube before evaporation under a nitrogen stream to a volume of 200 μl.
Fish muscle and liver were freeze-dried for 48 h then ground to obtain a fine powder. Then, 2 g dry weight muscle and 0.15 g dry weight liver were spiked with ISs in a 40-ml centrifuge glass tube and then extracted with 20 ml of hexane/acetone (80:20, v/v) in a Bransonic 2510 ultra-sonic bath (VWR) during 20 min. Extracts were centrifuged (5 min, 970 × g) and supernatants were collected. This procedure was performed twice and both extracts were combined, prior to concentration under a nitrogen stream. Then, the extracts were passed through Florisil cartridges for cleaning-up and eluted with 10 ml of hexane/diethyl ether (80:20 v/v). Phthalate diester extracts were then, concentrated under nitrogen stream to a volume of 200 μl, transferred into vials and the SrS was added before analysis.
Concentrations of seven monoesters (MMP, MEP, MiBP, MnBP, MBzP, MnOP and MEHP) and two oxidized by-products from phase I biotransformation of DEHP (MEOHP and MEHHP) were determined in the liver, bile and plasma. The method used derived from that developed by Blount et al. (2000), Silva et al. (2004) and Mortensen et al. (2005) for the determination of phthalate metabolites in serum and urine of rodents and humans.
First, 100 mg freeze-dried liver tissue was crushed with an Ultra-Turrax in 2 ml of ammonium acetate (1 M). Then, 500 μl of plasma or bile were diluted with 500 μl of ammonium acetate (1 M). A deconjugation step was realized (90 min, 37 °C) with β-glucuronidase from E. coli K12 (5 μl, 200 units ml−1) free from any detectable esterase activity upon phthalate diesters (Blount et al. 2000). The glucuronide conjugate of 4-MU, the 4-methylumbelliferyl-β-d-glucuronide, was used (140 mg by incubation) for controlling the deconjugation reaction. Thus, for each parent phthalate, total metabolites were quantified including deconjugated compounds.
After deconjugation, the samples were loaded onto SPE cartridges (Oasis HLB 3 ml/60 mg) and eluted with a basic buffer followed by an acid one. The acidified sample was then purified in a second SPE cartridge (Oasis HLB 6 ml/200 mg) and washed with acid buffer then, with H2O. The analytes were eluted by ACN then, ethyl acetate and the pooled eluate was evaporated and resuspended in ACN/H2O (10:90 v/v) before analysis. The complete procedure was detailed by Mortensen et al. (2005).
Analytical methods
Phthalates diesters were analyzed by high resolution gas chromatography/mass spectrometry (GC/MS) with a 7890 A GC coupled to a 5975 A MS (Agilent Technologies, Massy, France). The specific conditions of chromatography and quantification methods previously described by Teil et al. (2012) are presented in Appendices 1 and 2.
Phthalate monoesters were quantified as triplicates, by high performance liquid chromatography (1200 series; Agilent Technologies, Massy, France)–tandem mass spectrometry (6410-triple quadrupole MS, in negative ion mode for the electrospray source; Agilent Technologies) (HPLC-MS/MS), equipped with a silice upti-prep strategy column (100 A, C18-2, 2 × 100 mm, 2.2 μm; Interchim) heated at 40 °C. The source was in ESI negative mode (N2: 350 °C; gas flow: 660 l/h; capillary: 4,000 V) and the injected sample volumes were 10 μl. The LC gradient flow was applied as described by Silva et al. (2004) (Appendix 3). The parameters for LC–MS/MS detection and limits of quantification (LOQs) for roach tissues are indicated in Appendix 4.
Concentrations of phthalates and of their by-products were expressed in liver and muscle as ng/g of dry weight, in surface water as ng/l and in plasma and bile as ng ml−1. Correlations were expressed by the Spearman’s r coefficient and read in the table of Fisher and Yates (XL STAT Pro 7.01 Addinsoft, USA).
