
Abstract
Mercury (Hg) mobility at the sediment–water interface was investigated during a laboratory incubation experiment conducted with highly contaminated sediments (13 μg g-1) of the Gulf of Trieste. Undisturbed sediment was collected in front of the Isonzo River mouth, which inflows Hg-rich suspended material originating from the Idrija (NW Slovenia) mining district. Since hypoxic and anoxic conditions at the bottom are frequently observed and can influence the Hg biogeochemical behavior, a redox oscillation was simulated in the laboratory, at in situ temperature, using a dark flux chamber. Temporal variations of several parameters were monitored simultaneously: dissolved Hg (DHg) and methylmercury (MeHg), O2, NH4 +, NO3 - + NO2 -, PO4 3-, H2S, dissolved Mn2+, dissolved inorganic and organic carbon (DIC and DOC). Under anoxic conditions, both Hg (665 ng m2 day-1) and MeHg (550 ng m2 day-1) fluxed from sediments into the water column, whereas re-oxygenation caused concentrations of MeHg and Hg to rapidly drop, probably due to re-adsorption onto Fe/Mn-oxyhydroxides and enhanced demethylation processes. Hence, during anoxic events, sediments of the Gulf of Trieste may be considered as an important source of DHg species for the water column. On the contrary, re-oxygenation of the bottom compartment mitigates Hg and MeHg release from the sediment, thus acting as a natural “defence” from possible interaction between the metal and the aquatic organisms.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
In coastal marine areas, degradation of sedimentary organic matter (OM) is dependent on the oscillating redox conditions of surface sediments (Bouchet et al. 2011). Organic carbon remineralization drives the biogeochemical cycling of several elements (Canfield et al. 2005); among them, mercury (Hg) is characterized by a complex cycle (Merritt and Amirbahman 2009) which involves its transformation into methylmercury (MeHg), the most toxic and potentially bioaccumulable form of Hg to humans and wildlife (Clarkson 1998).
Under controlled laboratory conditions, it is possible to observe the effects of oxygen (O2) depletion on the biogeochemical behavior of the benthic compartment (Belias et al. 2007) and to assess both Hg mobility and the release of MeHg from sediments to the upper water column (Covelli et al. 2008; Emili et al. 2011; Koron and Faganeli 2012).
A suitable test area for these kinds of experiments is represented by the Gulf of Trieste (Northern Adriatic Sea), one of the most Hg-contaminated coastal areas in the world, where cinnabar (HgS)-rich material from the Idrija (Slovenia) mining district have been (Covelli et al. 2001) and are still being delivered (Horvat et al. 1999; Faganeli et al. 2003; Covelli et al. 2006, 2007) by the Isonzo River, the largest contributor of this metal into the area since the 16th century.
The Isonzo River is the main freshwater input in the Gulf, with an estimated annual flow rate ranging between 1.1–666 m3 s-1 (1998–2007 period; Comici and Bussani 2007) and fluvial inputs appear to control primary production in the Gulf, as the highest inputs of land-born nutrients, in particular, nitrate leaching from cropping areas of the Venezia Giulia plain (Cantoni et al. 2003) are associated with the highest river discharges (Malej et al. 1995). The Gulf is a shallow coastal basin with a maximum water depth of 25 m. Water circulation is anticlockwise and wind-driven superficial currents mostly influence the first 5 m of the water body (Stravisi 1983). Periodical hypoxic/anoxic conditions have been observed in the Gulf, as a consequence of high loadings of nutrients and OM and strong late summer water stratification (Faganeli et al. 1985, 1991; Kemp et al. 1999). This latter represents one of the key factors controlling in situ MeHg production at the sediment–water interface (SWI, Fitzgerald et al. 2007; Merritt and Amirbahman 2009).
Within this context, the aim of this study was to evaluate Hg and MeHg cycling at the SWI in the Gulf of Trieste under anoxic conditions, simulating an oxic/anoxic redox transition in a dark incubated benthic chamber, as previously reported for heavily Hg-contaminated coastal sites (Covelli et al. 2008; Emili et al. 2011). To this purpose, a volume of undisturbed marine sediment was sampled right in front of the Isonzo River mouth, where on the basis of previous research (Covelli et al. 2001; Hines et al. 2006), the highest Hg contents in the Gulf (up to about 20–30 μg g-1) were expected.
In addition, in order to investigate the potential Hg mobility, the speciation in the sampled sediment was determined by means of a selective sequential chemical extraction (Bloom et al. 2003) and the results from the sediment incubation were compared to similar investigations previously conducted in the area (Emili et al. 2011; Koron and Faganeli 2012).
Materials and methods
Sampling
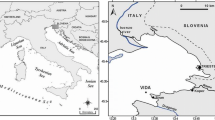
Bottom sediment and the overlying water were collected by a SCUBA diver in June 2010 at station D6 (Fig. 1), using a cylindrical Plexiglas chamber as a sampler (h = 25 cm, i.d. = 24 cm). The chamber was carefully transported to the laboratory, in order to prevent disturbance of the SWI. Here, the water overlying the sediment was drained off and the chamber was filled with bottom seawater collected at the same location.

The chamber was sealed and placed in a dark room at in situ temperature for incubation (Fig. 2). After 27 days, the system was reoxygenated by opening the chamber and leaving the water surface in contact with the atmosphere for 7 days. The water temperature throughout this period was kept constant at 23.0 ± 1 °C, which was the water temperature at the sampling day.

