
Abstract
Introduction
Obstructive sleep apnea (OSA) is a common disorder that is associated with impaired attention, memory and executive function. However, the mechanisms underlying such dysfunction are unclear. To determine the influence of sleep fragmentation and hypoxia, this study examined the effect of sleep fragmentation and hypoxia on cognition in OSA, while controlling for potentially confounding variables including sleepiness, age and premorbid intelligence.
Method
Participants with and without OSA (N = 150) were recruited from the general community and a tertiary hospital sleep clinic. All underwent comprehensive, laboratory-based polysomnography (PSG) and completed assessments of cognition including attention, short- and long-term memory and executive function. Structural equation modelling (SEM) was used to construct a theoretically-driven model to examine the relationships between hypoxia and sleep fragmentation, and cognitive function.
Results
Although after controlling for IQ, increased sleep disturbance was a significant predictor of decreased attention (p = 0.04) and decreased executive function (p = 0.05), controlling for age removes these significant relationships. No significant predictors of memory function were found.
Conclusions
The mechanisms underlying the effects of OSA on cognition remain to be defined. Implications are discussed in light of these findings.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Obstructive sleep apnea (OSA) is a common sleep disorder [1] characterized by repeated upper airway collapse resulting in intermittent hypoxia and arousals from sleep [2]. Previous studies, comparing individuals with and without OSA, have shown that OSA is associated with impaired cognitive function, particularly in the domains of attention, memory and executive function. In the attention domain, vigilance appears to be most affected [3–5], while in the memory domain, impairment is in delayed-visual and verbal memory [4, 6, 7], and in visuospatial/constructional memory [4]. Executive function impairments have been demonstrated in shifting, updating, inhibition, generativity and fluid reasoning [3–5, 8, 9]. Several domains of cognition appear unaffected by OSA, including language abilities [3, 4], immediate visual and verbal memory [7], visuospatial learning [7], and psychomotor functions [4].
The mechanisms underlying cognitive dysfunction in OSA remain undefined. In 2002, Beebe and Gozal [10] proposed a conceptual framework based on critical roles for sleep fragmentation and nocturnal hypoxia in the development of cognitive dysfunction in individuals with OSA. In their model, sleep is viewed as a necessary restorative process, regulating processes including reinforcing foundations for learning and memory [11], and modulating neuroendocrine demands [12]. Disruption of these processes due to sleep fragmentation leads to an inability of the body to return to a balanced state, impairing neural function. The model also postulates that blood gas abnormalities (including hypoxia) due to periodic obstructed breathing compound this damage. A proposal of this model is that both hypoxia and sleep fragmentation contribute to impaired cognition in OSA, however, it remains unclear whether or not this is correct.
Substantial evidence exists for a negative impact on cognition by sleep fragmentation and/or sleep deprivation [13–16]. Indeed, some authors argue that sleep fragmentation is the key mechanism underlying impaired cognition in OSA [15, 17]. Of note, is the finding that the nature of cognitive deficits in sleep deprived individuals is similar to individuals with OSA [13–15]. Impairment in attention and executive function is prominent amongst them [13–15]. It has also been noted that the deficits in attention in individuals with OSA are associated with indices of sleep fragmentation [18].
Other research suggests that intermittent hypoxia experienced throughout the night by individuals with OSA is primarily responsible for irreparable neural damage and lasting cognitive impairments [3, 10]. Such permanent damage might explain the finding that, even with effective treatment (viz. abolition of hypoxia and sleep fragmentation), individuals with OSA retain some decrements in memory and executive functioning [4, 7, 8]. Thus, while there is considerable evidence that both sleep fragmentation [19–23] and hypoxia [4, 6, 24] lead to cognitive dysfunction in OSA, their relative importance remains unclear.
In addition to hypoxia and sleep fragmentation, other inter-individual factors including age [25], premorbid intelligence and sleepiness should be considered in any assessment of cognitive function in individuals with OSA. This is because those individuals with OSA who have high intelligence may exhibit intact performance on neuropsychological assessments, as they can recruit additional cognitive resources or utilise cognitive strategies that aid in performance, acting as a ‘buffer’ against brain insult and injury (i.e. cognitive reserve) [26]. Similarly, individual differences in age and subjective sleepiness in OSA could modify performance on neurocognitive tests because age independently affects cognition [27], and excessive sleepiness lessens the ability to direct cognitive resources to attend to the task at hand [14, 25, 28, 29].
Despite the wealth of published evidence, most studies of the cognitive burden of OSA have compared OSA to non-OSA participants [4]. By contrast, few studies have explored the relationship between the severity of OSA (e.g. as quantified by the Apnoea Hypopnea Index, AHI) and the degree of deficits experienced, and none has explored the relative contribution of the two proposed mechanisms (i.e. sleep fragmentation AND hypoxia) within the same sample, whilst controlling for premorbid intelligence, age and daytime sleepiness [25]. Thus, this study is the first comprehensively to test the relationship between the severity of sleep apnea and of cognitive deficits. To do this, we utilized structural equation modeling (SEM), a powerful analytic technique that allows testing and estimation of causal relationships between multiple potential mechanisms of cognitive dysfunction, as well as exploration of the relative importance of inter-individual factors of age, premorbid IQ and sleepiness.