The ratio of MEHP to DEHP contents as ng g−1 dry weight in liver tissue was calculated. Ratios between DEHP metabolites: MEHHP/MEOHP and MEHP/MEOHP were calculated for plasma and liver.
The bioaccumulation factor was defined as BAF = C biota/C water (l/kg), where C biota is expressed as ng/kg dry weight and C water as ng/l.
Results and discussion
Method validation
The column separation obtained for the GC/MS (Fig. 2) and the LC/MS/MS (Fig. 3) methods were consistent with accurate quantification.

Chromatograms of phthalate diesters in a liver sample (above) and a standard solution (below)

Chromatograms of phthalate monoesters in liver, bile and plasma samples and a standard solution
Most of parent phthalates fragmentize with characteristic ions such as m/z 149 except DMP (m/z 163) and their fragmentation patterns allow a sensitive and selective detection in the Selected Ion Monitoring (SIM) mode. For their metabolites, the Multiple Reaction Monitoring mode (MRM) separates masses in two stages, making the instrument significantly more selective than a single quadrupole system (Fig. 4). As a result, even components that cannot be analyzed by conventional scan or SIM modes can be easily identified and quantified in the presence of complex matrices using MRM.

Details of the LC/MS/MS spectra for the different monoester compounds and umbelliferone
Quantification of phthalates and of their metabolites was carried out by calculating the response factor for each compound relative to the corresponding IS and concentrations were obtained using a linear regression analysis of the peak area versus the concentration ratio.
Performance in terms of linearity and dynamic range was checked by setting a suite of increasing concentrations for the different compounds (Fig. 5). Their results, summarized in Appendix 5, were satisfactory with an R 2 ranging from 0.989 to 0.999.

Details of MEOHP analyzed for a plasma sample
The instrumental detection limits (IDLs) were determined using the less concentrated standard solutions as a signal/noise ratio of 3 peak to peak (Appendices 2 and 4). They ranged from 2 to 17 pg for parent phthalates and from 0.3 to 6 pg, for their metabolites.
The LOQs corresponded to the concentration of a signal/noise ratio of 5 (peak to peak) (Appendices 2 and 4). The LOQs were compatible with fish concentrations and allowed quantification of all compounds.
Phthalate amounts detected in the blanks were presented in Appendix 6 and were consistent with compound determinations. Sample measurements were corrected by the concomitant procedural blanks for all compounds. The phthalate ubiquity constitutes a real problem through the treatment and analysis processes, requiring a careful check for blank concentrations. Special precautions were taken to avoid sample contamination as solvent quality and glassware cleaning procedures. Direct injection methods, using a column switching LC/MS/MS system and a SPE/HPLC should minimize the sample contamination during experimental procedures (Takatori et al. 2004). However, these methods include loading of matrices into the analytical system which may interfere with the determination accuracy.
Recoveries for the overall extraction procedure displayed satisfactory results (Appendix 6).
Roach contamination
All the phthalate diesters investigated were characterized in river water and also in liver and muscle with higher contents in liver than in muscle (Table 1). A correlation was observed only between water concentrations and liver contents (r = 0.961, p < 0.01). BAF values ranged from 2.9 E + 03 to 5.9 E + 04 in liver and were lower for muscle from 4.8 E + 02 to 2.2 E + 04. DnOP, the heaviest compound, displayed the highest BAF for both tissues. In the REACH program for the Registration, Evaluation, Authorisation and Restriction of Chemical substances in the Europe Union, entered into force on 1 June 2007, the bioaccumulation criteria for predicting accumulation in fish is BAF >2000 and for a highly bioaccumulative compound, BAF >5000 (Inoue et al. 2012).
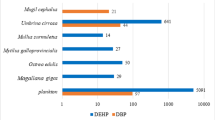
All the investigated phthalate metabolites, were characterized in nearly all tissues indicating the chemical impregnation level of roach from the river Orge (Table 2, Fig. 2).