Chamber used for the incubation experiment. The plexiglas cylinder is watertight and it is equipped with a magnetic stirrer which mixes water inside the chamber before each sampling. Water samples were periodically collected using a plastic syringe through a stopcock at the top. The average depth of sediments inside the chamber was 15 cm with about 10 cm of overlying water
During the incubation experiment, water samples were periodically collected from the chamber using a plastic syringe and the withdrawn volume of water was compensated by in situ collected bottom seawater. Prior to each sampling, the system was homogenized for 15 min by means of a stirrer driven by a stepper electric motor housed in a waterproof case. Coupling between the stirrer and the motor is provided by rare earth magnets. Rotation of the stirrer is kept to 5 rpm to prevent sediment resuspension while at the same time providing constant and effective mixing of the water column.
Analyses of the solid phase
Total mercury (THg) was determined by CV-AAS (Perkin-Elmer, AAnalyst 100-FIAS 100) employing NaBH4 3 % in NaOH 1 % for the reduction step, after a total decomposition of the sample with a mixture of HF + aqua regia (HNO3:HCl = 1:3 v.v.) in a closed microwave system (Milestone, MLS 1200). Accuracy was tested using a Certified Reference Material (PACS-2 marine sediment, NRCC). Results obtained (3.03 ± 0.30 mg kg-1 Hg; n = 3) were in good agreement with the certified value (3.04 ± 0.20 mg kg-1 Hg). The detection limit of the method was 0.13 μg g-1. The precision expressed as relative standard deviation of at least three determinations was at least <7 %.
For Hg speciation, a five-step selective sequential extraction (SSE) procedure described by Bloom et al. (2003) and subsequently adapted for sediments by Shi et al. (2005) was followed. Extractions were carried out using about 0.4 g of lyophilized and fine-ground surface sediment samples placed into 50 ml pretreated borosilicate glass centrifuge tubes. After the addition of each selective extractant (V = 20 ml), the sediment was subject to end-over-end shaking (t = 18 ± 4 h; at room temperature). The tubes were then centrifuged (3,000 rpm; t = 15 min) and the supernatant liquid was decanted and filtered through 0.45 μm pore size membrane filters (Millipore Millex-HA). Finally, 1 ml of HNO3 was added and the extract was placed in a clear borosilicate bottle and oxidized by adding 500 μl of BrCl. The solid residue was washed with the related extractant and the rinse was discarded. The four extractants used were: (1) Milli-Q water for “water-soluble Hg” (Hg-w); (2) 0.1 mol l-1 CH3COOH + 0.01 mol l-1 HCl for “human stomach acid soluble Hg” (Hg-h); (3) 1 mol l-1 KOH for “organo-chelated Hg” (Hg-O); (4) 12 mol l-1 HNO3 for “elemental/strongly complexed Hg” (Hg-e). In the last step (5), the residue was air dried and total digestion with aqua regia was performed at room temperature for the “mercuric sulfide” (Hg-s) fraction which comprises Hg sulfides and Hg immobilized by pyrite (FeS2). Filtered extractant solutions were employed as analytical blanks. Total dissolved Hg (DHg) in each extract was determined by CV-AFS (Brooks Rand, Model III) after a prereduction step with SnCl2 and single gold trap amalgamation. The obtained values were corrected for blanks. The method was verified for all samples by summing the ratio of extracted Hg in each phase (Hgx) to THg in the sample (∑Hgx/THg). The average extraction budget obtained from these ratios was about 80 %.
Analyses of the dissolved phase
Dissolved oxygen (O2) was determined by the Winkler method (Grasshoff et al. 1983) using an automated titration system (Mettler DL21). Determination of sulfides (H2S) was performed spectrophotometrically after trapping with Zn acetate (Grasshoff et al. 1983). Other parameters in the dissolved phase were determined on samples filtered through a Millipore Millex HA 0.45 μm filter. Dissolved inorganic carbon (DIC) and dissolved organic carbon (DOC) determinations were performed using a Shimadzu TOC 5000A analyzer. Analysis showed a variation coefficient < 2 %. The reproducibility of the method was between 1.5 % and 3 %. Dissolved manganese (Mn2+) was determined by GF-AAS (Perkin-Elmer AAnalyst 100-HGA 850) using Mg(NO3)2 as a matrix modifier. Analyses showed a variation coefficient < 4 %. Nutrients (NO2 -, NO3 -, NH4 + and PO4 3-) were determined according to Grasshoff et al. (1983) using a continuous flow segmented system (Bran-Luebbe, AAQuattro). Detection limits for nutrients were, respectively: 0.5 μmol l-1 for NO3 - + NO2 - as N, 0.4 μmol l-1 for NH4 + as N, 0.01 μmol l-1 for PO4 3- as P.
The determination of total DHg in the overlying water column was performed after an initial step of oxidation with BrCl (500 μl/100 ml sample). A prereduction using NH2OH-HCl (30 %, 250 μl), until the yellow color disappeared, was followed by reduction with SnCl2 and Au trapping. Analyses were performed by means of a Brooks Rand CV-AFS (Horvat et al. 1991).