Methods
Participants and protocol
In total, 150 adults (18 years and above) with and without OSA participated: 134 patients were diagnosed with OSA attending the Sleep Clinic of the West Australian Sleep Disorders Research Institute, Queen Elizabeth II Medical Centre (WASDRI-QEII), Western Australia, for diagnostic polysomnography between March 2009 and July 2011. Cognitive assessments were performed with these individuals after diagnosis but before starting treatment for OSA. To capture the full range of severity of OSA (from no disease to severe disease), an additional 16 community volunteers were recruited. These individuals were members of the West Australian Participant Pool of the University of Western Australia (WAPP-UWA), who completed a set of questionnaires on sleep health between October 2011 and October 2012, and who undertook cognitive assessment prior to a sleep study at the Centre for Sleep Science at the University of Western Australia. The study was approved by the Human Research Ethics Committees of Sir Charles Gairdner Hospital and the University of Western Australia. All participants provided written informed consent.
Measurements
Polysomnography (PSG)
required the participants to attend a full, overnight laboratory-based sleep study. Electrodes were attached according to the American Academy of Sleep Medicine (AASM) recommendations: electroencephalogram (EEG) electrode, electrooculogram (EOG) channels, electromyogram (EMG) (chin and legs), oxygen saturation via pulse oximetry (SpO2), electrocardiogram (ECG), thoracic and abdominal respiratory effort via belts, oral/nasal thermistor, nasal prongs and position sensor. Studies were analysed, and apnea-hypopnea index (AHI) and arousal index (ArI) were determined according to standard guidelines [30].
Attention and memory
were assessed using the Cognitive Drug Research System (CDR; United BioSource Corporation), a 30-min computerized battery of cognitive assessments [31, 32]. All CDR tasks were administered on a 15-in. screen laptop computer and included assessment of Attention (digit vigilance, simple and choice reaction time), long-term memory (delayed picture recognition, delayed word learning and delayed word recognition) and short-term memory (digit recall and visuo-spatial recall). Responses were provided either verbally or via a YES/NO response box.
Executive function
was assessed using the clock drawing task (CLOX 1 and 2) [33], the Controlled Oral Word Association task (COWA) and the Trail Making Test (TMT; Trails A and B) [34–36].
Premorbid intelligence
was assessed using the National Adult Reading Test-2 (NART-2), a widely accepted method of estimating premorbid IQ from current capacity, by testing reading capacity of irregular nouns [37]. The number of errors is used to estimate IQ using a regression equation, with fewer errors being associated with higher premorbid IQ. Irregular word reading ability has been shown both to correlate highly with full scale IQ [37] and to be resistant to decline due to dementia [38].
Subjective sleepiness
was assessed using the Epworth Sleepiness Scale (ESS) [39] including its separation into three factors: low, moderate and high situational somnificity [40].
All cognitive assessments were administered by graduate psychologists at the University of Western Australia, and all sleep study measurements were obtained and scored by trained PSG technicians.
Analyses
Data screening and descriptive analysis were performed using SPSS 20.0 for Windows [41].
Characterising hypoxia and sleep fragmentation
The degree of hypoxia was operationalised in two ways: (i) mean overnight SpO2 and (ii) minimum overnight SpO2. Footnote 1 The degree of sleep fragmentation was also represented in two ways: (i) sleep efficiency, the ratio of time spent asleep (total sleep time/the amount of time spent in bed) and (ii) proportion of arousals not associated with a 3 % oxygen desaturation (Total ODI3) [42], the latter being the number of times an individual’s SpO2 decreased to below 3 % of baseline. The purpose of this index was to model arousals not associated with a significant drop in oxygen, as significant respiratory events generally occur with an arousal making these two factors highly correlated.
Data screening
Little’s MCAR revealed that the data were missing completely at random, X 2(485) = 10.19, p = 1.00. The proportion of missing values was less than 10 %, thus expectation maximisation was used to replace missing values [43]. Data were assessed for normality. Many variables violated normality assumptions; hence, generalised least squares (GLS) was used to calculate the estimates of model fit [44]. Furthermore, incremental fit indices are reported, as these have been shown to be less sensitive to non-normality [44].
Confirmatory factor analysis
CFA is a confirmatory form of SEM that can be used to confirm a theorised model. SEM is an extension of multiple regression designed to test a set of hypothesised relationships between variables, that are estimated simultaneously [45]. It provides a mechanism through which to examine the relationships between hypothesized constructs, whilst controlling for individual differences, such as premorbid intelligence and sleepiness, and accounting for measurement error. SEM is particularly useful when examining hypothesized constructs such as memory, attention and executive function, as it can account for measurement error that naturally exists between the ‘pure’ construct and its measurement providing a stringent test of the latent structure [46–48]. The weights presented in the figures represent standardized regression weights, and the online supplement presents a correlation matrix of all variables included in the final model.