Whatever the tissue, MiBP was prevailing, followed by MnBP. Then, by decreasing importance order, the third metabolite varied, according to the considered tissues and was as follows: MEHHP in the plasma, MEP in the bile and MEHP in the liver (Fig. 6). Plasma metabolite concentrations were higher than those of bile and gave information about the systemic contamination of the roach. An investigation of DEHP hepatic metabolism in rainbow trout (Oncorhynchus mykiss), showed the prevalence of MEHP and of its glucuronide conjugate (41.6 % of all metabolites) in the liver that is in accordance with our findings (Barron et al. 1995).

Phthalate metabolites in roach tissues: liver, bile and plasma
However, up to now, phthalate metabolism was scarcely documented in fish as compared to the number of data reported for mammals. In that way, Frederiksen et al. (2010) in Denmark, found far lower (on average, 20 times) concentrations in human serum with a different distribution pattern : MEHP was prevailing (6.7 ng ml−1), followed by MEP (4.1 ng ml−1) then, MiBP (0.72 ng ml−1) and last, MnBP (0.43 ng ml−1). To explain these differences, it may be hypothesized that the exposure level is higher for fish than for humans and that metabolic abilities differ according to the species.
Among the metabolites studied, three were by-products of DEHP degradation: MEHP, MEHHP and MEOHP. MEHP was the major metabolite in bile and liver whereas in plasma, MEHHP prevailed. MEOHP was always minor whatever the roach tissue (Table 2).
Relationships between contents of parent phthalates and those of their metabolites, were investigated to assess the fate of these chemicals throughout roach metabolism (Tables 1 and 2). DiBP and the monoester MiBP displayed the highest contents in the liver. Silva et al. (2007) studied the fate of DnBP in rats after oral administration (100 mg/kg/j) and found MnBP as the prevailing metabolite in serum (40 μg ml−1) and the optimal biomarker of exposure to DnBP.
In roach liver, the MEHP/DEHP content ratio ranged from 0.04 to 0.2 (Table 3). In contrast, in the liver and plasma of rats, the ratio ranged from 3.7 to 8.8 was, indicating a complete conversion of DEHP to its metabolite (Sircar et al. 2008). Similarly, Takatori et al. (2004) reported in human serum MEHP/DEHP ratios of 2 to 4.7, since a large part of DEHP is absorbed in the intestine after hydrolysis to MEHP. However, in human cord blood, Latini et al. (2003) found DEHP and MEHP concentrations of 1.19 and 0.52 μg ml−1, corresponding to a ratio MEHP/DEHP of 0.4. These observations suggest discrepancies of diester hydrolysis activities among different species of vertebrates. In addition, Kessler et al. (2004) performed a toxico-kinetic investigation of DEHP administered per os in marmoset (rodent) and hypothesized the occurrence of a saturation mechanism. Thus, the DEHP biotransformation step in roach appeared similar to that in mammalian species, although the former presented lower esterase activity.
Next, for the phase I of biotransformation, the ratio between MEHHP/MEOHP and MEHP/MEOHP were 9 and 5 in plasma and 22 and 33 in liver, respectively (Table 2). Ye et al. (2014) found in marine medaka (Oryzias melastigma) exposed to DEHP that its predominant metabolites were MEHP and MEHHP whereas MEOHP was minor. Ratios of MEHHP to MEOHP and MEHP to MEOHP were 100. Similar results were reported in rainbow trout (O. mykiss) by Barron et al. (1995). In contrast, in humans, Frederiksen et al. (2013) showed that MEHHP and MEOHP were the major metabolites. Again, although DEHP phase I transformation pathways were similar in fish and in mammals, the importance of the different metabolite concentrations seemed to depend on the species.