For dissolved methylmercury (DMeHg) analyses all samples were distilled to separate MeHg from the water matrix (Horvat et al. 1993). Briefly, after an ethylating agent was added to each sample to form a volatile methyl-ethylmercury derivative, the volatile organomercury species were purged onto graphite carbon traps as a means of preconcentration and interference removal. The sample was then isothermally chromatographed, pyrolitically broken down to elemental Hg, and detected by CV-AFS (Bloom and Fitzgerald 1989). Sample results were corrected for distillation efficiency. Samples were analyzed by EPA Method 1630 (EPA 1998). The achieved detection limit was 0.0188 ng l-1, while recoveries ranged from 70 % to 112 %.
Results and discussion
Mercury content and speciation in sediments
The average total Hg (THg) concentration in the surficial sediment (0–3.5 cm) was about 13 μg g-1, which is lower than the levels previously reported for the same location (23.3 μg g-1 Covelli et al. 2001; >30 μg g-1 Hines et al. 2006). This result can be justified by the fact that this site is directly affected by riverine discharge which can vary with time, thus changing the accumulation rate and the associated Hg content in sediments. In the Gulf of Trieste, the highest Hg concentrations were found within the Isonzo River mouth and in near-shore sediments of the Italian sector, particularly alongside both sides of the river delta (Covelli et al. 2001). Moreover, the highest Hg concentrations were found to be associated with near-shore coarse sediments, whereas Hg concentrations sharply decrease with distance from the river mouth to offshore areas, where fine particles (mostly <16 μm) settle down.
Results from the SSE showed that by large, the predominant fraction in the D6 sediment was Hg-s, representing 76 % of the extracted Hg. Hg-s comprises mostly the poorly soluble cinnabar (α-HgS(s); K sp = 10-36.8) and metacinnabar (β-HgS(s); Ksp = 10-36.4), but also HgSe and HgAu complexed forms (Bloom et al. 2003).
Recent studies suggest that in addition to methylating bacteria activity and the pore water speciation of DHg, the solid-phase speciation of Hg in sediments is a key factor controlling methylation rates (Jonsson et al. 2012). The authors reported a wide range of Hg methylation rate constants determined in estuarine sediments, depending on the chemical form of the added isotopic tracer. They concluded that the incorporation of Hg(II) in less soluble phases, such as cinnabar and metacinnabar, decreases the availability of Hg(II) for methylation processes and, consequently, the formation of MeHg in estuarine sediments. However, the same authors recognized that in reducing sediments, both cinnabar and metacinnabar could be dissolved to a significant rate, thus providing Hg(II) to methylating bacteria and enhancing MeHg formation.
Cinnabar from the Idrija Hg mine, inflowing from the Isonzo River and deposited at the Isonzo River mouth, is responsible for the predominance of the Hg-s fraction in the D6 surface sediment. Separation of Hg phases performed by means of a solid-phase Hg thermodesorption technique to quantify cinnabar (HgS) and non-cinnabar Hg compounds in sediments of the Isonzo River and the Gulf showed the predominance of microcrystalline red cinnabar, prevalently bound to the coarse fraction, which was higher than 90 % at D6 (Biester et al. 2000). Although sampling and analyses were conducted on different samples and in different times, the results seem to be quite concordant with this study, taking into account minor discrepancies due to the different analytical approaches.
The second most abundant fraction in the D6 site was Hg-e, which accounts for 23 % of the extracted Hg. This fraction can also be considered as poorly mobile. However, it is often considered as a good estimate of the free Hg(0) present in the sediment matrix, although some interferences with Hg(I), amorphous organo-sulfur and crystalline Fe/Mn oxide phases could lead to an overestimation (Bloom et al. 2003).
The remaining 1 % is constituted by the sum of Hg-w (0.08 %), Hg-h (0.03 %) and Hg-o (0.77 %). Regarding Hg mobility and bioavailability, the most concerning fractions are the first two (Hg-w, Hg-h) which comprise highly soluble and easily exchangeable compounds (i.e. HgCl2, HgSO4 and HgO). The sum of these fractions in D6 accounts for just 0.11 % of the extracted Hg, which can be considered negligible. The organo-chelated Hg fraction (Hg-o), which has a moderate mobility, is also extremely low in terms of percentage, and it is not expected to give a significant contribution to Hg mobility in this sediment. This fraction includes Hg complexed with humic, fulvic and amino acids, living and dead biota and the relative small fraction of methylated species. Bloom et al. (2003) reported a positive correlation between the Hg-o fraction in sediments and MeHg content, but emphasized how each sediment type has a different methylation potential. If redox conditions change, the slow dissolution of Hg-binding solid phases would occur, followed by the consequent release of Hg into porewaters, where it can reprecipitate in other phases (such as complexes with OM and Fe/Mn oxyhydroxides) and, thus, contribute to the Hg-o and Hg-e fractions.
Variability of chemical parameters in the incubated chamber
An O2 concentration < 32 μmol l-1 (1 mg l-1) was considered the limit for the hypoxic/anoxic transition. In this experiment, after 7 days of incubation, O2 concentration in the benthic chamber was no more detectable (Fig. 3). Oxygen was resupplied to the system by opening the benthic chamber after 27 days of incubation. As a consequence, O2 concentration quickly rose to 142 μmol l-1, reaching a maximum of 291 μmol l-1 , which was only 25 % lower than the concentration value observed at the beginning of the experiment.