CFA was used to evaluate the theoretical models (Figs. 1 and 2). The fit of the models was compared through examination of the chi-square difference test and incremental model fit indices. ‘Good fit’ indicates that the hypothesised model generates a sample covariance matrix that is similar to the observed covariance matrix inherent in the data set: that is the model is a good explanation of the variance in the observed data [49]. Good fit is indicated by a non-significant chi-square value, indicating that the observed and hypothesised models are similar, and fit indices are within recommended guidelines (Table 2 foot note). Particular emphasis is placed on the values of the comparative fit index (CFI): Standardized Root Mean Squared Residual (SRMR) and Root Mean Square Error of Approximation( RMSEA), as the power and robustness of these particular indices have previously been demonstrated [46–48, 50]. The factor structure of the models was evaluated using AMOS 18.0 for Windows Version 2.0 [51]. The correlation matrix is provided in the online appendices, Appendix S1.

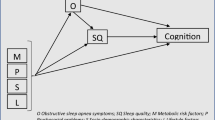
Model I-The sleep fragmentation (Sleep fragment) and oxygen desaturation (Hypoxia) model showing the relationship between nocturnal disturbance and cognitive constructs: attention, long-term memory, short-term memory and executive function, controlling for IQ. All weights are presented as standardised regression weights. Note. This model was also explored controlling for daytime sleepiness (using the ESS). ESS scores produced poor fit and were removed

Model II-The sleep fragmentation (Sleep fragment) and oxygen desaturation (Hypoxia) model showing the relationship between nocturnal disturbance and cognitive constructs: attention, long-term memory, short-term memory and executive function, controlling for IQ and age. All weights are presented as standardised regression weights. Note. This model was also explored controlling for daytime sleepiness (using the ESS). ESS scores produced poor fit and were removed
Four models were analysed. The first model examined the interrelationships between hypoxia/sleep fragmentation and cognitive function, controlling for premorbid IQ. The second model repeated this analysis, but controlled for premorbid IQ and age. A third model examined the interrelationships between AHI and cognitive function by replacing the measures of hypoxia and sleep fragmentation with AHI. This was performed because AHI is the primary measure of OSA disease severity used in current clinical practice [52, 53], and the utility of this single measure to predict cognitive dysfunction is disputed [52, 53]. The fourth model assessed the role of attention in mediating cognitive deficits in the domains of short- and long-term memory and executive function. This model was examined because cognitive deficits in OSA have been proposed to be due to attention difficulties as a consequence of sleep fragmentation [15].
Results
Descriptive data and preliminary analyses
Participants were aged 52.4 ± 13.1 years (range 19 to 82 years), BMI was 33.0 ± 7.8 kg/m2 (range 15.5 to 57.5), AHI was 37.1 ± 27.5 events/h (range 0 to 154), had received 11.9 ± 3.1 years (range 6 to 23) of education, and 69 (46 %) were female. Descriptive statistics for cognitive and sleep apnea indices are provided in Table 1.
Compared to the clinical sample (n = 134), the community sample (n = 16) was younger (p = 0.001) and had more years of education (p = 0.003). As anticipated, they had lower BMI (p = 0.003) and lower AHI (p = 0.001). There were no differences between the groups in estimated premorbid intelligence (p = 0.234). For descriptive statistics on these variables, and for the sample as a whole, see Table 2. Age and education were accounted for within the SEM analysis.
Model I: Hypoxia/sleep fragmentation vs. cognitive function
Preliminary assessment of model weights showed that neither sleepiness nor somnificity factors [40] was related to any cognitive domain (p > 0.05 for all) (Supplementary Table 2),Footnote 2 whilst premorbid intelligence was related to the cognitive domains (p < 0.05) (Fig. 1). Thus, sleepiness/somnificity was removed from subsequent models whilst premorbid intelligence was retained.
The final model (Fig. 1) was a good account of the relationships between sleep fragmentation (i.e. proportion non-ODI3 arousals and sleep efficiency) and hypoxia (i.e. mean and lowest SpO2) and cognitive function (Fig. 1). Specifically, good model fit was indicated by a non-significant chi-square statistic, X 2(79) = 91.52, p = 0.159, and incremental fit indices were within recommended guidelines (Table 3).
The strength of the interrelationships between sleep fragmentation, hypoxia and each cognitive factor (short- and long-term memory, attention and executive function) was assessed by examination of the beta weights (Fig. 1). Because the sleep fragmentation and hypoxia factors were highly correlated (r = 0.88) some paths from these factors to cognition had beta weights >1.0 [54, 55], model fit was assessed when sleep fragmentation and hypoxia were combined into a single construct, ‘sleep disturbance’ (Supplementary Fig. 1). This model included measures of ArI as a proportion of total ODI3, sleep efficiency, mean and lowest SpO2 loading into a latent construct termed ‘sleep disturbance’ and examined the relationship between these constructs and cognition. Although chi-square model fit was non-significant, the Akaike Information Criterion (AIC) indicated that the one factor model (i.e. sleep disturbance) provided a poorer fit to the data than the original model. Thus, we retained the model separating sleep fragmentation and hypoxia.
The beta weights of Model I revealed a significant relationships between sleep fragmentation and both attention (r = 1.16, p = 0.04) and executive function (r = 1.08, p = 0.03), after controlling for premorbid intelligence (Fig. 1), indicating that increased sleep fragmentation is significantly and positively related to increased difficulty with attention and with executive function.