In addition, relationships between DEHP and its oxidized metabolites were investigated (Table 3). Only, MEOHP contents were correlated with those of DEHP (r = 0.914, p < 0.05). Frederiksen et al. (2010) stated that organ contents of oxidized metabolites were a better indicator of parent phthalate impact than that of monoesters. In that respect, Kato et al. (2004) stated that monoesters were easily formed from parent phthalate hydrolysis and were less accurate markers of phthalate contamination than the oxidized metabolites. Indeed, although we used ultrapure reagents free from any esterase activities, the occurrence of monoester formation from parent compounds throughout laboratory procedures, could not be excluded. Thus, the determination of DEHP oxidized metabolites is routinely used mainly in human urine as markers of human daily phthalate exposure. However, at present, oxidized metabolite quantification is not currently used for phthalates other than DEHP (German Federal Environmental Agency 2011) and should be a pertinent approach for further developments.
Conclusion
Our study constitutes the first approach of phthalate metabolism in wild fish which up to now was poorly, documented as compared to rodents and man.
At present, the investigation of degradation products in target organs for exposure level assessment of the biota is developing. The simultaneous characterization of parent contaminants together with their by-products, allows knowledge improvements about the metabolic impact of these chemicals upon organisms, that could not necessarily be given by bioaccumulation estimation alone. Indeed, determination of phthalate metabolites in target tissues of roach, contributes to explore the systemic impregnation level.
Our investigation upon field samples, gives indication of the pollutant impact under biocenotic conditions. It recommends main guidelines for further investigation that should also explore phase II conjugates for a complete consideration of contaminant toxicity. It provides reliable variables that might be integrated to conceptual models for management of aquatic ecosystem quality.
References
Agency GFE (2011) Substance monograph: phthalates New and updated reference values for monoesters and oxidizes metabolites in urine of adults and children. Bundesgesundheitsblatt 54:770–785
Barron MG, Albro PW, Hayton WL (1995) Biotransformation of di(2-ethylhexyl) phthalate by rainbow trout. Environ Toxicol Chem 14:873–876
Blount BC, Milgram KE, Silva MJ, Malek NA, Reidy JA, Needham LL, Brock JW (2000) Quantitative detection of eight phthalate metabolites in human urine using HPLC–APCI–MS/MS. Anal Chem 72:4127–4134
Erkekoglu P, Rachidi W, Yuzugullu OG, Giray B, Favier A, Ozturk M, Hincal F (2010) Evaluation of cytotoxicity and oxidative DNA damaging effects of di(2-ethylhexyl)-phthalate (DEHP) and mono(2-ethylhexyl)-pthalate (MEHP) on MA-10 Leydig cells and protection by selenium. Toxicol Appl Pharmacol 248:52–62
Frederiksen H, Jorgensen N, Andersson AM (2010) Correlations between phthalate metabolites in urine, serum and seminal plasma from young Danish men determined by isotope dilution liquid chromatography tandem mass spectrometry. J Anal Toxicol 34:400–410
Frederiksen H, Kolstrup Søgaard Nielsen J, Aarøe Mørckorgensen T, Winton Hansen P, Fangel Jensen J, Nielsen O, Andersson AM, Knudsen LE (2013) Urinary excretion of phthalate metabolites, phenols and parabens in rural and urban Danish mother–child pairs. Int J Hyg Environ Health 216:772–783
Gerbron M, Geraudie P, Fernandes D, Rotchell JM, Porte C, Minier C (2014) Evidence of altered fertility in female roach (Rutilus rutilus) from the River Seine (France). Environ Pollut 191:58–62