Evolution of dissolved O2 concentration, pH, Eh, DIC and DOC, nutrients (NO2 - + NO3 -, NH4 + and PO4 3-), Mn2+, H2S, DHg and DMeHg in the benthic chamber during the incubation experiment at D6 (oxic, anoxic and reoxigenation phases)
Oxic conditions at t 0 were associated to a positive Eh value (125 mV). From t 3, Eh appeared negative (-14 mV), and it rapidly fell down to -286 mV (t 6) just before the disappearance of oxygen. In anoxic conditions, Eh remained constant (about -360 mV). From t 16 on, when the system started to recover oxic conditions, Eh values turned back to positive values in a couple of days.
The pH value decreased from 8.27 at t 0 to 7.75, when anoxic conditions were established. During anoxia (t7–t14), pH decreased only slightly, falling to 7.66 before reoxygenation. When the system shifted back to oxic conditions, pH sharply increased up to 8.39, slightly higher than at the beginning of the experiment.
During the oxic/anoxic transition, O2 is consumed near the SWI due to early diagenetic processes involving OM. The remineralization follows the well-established sequence of reactions controlled by the preferential use of the electron acceptor that yields the highest amount of free energy for the bacterially mediated oxidation of OM (Froelich et al. 1979). These processes were clearly evident by following nitrogen (N) species behavior during incubation.
At the beginning of the experiment, nitrate (NO3 -) was, as expected, the predominant N inorganic form present in the water column (≈66 %). As incubation proceeded, NO2 - and NO3 - were rapidly consumed, in parallel with O2 depletion, as alternative electron acceptors, falling from a starting concentration of 7.25 μmol l-1 to <0.5 μmol l-1 after 5 days. Following reoxygenation, these redox-sensitive species were effectively regenerated, quickly rising in concentration to a maximum of 15.6 μmol l-1 at the end of the experiment.
Ammonium (NH4 +) was promptly released into the water column, rising from the initial concentration of 3.55 μmol l-1 to around 300 μmol l-1 during anoxia, and showed a slow decrease after O2 resupply. The simultaneous decrease of NO3 - concentration and increase of NH4 + is consistent with the bacterial or enzyme reduction of N species in the system. Throughout the experiment, NH4 + was by far the dominant species in the system, being its concentration at the end of incubation one order of magnitude greater than the (NO3 - + NO2 -) concentration. However, nitrites and nitrates conversion alone does not justify the high NH4 + concentration measured in the water column, thus suggesting that the rapid degradation of N-rich soluble organic compounds in the sediment was the primary process responsible for the relevant release of NH4 + into the water column (Emili et al. 2011).
Phosphate (PO4 3-) recycling from the sediment to the water column was also related to the redox transition. Phosphate concentration was extremely low, increasing from 0.01 μmol l-1 at t 0 to a maximum of about 30 μmol l-1 before reoxygenation. Following oxygen depletion, the progressive shift to anoxic conditions leads to reduction of Fe(III) to Fe(II), thus determining the transformation of insoluble FePO4 into more soluble Fe3(PO4)2, releasing in turn PO4 3- into the overlying water. When oxygen was resupplied, PO4 3- scavenging from the water column was quick, and at the end of the experiment, PO4 3- concentration was 3.82 μmol l-1, higher than t 0 but one order of magnitude lower than the average concentration during anoxia. Removal from the water column probably proceeded with the formation of Fe(III) oxyhydroxides, as previously suggested (Ogrinc and Faganeli 2006). The significant positive correlation (r = 0.820, p < 0.0001, n = 21) found between NH4 + and PO4 3- suggests a common origin of these species from sedimentary OM degradation.
Reduction of sulfate produced dissolved sulfide (H2S), which rose from 0.36 μmol l-1 at t 0 to a maximum of 75 μmol l-1 after 20 days of incubation. After that, H2S concentration decreased, reaching undetectable levels after reoxygenation. As expected, sulfide was significantly correlated with PO4 3- (r = 0.850, p < 0.0001, n = 15) and NH4 + (r = 0.724, p < 0.003, n = 15), given that sulfate reduction is one of the main processes of microbial degradation of OM in sediments. Sulfide was also significantly correlated with both pH (r = -0.756, p < 0.002, n = 15) and Eh (r = -0.898, p < 0.0001, n = 15). Sulfide scavenging from the water column before reoxygenation suggests the precipitation of insoluble sulfides (Benoit et al. 1999; Mason et al. 2006) and the consequent removal of S-binding species from solution, such as Hg, due to its high degree of pyritization (Huerta-Diaz and Morse 1992).
DIC steadily increased during the experiment (up to 5.78 mmol l-1), as a consequence of OM respiration by the benthic compartment, showing a slight decrease following reoxygenation. Throughout the experiment, DOC exhibited low concentration (0.6 mmol l-1 on average) and did not follow a clear trend. DOC represents a fraction of dissolved organic matter (DOM) and inorganic Hg can form stable complexes in aquatic media with DOM (Benoit et al. 2001). The interaction between DOC and Hg prevails in systems where its concentration is higher than 5 mg l-1 (0.42 mmol l-1; Hissler et al. 2006). While the average concentration of DOC in the benthic chamber was above this threshold, the lack of correlation between DOC and Hg suggests that OM was not the primary control on Hg availability for methylation in the water column, as expected in sulfidic environments (Benoit et al. 2001).
Manganese was already released from the sediment at the beginning of the experiment, when O2 depletion was slowly ongoing. The quick drop in concentration following reoxygenation suggests that Mn oxides were formed, possibly removing other solutes from solution, MeHg included, by coprecipitation and/or adsorption.