Model II: Controlling for age
The model above was also examined controlling for age (Fig. 2). This analysis was conducted separately in order to allow examination of the relationships prior to the removal of age from the model, as age was likely to be strongly associated with disease duration.
This model was a good account of the relationships between sleep fragmentation (i.e. arousals as a proportion of total ODI3 and sleep efficiency) and hypoxia (i.e. mean and lowest SpO2) and cognitive function (Fig. 2). Specifically, good model fit was indicated by a non-significant chi-square statistic, X 2(90) = 103.72, p = 0.153, and incremental fit indices were within recommended guidelines (Table 3).
When age was added, however, there was no longer a significant relationship between either of the sleep disturbance factors and any cognitive factor. That is, the relationship between sleep fragmentation, attention and executive function was no longer found.
Model III: AHI
The model fit was also assessed when hypoxia and sleep fragmentation were replaced by AHI. For this model (Supplementary Fig. 2), chi-square was statistically significant, X 2(57) = 86.31, p = 0.007, and incremental fit indices did not meet required cut points (CFI = 0.39, RMSEA = 0.06, SRMR = 0.08). Such findings indicate that severity of AHI does not predict the level of cognitive performance in OSA.
Model IV: Cognitive dysfunction mediated by attention
A model was also developed to test the hypothesis that cognitive deficits in executive function and memory are mediated by attention (Supplementary Fig. 3). This model would not converge; hence, this model provided a poor account of the data and could not be explored further.
Discussion
This study used structural equation modelling to examine the effects on cognition of sleep fragmentation and hypoxia in OSA, while controlling for the potential confounding influences of age, premorbid intelligence, mood and sleepiness.
Before controlling for age, greater sleep fragmentation significantly predicted poorer executive function and attention, whereas hypoxia was unrelated to cognition. After controlling for age, sleep fragmentation joined hypoxia in failing to predict level of cognitive performance in any domain (attention, language, executive function or episodic memory).
This is not the first study to report no relationship between the severity of cognitive deficits and the severity of OSA (see studies reviewed by Aloia et al. [6] and presented in Table 3 of their publication). Previous studies, using the AHI, have explained their finding by arguing that AHI is a poor index of OSA severity, at least as it relates to the cognitive burden of the disorder [53]. Other authors have suggested that the lack of a relationship is due to the lack of control of the inter-individual factors of age, premorbid IQ, daytime sleepiness or attention [15, 26, 53, 56].
This study addressed all of these methodological criticisms and still failed to find a relationship between OSA severity and cognition. What should be made of such findings?
The simplest explanation for the results of the present and other similar studies [6, 7] is that there is no dose–response relationship in OSA. That is to say, over a critical threshold there will be neural damage and cognitive dysfunction, but this will not increase with increasing severity of sleep fragmentation or hypoxia.
However, given that cognitive performance is related to time spent without oxygen in other instances of hypoxia (e.g. climbing at altitude [57] and carbon monoxide poisoning [58]) and longer time exposed to sleep deprivation results in greater cognitive dysfunction [28], this account seems unlikely. An alternative hypothesis is that hypoxia or sleep fragmentation is not captured appropriately and/or that the correct mechanisms of harm are not being captured.
An individual’s level of hypercapnia is closely linked to their level of hypoxia, and excessive levels of carbon dioxide are found in individuals with OSA [30, 59]. Yet, hypercapnia is not routinely explored in OSA-cognition research, despite being included as a potential mechanism of harm in the dominant model of cognitive harm in OSA [10]. Further, there is evidence that the degree of hypercapnia correlates with overall cognitive impairment [60].
Alternatively, routine indices of sleep fragmentation (ArI) and hypoxia (SpO2) may be insufficiently sensitive to the extent of sleep disturbance or the length and depth of hypoxia [61]. An individual with OSA may exhibit many small fragmentations of sleep architecture, shallow or lengthy oxygen desaturations, not captured by standard indices. Novel measures that capture more detail about sleep fragmentation [62] or describe the length and depth of desaturation (e.g. Integrated Area of Desaturation) [61] may demonstrate greater sensitivity to a dose–response relationship in OSA.
The impact of inter-individual differences: Age and IQ
The final model showed that, when premorbid IQ was accounted for, more severe sleep fragmentation, but not hypoxia, was significantly associated with poorer attention and executive dysfunction, but not with poorer memory. However, adding age resulted in these relationships becoming non-significant. A simple explanation for this finding is that age accounts for all of the cognitive deficits seen in OSA. This is unlikely, as case-control studies (with participants matched for age) reveal clear evidence of cognitive deficits in OSA [4]. Furthermore, not all individuals experience age-related cognitive decline, nor are all individuals affected equally. Indeed, many older adults stay cognitively healthy [63–65]. Given that there is no good measure of disease duration in OSA, and that older individuals are likely to have experienced OSA for longer, age itself is confounded with disease duration. By controlling for age, it is possible that the variance of interest in an exploration of the mechanisms of cognitive harm in OSA has been discarded. One of the challenges for the field of sleep medicine, therefore, is to develop sound methods of estimating or measuring disease duration.