Inoue Y, Hashizume N, Yoshida T, Murakami H, Suzuki Y, Koga Y, Takeshige R, Kikushima E, Yakata N, Otsuka M (2012) Comparison of bioconcentration and biomagnification factors for poorly water-soluble chemicals using common carp (Cyprinus carpio L.). Arch Environ Toxicol 63:241–248
Kato K, Silva MJ, Reidy JA, Hurtz ID, Malek NA, Needham LL, Nakazawa H, Barr DB, Calafat AM (2004) Mono-(2-ethyl-5-hydroxyhexyl) phthalate and mono-(2-ethyl-5-oxohexyl) phtalate as biomarkers for human exposure assessment to di(2-ethylhexyl) phthalate. Environ Health Perspect 112:327–330
Kessler W, Numtip W, Grote K, Csanady GA, Chahoud I, Filser JG (2004) Blood burden of di(2-ethylhexyl) phthalate and its primary metabolite mono(2-ethylhexyl)phthalate in pregnant and nonpregnant rats and marmosets. Toxicol Appl Pharmacol 195:142–153
Latini G, De Felice C, Presta G, Del Vecchio A, Paris I, Ruggieri F, Mazzeo P (2003) In Utero exposure to di-(2-ethylhexyl) phthalate and duration of human pregnancy. Environ Health Perspect 111:1783–1785
Minier C, Caltot G, Leboulanger F, Hill EM (2000) An investigation of the incidence of intersex fish in Seine-Maritime and Sussex regions. ANALUSIS 28:801–806
Mortensen GK, Main KM, Andersson AM, Leffers H, Shakkebaek NE (2005) Determination of phthalate monoesters in human milk, consumer milk, and infant formula by tandem mass spectrometry (LC-MS/MS). Anal Bioanal Chem 382:1084–1092
Silva MJ, Barr DB, Reidy JA, Kato K, Malek NA, Hodge CC (2003) Glucuronidation patterns of common urinary and serum monoester phthalate metabolites. Arch Toxicol 77:561–567
Silva MJ, Slakman AR, Reidy JA, Preau JL Jr, Herbert AR, Samandra E, Needham LL, Calafat AM (2004) Analysis of human urine for fifteen phthalate metabolites using automated solid-phase extraction. Chromatogr B 805:161–167
Silva MJ, Samandar E, Reidy JA, Hauser R, Needham LL, Calafat AM (2007) Metabolite profiles of di-n-butyl phthalate in humans and rats. Environ Sci Technol 41:7576–7580
Sircar D, Albazi SJ, Atallah Y, Pizzi W (2008) Validation and Application of an HPLC Method for determination of di (2-ethylhexyl) phthalate and mono (2-ethylhexyl) phthalate in liver samples. J Chromatogr Sci 46:627–631
Staples CA, Peterson DR, Parkerton TF, Adam WJ (1997) The Environmental fate of phthalates esters. A literature review. Chemosphere 35:667–749
Takatori S, Kitagawa Y, Kitagawa M, Nakazawa H, Hori S (2004) Determination of di(2-ethylhexyl)phthalate and mono(2-ethylhexyl)phthalate in human serum using liquid chromatography–tandem mass spectrometry. J Chromatogr B 804:397–401
Teil MJ, Tlili K, Blanchard M, Chevreuil M, Alliot F, Labadie P (2012) Occurrence of polybrominated diphenyl ethers, polychlorinated biphenyls, and phthalates in freshwater fish from the Orge River (Ile-de France). Arch Environ Contam Toxicol 63:101–113
Ye T, Kang M, Huang K, Fang C, Chen Y, Shen H, Dong S (2014) Exposure to DEHP and MEHP from hatching to adulthood causes reproductive dysfunction and endocrine disruption in marine medaka (Orysias melastigma). Aquat Toxicol 146:115–126
Acknowledgments
This work was supported by the PIREN-SEINE Research Programme. The French National Agency for Water and Aquatic Environments (ONEMA) provided assistance for electric fishing. We thank Catherine Bourges and Annie Desportes for their technical assistance.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible editor: Roland Kallenborn
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOCX 43 kb)
Rights and permissions
About this article

Cite this article
Valton, A.S., Serre-Dargnat, C., Blanchard, M. et al. Determination of phthalates and their by-products in tissues of roach (Rutilus rutilus) from the Orge river (France). Environ Sci Pollut Res 21, 12723–12730 (2014). https://doi.org/10.1007/s11356-014-3213-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-014-3213-0