Behavior of mercury species in the benthic chamber
Total DHg was present in the water column at t 0 (2.1 ng l-1), and its concentration in the benthic chamber steadily increased with the progressive O2 depletion (Fig. 3). After 5 days of incubation, DHg concentration rose to 15.6 ng l-1, further increasing during the anoxic phase, up to 68.6 ng l-1 at t 11. After peaking, the concentration of DHg rapidly decreased to about 30 ng l-1 before reoxygenation. Dissolved MeHg (DMeHg), which was almost undetectable (0.03 ng l-1) at the beginning of the experiment, paralleled DHg, with a smoother behavior in the intermediate phase of anoxia, when its concentration hit a plateau of about 39 ng l-1. After 20 days of incubation, still under anoxic conditions, DMeHg concentration decreased, falling to about 22 ng l-1 before reoxygenation
A positive correlation was found between DHg, H2S (r = 0.898, p < 0.0001, n = 15) and DMeHg (r = 0.935, p < 0.00001, n = 21). DMeHg was also positively correlated with H2S (r = 0.786, p < 0.001, n = 14), suggesting that Hg methylation by sulfate-reducing bacteria (Compeau and Bartha 1985; Gilmour and Henry 1991) was active in the incubated sediment. Hypoxic/anoxic conditions favored methylation processes, as long as H2S concentration in the water column was below 80 μmol l-1. After reaching the optimum value in sulfate reduction and the corresponding methylation processes, the formation of insoluble sulfides buffered H2S and Hg, removing them from solution. The corresponding disappearance of DMeHg suggests coprecipitation with sulfides and possible demethylation by reductive pathway (Hines et al. 2006).
When O2 was resupplied to the system, DHg quickly dropped below 10 ng l-1, finally returning to concentration values (3.8 ng l-1) similar to the starting concentration at t 0. DMeHg quickly disappeared from the water column, decreasing to 0.3 ng l-1 at the end of the experiment.
The decrease of DHg and DMeHg in reoxigenated conditions could be due to the coprecipitation with newly formed authigenic Fe and Mn oxides, which are scavengers of soluble metal compounds in oxic surface sediments (Gagnon et al. 1997; Muresan et al. 2007). The decreasing percentage of DMeHg to DHg (from 76 % at t 14 to 7.5 % at t f), following reoxygenation, also suggests an important role played by demethylation in oxic conditions (Hines et al. 2006).
Comparison with previous incubation experiments in the Gulf of Trieste
These results were compared to previous incubation and reoxygenation experiments (Emili et al. 2011; Koron and Faganeli 2012) conducted in the Gulf of Trieste along the southward Isonzo River mouth–Piran transect (Fig. 1), which is characterized by a decrease of Hg concentration in sediments, related to the progressive distance from the source of contamination (Covelli et al. 2001). The CZ station (Emili et al. 2011) is roughly located 12 km away from the river mouth, with an average water depth of 21 m. Stations E and F1 (Koron and Faganeli 2012) are located in the southern part of the Gulf, in Slovenian waters: F1 is approximately located 2.5 km off the coastal city of Piran at an average water depth of 22 m; E is located in front of the Marine Biological Station of Piran, at a water depth of 8 m.
Mercury concentration in CZ was determined by Hines et al. (2000) to range between 0.8 and 0.9 μg g-1, whereas it accounted for 0.6 μg g-1 at station E and 0.2 μg g-1 at station F1 (Covelli et al. 2001), far less than the average 13 μg g-1 determined in this study for D6 (0–3.5 cm). Methylmercury concentration varied between 0.21 and 0.48 ng g-1 at sites E and F1 (Koron and Faganeli 2012), similar to what observed in CZ (<0.5 ng g-1; Hines et al. 2006) but lower than D6 (about 2 ng g-1; Hines et al. 2006). Overall, MeHg accounted for a minimal percentage of total Hg in sediments (<0.1 %).
O2 consumption in the benthic chamber followed different dynamics in the selected sites. At station E (May 2009; 20 °C), O2 (t 0 = 469 μmol l-1) was consumed in only 4 days, whereas at CZ (November 2007; 20 °C; t 0 = 393 μmol l-1 O2), it was depleted in 7 days, similar to D6 (t 0 = 469 μmol l-1 O2). Conversely, at F1 (May 2010; 20 °C), O2 consumption (t 0 ≈ 500 μmol l-1) took about 15 days. In all cases, O2 was resupplied to the system after 16 days of incubation, by opening the chamber lid and letting O2 diffuse into water. Since differences in the initial O2 concentration among the three sites do not seem to be significant, it is suggested that mineralization of OM progressed more slowly at F1.
Patterns of H2S, DHg and DMeHg (Fig. 4) proceeded in parallel at CZ and F1, peaking during the anoxic phase of the experiment and quickly disappearing following reoxygenation, similarly to D6. At station E, on the other hand, the coupling of DHg and DMeHg was not well correlated with H2S.