The choice of measures of sleep fragmentation and hypoxia in the present study was guided by indices that are routinely produced by most sleep software and that have previously been shown to be related to aspects of cognition [6]. Statistical modelling showed that measures of hypoxia and sleep fragmentation exhibited a large degree of covariation, indicating shared underlying variance, even after calculating an index of arousals independent of 3 % oxygen desaturation. The importance of this covariance was investigated by modifying the original model such that sleep fragmentation and hypoxia were considered as a single factor by: (i) loading all measured variables from the latent factors ‘sleep fragmentation’ and ‘hypoxia’ onto a single factor (termed sleep disturbance) and (ii) replacing sleep fragmentation and hypoxia with AHI. In both cases, the resultant model fits were not improved relative to the model that utilised separate constructs of hypoxia and sleep fragmentation, suggesting that separating out these components of potential harm was the correct approach.
The present study was also careful to consider the potentially important confounding influences of premorbid intelligence and sleepiness in assessing cognitive function in individuals with OSA. The finding that premorbid intelligence was positively related to cognition was expected and underscores the importance of controlling for this when evaluating cognition in OSA [26, 36, 66, 67]. In contrast, analysis of subjective sleepiness revealed that neither ESS total nor factors of situational somnificity were related to cognition. Such findings support studies reporting that subjectively measured sleepiness and somnificity are not related to cognition in OSA [68]. One reason for this might be that individuals with chronic sleep restriction, as occurs in OSA, can become desensitized to the feeling of sleepiness [14].
The role of attention in mediating cognitive deficits in OSA warranted a separate analysis, given the proposal that sleep fragmentation might cause impaired attention which, of itself, could lead to impaired memory and executive function [15, 17, 69]. However, the resultant model fit was poor, suggesting that attention deficits did not explain the effect of sleep fragmentation on attention and executive function.
Cognitive damage, but no relationship to disease severity
Despite growing evidence that OSA is associated with cognitive impairment [4], the findings of the present study, and those of meta-analyses and systematic reviews, have revealed no consistent evidence that the degree of sleep fragmentation or hypoxia is related to the degree of cognitive dysfunction in OSA [6, 7]. There are several possible reasons for this lack of association.
First, it is possible that the cognitive dysfunction in OSA is due to a mechanism not captured in the models presented in the current study. One such mechanism could be recruitment of additional brain regions to compensate for poor cognitive performance. Consistent with this hypothesis, Ayalon et al. [66] reported that individuals with OSA recruit additional brain areas, not typically recruited during a verbal learning task, and Castronovo et al. [67] reported increased activation in the left frontal cortex, medial precuneus and hippocampus in OSA patients, whilst performance was at the same level as controls.
Second, individual differences might play a more critical role than previously credited [14]. Such a concept arises from emerging research indicating that the relationship between disease severity and cognitive dysfunction is the product of a multitude of vulnerability and protective factors [10, 70], of which sleep fragmentation, hypoxia and cognitive reserve are only three aspects [14, 71, 72]. These other facets could include duration of disease (as noted above) [72], genetic vulnerability (e.g. apolipoprotein e4 genotype) [71, 72], the role of the blood brain barrier [73] and cerebral blood flow [74].
Third, the present study examined attention, memory and executive function as whole domains, rather than exploring separate facets of these domains [7, 8]. Cognitive domains such as executive function are not unitary constructs and should be considered umbrella terms for a range of different cognitive capacities [75–77]. For example, executive function can be divided into shifting, updating, inhibition, generativity and fluid reasoning [75–77]. Decomposing each cognitive domain, and then exploring the contribution of a range of risk and protective factors specific to OSA, could enable a more targeted analysis of the impact of OSA on cognition.
Last, as mentioned briefly before, measurement of hypoxia through SpO2 may be a poor proxy measure of the underlying mechanism by which hypoxia causes harm, namely oxidative stress [78, 79], particularly given that oximetry measured below 80 % SpO2 is not particularly reliable or accurate [80]. Animal models suggest that oxidative stress and apoptosis-related neural injury might be measured more directly by Brain Derived Neurotrophic Factor (BDNF) [81], thioredoxin [78] or NADPH Oxidase [79]. It remains unclear whether such measurements would be of value in individuals with OSA.
Conclusions
Our findings demonstrate that the relationship of cognitive deficit to sleep fragmentation and hypoxia in OSA is still unclear. That OSA causes cognitive impairment is not in doubt. However, until disease duration can be measured and specific mechanisms of harm, the nature of protective and risk factors, and their relationship to specific aspects of cognitive ability are established, there is no basis on which to identify who is most at risk of cognitive decline or how to intervene most effectively.
Notes
Other indices of sleep fragmentation (e.g. TST, ArI during REM) and the following measures of hypoxia: CT90, Lowest SaO2 during REM, Mean SaO2 during REM, were considered and tested. None accounted for sufficient variance in cognitive performance to be included.