At all three sites, DHg increased during anoxia, peaking at 95 ng l-1 at F1, 44 ng l-1 at E, and 11 ng l-1 at CZ, compared to 69 ng l-1 at D6. DMeHg peaked at about 100 ng l-1 at F1, 21 ng l-1 at E, and 12 ng l-1 at CZ, compared to 39 ng l-1 at D6. The percentage of DHg as DMeHg in the benthic chamber reached a maximum of 100 % during anoxia in CZ (Emili et al. 2011) and F1 (Koron and Faganeli 2012), 94 % in D6, and 50 % in E (Koron and Faganeli 2012). Thus, DMeHg was the dominant form of Hg in the water column during the anoxic phase of the incubation experiments. This evidence poses a major concern regarding the possible transfer of Hg to the aquatic trophic chain. However, such observations were made under laboratory conditions, simulating a persistent and extreme anoxic event and not the normal environmental conditions.
Scavenging of Hg species was evident at D6 and E during anoxia, while it only occurred after reoxygenation at both CZ and F1. At D6, such scavenging ran in parallel with H2S removal, while at station E, such relation was less evident.
For stations E and F1, Koron and Faganeli (2012) identified the dissolution of Fe and Mn oxyhydroxides due to sediment anoxia as a major control in MeHg release from sediments. At the same time, co-precipitation of Hg species with Fe and Mn and intensive demethylation were suggested as factors explaining the rapid decrease of DHg and MeHg following reoxygenation.
Benthic fluxes of mercury species and factors influencing mercury cycling in the incubation experiments
Benthic fluxes for D6 were calculated from linear regression of the variation of solutes concentration with incubation time, according to Covelli et al. (2008) and Emili et al. (2011), and compared to CZ (Emili et al. 2011), E and F1 (Koron and Faganeli 2012).
As shown in Table 1, DHg fluxes under anoxic conditions were quite similar among D6, E and F1 (about 665 ng m-2 day-1), despite a much higher THg content in D6. At CZ, DHg flux was one order of magnitude lower than at the other sites (71 ng m-2 day-1). DMeHg fluxes were highest in E (577 ng m-2 day-1) and D6 (550 ng m-2 day-1), followed by F1 (361 ng m-2 day-1) and CZ (238 ng m-2 day-1).
It appears that THg content in sediments alone cannot predict the flux of DHg and DMeHg from the solid phase to the upper water column during an oxic–anoxic transition. DHg and DMeHg fluxes are for the most part comparable throughout the Gulf transect, despite a wide range of Hg concentration. (0.2–13 μg g-1) in sediments.
Speciation analysis by SSE showed that most of sedimentary Hg in D6 is present in highly insoluble forms. Such information is lacking for the other sites but, based on their location, further away from the Isonzo River mouth where cinnabar-rich coarser particulate matter tends to settle (Biester et al. 2000), it is probable that more labile forms should be present (Covelli et al. 2001).
As previously noted (Emili et al. 2011), sediment incubation and the persistence of anoxic conditions favor Hg and MeHg release from sediments to the water column. In spite of the high percentage of the cinnabar component, sediments of D6 become a source of MeHg to the water column if anoxic conditions occur.
Besides, in accordance with the SSE results, THg in the D6 sediments exhibited a strong binding to the solid phase, limiting its dissolution into porewaters and, such, its availability.
The partitioning of Hg between the solid and the dissolved phase can be described in terms of the log K D value (Bloom et al. 1999), where K D is expressed in l kg-1:
[Hg]s is the solid-phase concentration of Hg (in ng kg-1), whereas [Hg]pw is the pore–water concentration of Hg (in ng l-1).
Low K D values are associated with an enhanced release into porewaters from the solid phase, while higher values are indicative of a stronger binding of the species to the sedimentary matrix. The log K D for THg in D6 (0–3.5 cm) accounted for 6.12 at t 0 and 6.27 at t f. Such values are higher than those reported by Covelli et al. (1999) for the central part of the Gulf of Trieste (station AA1) and by Hines et al. (2006) for the lower Isonzo River.
Conclusions
Hypoxic and anoxic conditions play a significant role in the remobilization of Hg from sediments at the Isonzo River mouth in the Gulf of Trieste. The increasing sulfide concentration in the benthic chamber is positively correlated with the parallel increase in MeHg levels, in accordance with methylation being related to sulfate reduction in anoxic sediments.
However, total Hg concentration in sediments is not the primary factor determining Hg mobility and transformation, such as methylation. In sediments, mercury speciation seems to play a key role, especially where mining activity was the main source of Hg into the environment.
The higher concentration of Hg in sediments of the Isonzo River mouth, compared to other sites in the Gulf where Hg content was up to two orders of magnitude lower, did not result in a higher methylmercury flux, in spite of significant anoxic conditions established in the incubated chamber.
Although extreme anoxic events are unlikely at the Isonzo River mouth, due to shallowness and water circulation, if bottom sediments were dredged and discharged in environments more inclined to such extreme oxygen depletion, we should not expect significantly higher MeHg release into the water column.
Besides, the observed effects of sediment reoxygenation on the solutes concentration suggest that reversion to oxic conditions removes Hg species from the water column, most probably through precipitation and coprecipitation with Fe and Mn oxyhydroxides. These findings are of paramount importance to understand Hg cycling at the SWI during hypoxic/anoxic events in this highly contaminated environment and to foresee the fate of Hg species following the restoration of the normal oxygenated conditions of bottom waters. However, field observations generally lead to a more correct quantification of the production and mobility of mercury species in natural settings. This kind of research should therefore be strongly encouraged and pursued in conjunction with laboratory experiments.
References
Belias C, Dassenakis M, Scoullos M (2007) Study of the N, P and Si fluxes between fish farm sediment and seawater. Results of simulation experiments employing a benthic chamber under various redox conditions. Mar Chem 103:266–275
Benoit JM, Gilmour CC, Mason RP, Heyes A (1999) Sulphide controls on mercury speciation and the bioavailability in sediment porewaters. Environ Sci Technol 33:951–957
Benoit JM, Mason RP, Gilmour CC, Aiken GR (2001) Constants for mercury binding by dissolved organic matter isolates from the Florida Everglades. Geochim Cosmochim Acta 65:4445–4451
Biester H, Gosar M, Covelli S (2000) Mercury speciation in sediments affected by dumped mining residues in the drainage area of the Idrija mercury mine. Environ Sci Technol 34:3330–3336
Bloom N, Fitzgerald WF (1989) Determination of volatile mercury species at the picogram level by low-temperature gas chromatography with cold-vapour atomic fluorescence detection. Anal Chim Acta 208:151–161
Bloom NS, Gill GA, Cappellino S, Dobbs C, McShea L, Driscoll C, Mason R, Rudd J (1999) Speciation and cycling of mercury in Lavaca Bay, Texas, sediments. Environ Sci Technol 33:7–13
Bloom NS, Preus E, Katon J, Hiltner M (2003) Selective extractions to assess the biogeochemically relevant fractionation of inorganic mercury in sediments and soils. Anal Chim Acta 479:233–248
Bouchet S, Bridou R, Tessier E, Rodriguez-Gonzalez P, Monperrus M, Abril G, Amouroux D (2011) An experimental approach to investigate mercury species transformations under redox oscillations in coastal sediments. Mar Environ Res 71:1–9
Canfield DE, Kristensen E, Thamdrup B (2005) Aquatic geomicrobiology. Adv Mar Biol 48:1–599
Cantoni C, Cozzi S, Pecchiar I, Cabrini M, Mozetič P, Catalano G, Fonda Umani S (2003) Short-term variability of primary production and inorganic nitrogen uptake related to the environmental conditions in a shallow coastal area (Gulf of Trieste, N Adriatic Sea). Oceanol Acta 26:565–575
Clarkson TW (1998) Human toxicology of mercury. J Trace Elem Exp Med 11:303–317
Comici C, Bussani A (2007) Analysis of the Isonzo River discharge (1998–2005). Boll Geof Teor Appl 48:435–454
Compeau G, Bartha R (1985) Sulphate-reducing bacteria: principal methylators of mercury in anoxic estuarine sediment. Appl Environ Microbiol 50:498–502
Covelli S, Faganeli J, Horvat M, Brambati A (1999) Porewater distribution and benthic fluxes measurements of mercury and methylmercury in the Gulf of Trieste (Northern Adriatic Sea). Estuar Coast Shelf Sci 48:415–428
Covelli S, Faganeli J, Horvat M, Brambati A (2001) Mercury contamination of coastal sediments as the result of long-term cinnabar activity (Gulf of Trieste, northern Adriatic Sea). Appl Geochem 16:541–558
Covelli S, Piani R, Kotnik J, Horvat M, Faganeli J, Brambati A (2006) Behaviour of Hg species in a microtidal deltaic system: the Isonzo River mouth (northern Adriatic Sea). Sci Total Environ 368:210–223
Covelli S, Piani R, Acquavita A, Predonzani S, Faganeli J (2007) Transport and dispersion of particulate Hg associated to a river plume in coastal Northern Adriatic environments. Mar Pollut Bull 55:436–450
Covelli S, Faganeli J, De Vittor C, Predonzani S, Acquavita A, Horvat M (2008) Benthic fluxes of mercury species in a lagoon environment (Grado lagoon, Northern Adriatic Sea, Italy). Appl Geochem 23:529–546
Emili A, Koron N, Covelli S, Faganeli J, Acquavita A, Predonzani S, De Vittor C (2011) Does anoxia affect mercury cycling at the sediment–water interface in the Gulf of Trieste (northern Adriatic Sea)? Incubation experiments using benthic flux chambers. Appl Geochem 26:194–204
EPA (1998) Method 1630. Methylmercury in water by distillation, aqueous ethylation, purge and trap, and CVAFS. Draft. March 1998
Faganeli J, Avčin A, Fanuko N, Malej A, Turk V, Tušnik P, Vrišer B, Vukovič A (1985) Bottom layer anoxia in the central part of the Gulf of Trieste in the late summer of 1983. Mar Pollut Bull 16(2):75–78
Faganeli J, Pezdic J, Ogorelec B, Herndl GJ, Dolenec T (1991) The role of sedimentary biogeochemistry in the formation of hypoxia in shallow coastal waters (Gulf of Trieste, Northern Adriatic). In: Tyson RV and Pearson TH (eds) Modern and ancient continental shelf anoxia. Geological Society, London, Spec. Publ. 58, pp 107–117
Faganeli J, Horvat M, Covelli S, Fajon V, Logar M, Lipej L, Cermelj B (2003) Mercury and methylmercury in the Gulf of Trieste (northern Adriatic Sea). Sci Total Environ 304:315–326
Fitzgerald WF, Lamborg CH, Hammerschmidt CR (2007) Marine biogeochemical cycling of mercury. Chem Rev 107:641–662