All supplementary tables are available from Research Gate: https://www.researchgate.net/publication/261676388_Cognition_and_nocturnal_disturbance_in_OSA_the_importance_of_accounting_for_age_and_premorbid_intelligence#share
References
Young T, Peppard PE, Gottlieb DJ (2002) Epidemiology of obstructive sleep apnea. Am J Respir Crit Care Med 165(9):1217–1239
Kryger MH (2010) Atlas of clinical sleep medicine. Saunders Elsevier, Philadelphia
Beebe DW, Groesz L, Wells C, Nichols A, McGee K (2003) The neuropsychological effects of obstructive sleep apnea: a meta-analysis of norm-referenced and case-controlled data. Sleep 26:298–307
Bucks R, Olaithe M, Eastwood P (2012) Neurocognitive function in obstructive sleep apnoea: A meta-review. Respirology 18(1):61–70
Fulda S, Schulz H (2003) Cognitive dysfunction in sleep-related breathing disorders: a meta-analysis. Sleep Res Online 5:19–51
Aloia MS, Arnedt T, Davis JD, Riggs RL, Byrd D (2004) Neuropsychological sequelae of obstructive sleep apnea-hypopnea syndrome: a critical review. J Int Neuropsychol Soc 10:772–785
Wallace A, Bucks RS (2012) Memory and Obstructive Sleep Apnoea: A Meta-Analysis. Sleep 36(2):203–220
Olaithe M, Bucks R (2013) Executive dysfunction in OSA before and after treatment: a meta-analysis. Sleep 36:1297–1305
Saunamäki T, Jehkonen M (2007) A review of executive functions in obstructive sleep apnea syndrome. Acta Neurol Scand 115:1–11
Beebe DW, Gozal D (2002) Obstructive sleep apnea and the prefrontal cortex: Towards a comprehensive model linking nocturnal upper airway obstruction to daytime cognitive and behavioral deficits. J Sleep Res 11:1–16
Poe G, Walsh CM, Bjorness TE (2010) Cognitive neuroscience of sleep. Prog Brain Res 185:1–19
Everson CA (1995) Functional consequences of sustained sleep deprivation in the rat. Behav Brain Res 69:43–54
Alhola P, Polo-Kantola P (2007) Sleep deprivation: Impact on cognitive performance. Neuropsychiatr Dis Treat 3:553–567
Durmer JS, Dinges DF (2005) Neurocognitive Consequences of Sleep Deprivation. Seminars in Neurology 25(1):117–129
Verstraeten E, Cluydts R, Pevemagie D, Hoffman G (2004) Executive function in sleep apnea: controlling for attentional capacity in assessing executive attention. Sleep 27:685–693
Stepanski EJ (2002) The effect of sleep fragmentation on daytime function. Sleep 25:268–276
Verstraeten E, Cluydts R (2004) Executive control of attention in sleep apnea patients: Theoretical concepts and methodological considerations. Sleep Med Rev 8:257–267
Ayalon L, Ancoli-lsrael S, Aka AA, McKenna BS, Drummond SP (2009) Relationship between obstructive sleep apnea severity and brain activation during a sustained attention task. Sleep: J Sleep Sleep Disord Res 32:373–381
Bartlett D, Rae C, Thompson C, Byth K, Joffe D et al (2004) Hippocampal area metabolites relate to severity and cognitive function in obstructive sleep apnea. Sleep Med 5:593–596
Chiang AA (2006) Obstructive sleep apnea and chronic intermittent hypoxia: a review. Chin J Physiol 49:234–243
Hopkins RO, Kesner RP, Goldstein M (1995) Memory for novel and familiar spatial and linguistic temporal distance information in hypoxic subjects. J Int Neuropsychol Soc 1:454–468
Naismith S, Winter V, Gotsopoulos H, Hickie I, Cistulli P (2011) Neurobehavioral functioning in obstructive sleep apnea: Differential effects of sleep quality, hypoxemia and subjective sleepiness. J Clin Exp Neuropsychol 26:43–54
Gale SD, Hopkins RO (2004) Neuroimaging and neuropsychological findings following carbon monoxide poisoning and obstructive sleep apnea. J Int Neuropsychol Soc 10:60–71
Saunamäki T, Himanen S-L, Polo O, Jehkonen M (2010) Executive dysfunction and learning effect after continuous positive airway pressure treatment in patients with obstructive sleep apnea syndrome. Eur Neurol 63:215–220
Naismith S, Winter V, Gotsopoulos H, Hickie I, Cistulli P (2004) Neurobehavioral functioning in obstructive sleep apnea: Differential effects of sleep quality, hypoxemia and subjective sleepiness. J Clin Exp Neuropsychol 26:43–54
Alchanatis ZN, Deligiorgis N, Amfilochiou A, Dionellis G et al (2005) Sleep apnea-related cognitive deficits and intelligence: an implication of cognitive reserve theory. J Sleep Res 14:69–75
Alchanatis ZN, Deligiorgis N, Chroneou A, Soldatos C et al (2008) Comparison of cognitive performance among different age groups in patients with obstructive sleep apnea. Sleep Breathing 12:17–24
Durmer JS, Dinges DF (2005) Neurocognitive consequences of sleep deprivation. Semin Neurol 25:117–129
Guilleminault C, Brooks SN (2001) Excessive daytime sleepiness; a challenge for the practising neurologist. Brain 124:1482–1491