Froelich PN, Klinkhammer GP, Bender ML, Luedtke NA, Heath GR, Cullen D, Dauphin P, Hammond D, Hartman B, Maynard V (1979) Early oxidation of organic matter in pelagic sediments of the eastern Atlantic: suboxic diagenesis. Geochim Cosmochim Acta 43:1075–1090
Gagnon C, Pelletier M, Mucci A (1997) Behaviour of anthropogenic mercury in coastal marine sediments. Mar Chem 59:159–176
Gilmour CC, Henry EA (1991) Mercury methylation in aquatic systems affected by acid deposition. Environ Pollut 71:131–169
Grasshoff K, Ehrhardt M, Kremling K (1983) Methods of seawater analysis, 2nd edn. Weinheim, Verlag Chemie
Hines ME, Horvat M, Faganeli J, Bonzongo JCJ, Barkay T, Major EB, Scott KJ, Bailey EA, Warwick JJ, Lyons WB (2000) Mercury biogeochemistry in the Idrija River, Slovenia, from above the mine into the Gulf of Trieste. Environ Res 83:129–139
Hines ME, Faganeli J, Adatto I, Horvat M (2006) Microbial Mercury transformations in marine, estuarine and freshwater sediment downstream of the Idrija Mercury Mine, Slovenia. Appl Geochem 21:1924–1939
Hissler C, Probst JL, Mortatti J (2006) Annual inorganic mercury speciation in river water disturbed by chlor-alkali effluents: role and competition of ligands (Cl-, Br-, DOC). Geochim Bras 20:133–147
Horvat M, Miklavčič V, Pihlar B (1991) Determination of total mercury in coal fly ash by gold amalgamation cold vapour atomic absorption spectrometry. Anal Chim Acta 243:71–79
Horvat M, Bloom NS, Liang L (1993) Comparison of distillation with other current isolation methods for the determination of methyl mercury compounds in low level environmental samples: Part II. Water. Anal Chim Acta 282:153–168
Horvat M, Covelli S, Faganeli J, Logar M, Mandic V, Rajar R, Sirca A, Zagar D (1999) Mercury in contaminated coastal environments; a case study: the Gulf of Trieste. Sci Total Environ 237(238):43–56
Huerta-Diaz MA, Morse JW (1992) Pyritization of trace metals in anoxic marine sediments. Geochim Cosmochim Acta 56:2681–2702
Jonsson S, Skyllberg U, Nilsson MB, Westlund P, Shchukarev A, Lundberg E, Björn E (2012) Mercury Methylation Rates for Geochemically Relevant Hg(II) Species in Sediments. Environ Sci Technol 46:11653–11659
Kemp M, Faganeli J, Puskaric S, Smith EM, Boynton WR (1999) Pelagic–benthic coupling and nutrient cycling. In: Malone TC et al (eds) Ecosystems at the land–sea margin: drainage basin to coastal sea. AGU, Washington DC, USA, pp 295–339
Koron N, Faganeli J (2012) Benthic fluxes of mercury during redox changes in pristine coastal marine sediments from the Gulf of Trieste (northern Adriatic Sea). J Soils Sediments 12:1604–1614
Malej A, Mozetic P, Malacic V, Terzic S, Ahel M (1995) Phytoplankton responses to freshwater inputs in a small semi-enclosed gulf (Gulf of Trieste, Adriatic Sea). Mar Ecol Prog Ser 120:111–121
Mason R, Kim E-H, Cornwell J, Heyes D (2006) An examination of the factors influencing the flux of mercury, methylmercury and other constituents from estuarine sediment. Mar Chem 102:96–110
Merritt KA, Amirbahman A (2009) Mercury methylation dynamics in estuarine and coastal marine environments—a critical review. Earth Sci Rev 96:54–66
Muresan B, Cossa D, Jezequel D, Prevot F, Kerbellec S (2007) The biogeochemistry of mercury at the sediment–water interface in the Thau Lagoon: 1. Partition and speciation. Estuar Coast Shelf Sci 72:472–484
Ogrinc N, Faganeli J (2006) Phosphorus regeneration and burial in near-shore marine sediments (the Gulf of Trieste, northern Adriatic Sea). Estuar Coast Shelf Sci 67:579–588
Shi J, Liang L, Jiang G, Jin X (2005) The speciation and bioavailability of mercury in sediments of Haihe River, China. Environ Int 31:357–365
Stravisi F (1983) The vertical structure annual cycle of the mass field parameters in the Gulf of Trieste. Boll Oceanol Teor Appl 1:239–250
Acknowledgments
The authors would like to thank Michele Giani and Cinzia De Vittor of OGS Trieste for H2S and DIC analyses, Daniela Berto of ISPRA Chioggia for DOC analyses, and Brenda Lasorsa of Battelle Marine Sciences Laboratory Sequim, WA for DMeHg analyses. Luis Carrasco kindly acknowledges a predoctoral fellowship (programa I3P) from the Consejo Superior de Investigaciones Científicas (CSIC, Spain).
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible editor: Vera Slaveykova
Rights and permissions
About this article
Cite this article
Emili, A., Carrasco, L., Acquavita, A. et al. A laboratory-incubated redox oscillation experiment to investigate Hg fluxes from highly contaminated coastal marine sediments (Gulf of Trieste, Northern Adriatic Sea). Environ Sci Pollut Res 21, 4124–4133 (2014). https://doi.org/10.1007/s11356-013-2225-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-013-2225-5