AASM (2007) The AASM manual for the scoring of sleep and associated events; rules, terminology and technical specifications. Iber C, Ancoli-Israeli S, Chesson AL, Quan SF (ed) American Academy of Sleep Medicine, Illinois
Wesnes KA (2000) Predicting, assessing, differentiating and treating the dementias: experience in MCI and various dementias using the CDR computerised cognitive assessment system. In: Vellas B, Fitten LI (eds) Research and practice in Alzheimer’s disease. Serdi, Paris, pp 59–65
Van Den Goor JM, Saxby BK, Tijssen TG, Wesnes KA, de Mol BA et al (2008) Improvement of cognitive test performance in patients undergoing primary CABG and other CPB-assisted cardiac procedures. Perfusion 23:267–273
Royall DR, Cordes JA, Polk M (1998) CLOX: an executive clock drawing task. J Neurol Neurosurg Psychiatry 64:588–594
Partington JE, Leiter RG (1949) Partington's pathway test. Psychol Serv Center Bull 1:9–20
Reitan RM (1958) Validity of the trail making test as an indicator of organic brain damage. Percept Mot Skills 8:271–276
AASM (1999) Sleep-related breathing disorders in adults: Recommendations for Syndrome Definition and Measurement Techniques in Clinical Research. Sleep 22:667–689
Nelson HE, Willison J (1991) National Adult Reading Test Manual. NFER-Nelson, Wilson
McGurn B, Starr JM, Topfer JA, Pattie A, Whiteman MC et al (2004) Pronunciation of irregular words is preserved in dementia, validating premorbid IQ estimation. Neurology 62:1184–1186
Johns MW (1991) A new method for measuring daytime sleepiness: the Epworth sleepiness scale. Sleep 14:540–545
Olaithe M, Skinner TC, Clarke J, Eastwood P, Bucks RS (2012) Can we get more from the Epworth Sleepiness Scale than just one score? a confirmatory factor analysis of the ESS. Sleep Breathing 17:763–769
IBM (2012) IBM SPSS Statistics for Windows. 21.0 ed. IBM Corporation, Armonk
Sulit L, Siorfer-Isser A, Kirchner L, Redline S (2006) Differences in polysomnography predictors for hypertension and impaired glucose tolerance. Sleep 29:777–783
Cohen J, Cohen P (1983) Applied multiple regression/correlation analysis for the behavioral sciences Hillsdale. Erlbaum, NJ
Lei M, Lomax RG (2005) The Effect of Varying Degrees of Nonnormality in Structural Equation Modeling. Struct Equ Model 12:1–27
Ullman JB, Bentler PM (2012) Structural equation modeling. In: Weiner IB (ed) Handbook of psychology, 2nd edn. John Wiley & Sons, Wiley Online Library
Iaccobucci D (2010) Structural equation modelling: Fit indices, sample size, and advanced topics. J Consum Psychol 20:90–98
Byrne BM (2010) Structural Equation Modelling with AMOS: Basic concepts, application and programming (2nd Ed.). Structural Equation Modelling with AMOS: Basic concepts, application and programming. Taylor & Francis Group, New York
MacCallum RC, Austin JT (2010) Applications of structural equation modelling in psychology research. Annu Rev Psychol 51:201–226
Schreiber JB, Stage FK, King J, Nora A, Barlow EA (2006) Reporting structural equation modeling and confirmatory factor analysis results: a review. J Educ Res 99:323–337
Hu L, Bentler PM (1999) Cutoff criteria for fit indexes in covariance structure analysis: Conventional criteria versus new alternatives. Struct Equ Model 6:1–55
IBM (2012) IBM AMOS Statistics for Windows 21.0 ed. IBM Corporation, Armonk
Kribbs NB, Pack AI, Kline LR, Getsy JE, Schuett JS et al (1993) Effects of one night without nasal CPAP Treatment on sleep and sleepiness in patients with obstructive sleep apnea. Am Rev Respir Disord 147:1162–1168
Tsai JCG (2010) Neurological and neurobehavioural sequelae of obstructive sleep apnea. Neurorehabilitation 26:85–94
Deegan JJ (1978) On the occurrence of standardized regression coefficients greater than one. Educ Psychol Meas 38:873–888
Joreskog KG (1999) How large can a standardized coefficient be. Unpublished report: SSI Central, Inc. pp. http://www.ssicentral.com/lisrel/techdocs/HowLargeCanaStandardizedCoefficientbe.pdf. June 2013
Alchanatis ZN, Deligiorgis N, Liappas I, Chroneou A et al (2008) Comparison of cognitive performance among different age groups in patients with obstructive sleep apnea. Sleep Breathing 12:17–24
Regard M, Oelz O, Brugger P, Landis M (1989) Persistent cognitive impairment in climbers after repeated exposure to extreme altitude. Neurology 39:210–213
Hopkins RO, Woon FLM (2006) Neuroimaging, cognitive, and neurobehavioral outcomes following carbon monoxide poisoning. Behav Cognitive Neurosci Rev 5:141–155
Kaw R, Hernandez AV, Walker E, Aboussouan L, Mohlesi B (2009) Determinants of hypercapnia in obese patients with obstructive sleep apnea: a systematic review and meta-analysis of cohort studies. CHEST 136:787–796
Findley LJ, Barth JT, Powers DC, Wilhoit SC, Boyd DG et al (1986) Cognitive impairment in patients with obstructive sleep apnea and associated hypoxemia. Chest 90:686
Asano K, Takata Y, Usui Y, Shiina K, Hashimura Y et al (2009) New index for analysis of polysomnography, 'integrated area of desaturation', is associated with high cardiovascular risk in patients with mild to moderate obstructive sleep apnea. Respiration 78:278–284
Pepin JL, Delavie N, Pin I, Deschaux C, Argod J et al (2005) Pulse transit time improves detection of sleep respiratory events and microarousals in children. CHEST 127:722–730
Hultsch DF, Hertzog C, Small BJ, Dixon RA (1999) Use it or lose it: Engaged lifestyle as a buffer of cognitive decline in aging? Psychol Ageing 14:245–263
Kramar AF, Erikson KI, Colcombe J (2006) Exercise, cognition, and the aging brain. J Appl Physiol 101:1237–1242
Norbury R, Craig M, Cutter WJ, Whitehead M, Murphy D (2004) Oestrogen: brain ageing, cognition and neuropsychiatric disorder. Menopause Int 10
Ayalon L, Ancoli-Israel S, Klemfuss Z, Shalauta MD, Drummond SPA (2006) Increased brain activation during verbal learning in obstructive sleep apnea. Neuroimage 31:1817–1825
Castronovo V, Canessa N, Ferini-Strambi L, Aloia MS, Consonni M et al (2009) Brain activation changes before and after PAP treatment in obstructive sleep apnea. Sleep 32:1161–1172
Incalzi RA, Marra C, Salvigni BL, Petrone A, Gemma A et al (2004) Does cognitive dysfunction conform to a distinctive pattern in obstructive sleep apnea syndrome? J Sleep Res 13:79–86
Verstraeten E (2007) Neurocognitive effects of obstructive sleep apnea syndrome. Curr Neurol Neurosci Rep 7:161–166
Beebe DW (2006) Neurobehavioral effects of obstructive sleep apnea: an overview and heuristic model. Curr Opin Pulm Med 11:494–500
Zimmerman ME, Aloia MS (2012) Sleep-disordered breathing and cognition in older adults. Curr Neurol Neurosci Rep 12:537–546
Casseli RJ (2008) Obstructive sleep apnea, apolipoprotein E e4, and mild cognitive impairment. Sleep Med 9:816–817
Lim DC, Pack AI (2013) Obstructive sleep apnea and cognitive impairment: Addressing the blood-brain barrier. Sleep Med Rev 18(1):35–48
Kiralti PO, Demir AU, Volkan-Salanci B, Demir B, Sahin A (2010) Cerebral blood flow and cognitive function in obstructive sleep apnea syndrome. Hell J Nucl Med 13:138–143
Miyake A, Friedman NP, Emerson MJ, Witzki AH, Howerter A (2000) The Unity and Diversity of Executive Functions and Their Contributions to Complex “Frontal Lobe” Tasks: A Latent Variable Analysis. Cogn Psychol 41:49–100
Lezak MD, Howieson DB, Loring DW (2004) Neuropsychological assessment. Oxford University Press, New York
Fisk JE, Sharp CA (2004) Age-related impairment in executive functioning: Updating, inhibition, shifting, and access. J Clin Exp Neuropsychol 26:874–890
Nair D, Dayyat EA, Zhang SX, Wang Y, Gozal D (2011) Intermittent Hypoxia-Induced Cognitive Deficits Are Mediated by NADPH Oxidase Activity in a Murine Model of Sleep Apnea. PLoS One 6: Epub Ahead of Publication
Guo Q, Yan W, Qing L, Min L, Huan W (2013) Levels of thioredoxin are related to the severity of obstructive sleep apnea: Based on oxidative stress concept. Sleep Breathing 17:311–316
Razi E, Akbari H (2006) A comparison of arterial oxygen saturation measured both by pulse oximeter and arterial blood gas analyzer in hypoxemic and non-hypoxemic pulmonary diseases. Turkish Repir J 7:43–47
Xie H, Yung W (2012) Chronic intermittent hypoxia-induced deficits in synaptic plasticity and neurocognitive functions: a role for brain-derived neurotrophic factor. Acta Pharmacol Sin 33:5–10
Disclosures
The authors were not provided with any financial support for this research.
Conflicts of interest
PE is a consultant and a member of the Medical Advisory Board for Apnex Medical Inc., and a member of the Medical and Industry Advisory Board, Sleep for Health and Safety Inc. DH is a consultant and a member of the Medical Advisory Board of Apnex Medical Inc., and conducts paid speaking engagements for Philips Respironics.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Figure 1
(DOCX 38 kb)
Supplementary Figure 2
(DOCX 35 kb)
Supplementary Figure 3
(DOCX 35 kb)
Supplementary Table 1
(DOCX 18 kb)
Supplementary Table 2
(DOCX 14 kb)
Rights and permissions
About this article

Cite this article
Olaithe, M., Skinner, T.C., Hillman, D. et al. Cognition and nocturnal disturbance in OSA: the importance of accounting for age and premorbid intelligence. Sleep Breath 19, 221–230 (2015). https://doi.org/10.1007/s11325-014-1000-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11325-014-1000-2