
Abstract
Introduction
The Metabolomics Standards Initiative has recommended four categories for metabolite assignments in NMR-based metabolic profiling studies. The “putatively annotated compound” category is most commonly reported by metabolomics investigators. However, there is significant ambiguity in reliability of “putatively annotated compound” assignments, which can range from low confidence made on minimal corroborating data to high confidence made on substantial corroborating data.
Objectives
To introduce a new ranking system, Rank and AssigN Confidence to Metabolites (RANCM), to assign confidence levels to “putatively annotated compound” assignments in NMR-based metabolic profiling studies.
Methods
The ranking system was constructed with three confidence levels ranging from Rank 1 for the lowest confidence assignment level to Rank 3 for the highest confidence assignment level. A decision tree was constructed to guide rank selection for each metabolite assignment.
Results
Examples are provided from experimental data demonstrating how to use the decision tree to make confidence level assignments to “putatively annotated compounds” in each of the three rank levels. A standard Excel sheet template is provided to facilitate decision-making, documentation and submission to data repositories.
Conclusion
RANCM is intended to reduce the ambiguity in “putatively annotated compound” assignments, to facilitate effective communication of the degree of confidence in “putatively annotated compound” assignments, and to make it easier for non-experts to evaluate the significance and reliability of NMR-based metabonomics studies. The system is straightforward to implement, based on the most common datasets collected in NMR-based metabolic profiling studies, and can be used with equal rigor and significance with any set of NMR datasets.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Metabolomics as a discipline has existed in some fashion (though without the moniker) since the middle of the twentieth century but it was first formally described as such in 1999 (Nicholson et al. 1999). The rapid development and expansion of the field over the next two decades has spawned large efforts to standardize reporting of metabolomics data (Sumner et al. 2007) and the creation of large-scale data repositories to collect raw data from metabolomics studies, e.g. the internationally supported MetaboLights database (Haug et al. 2013) and the United States National Institutes of Health funded Metabolomics Workbench database (Sud et al. 2016). A comprehensive set of recommendations regarding standardization of all aspects of metabolomics studies, including experimental design, sample preparation, data collection, data analysis and data reporting, was established by the Metabolomics Standards Initiative (MSI) group and published in 2007 (Sumner et al. 2007). Despite these early efforts, debate continues with respect to universal reporting standards (Creek et al. 2014; Salek et al. 2013). Standardized guidelines for reporting metabolite assignments from experimental NMR data, while addressed by the MSI recommendations, remain inadequately defined, and standardized reporting of metabolite assignments has not been universally adopted by the metabolomics community.
One of the earliest efforts to create a resource to aid in metabolite identification in metabolomics studies was the METabolite LINk (METLIN) database, established in 2005, which compiled mass spectrometry data on metabolite standards to provide a resource for investigators identification of metabolites in mass-spectrometry based metabolomics studies (Guijas et al. 2018; Smith et al. 2005). The Human Metabolite Data Base (HMDB) followed soon after in 2007 (Wishart et al. 2018, 2013, 2009, 2007), which was established as a free public database that included one-dimensional and two-dimensional NMR spectra of reference metabolites, in addition to mass spectrometry data, and biological, chemical, and clinical information about the metabolites. The HMDB has also been integrated into other metabolite analysis programs such as ChenomX (Edmonton, Alberta, Canada) (https://www.chenomx.com/). Another valuable resource for identification of metabolites in NMR based metabolic profiling studies is the BMRB, which was established in 2008 (Ulrich et al. 2008). More recently, powerful tools for analysis of two dimensional homonuclear and heteronuclear NMR data in support of making metabolite assignments in complex biological samples have been introduced (Bingol et al. 2014), in particular, the Complex Mixture Analysis by NMR, i.e. COLMAR (Bingol et al. 2016).
Following the development of the METLIN and HMDB online databases, over the course of several years (approximately from 2005 to 2007), the MSI’s Chemical Analysis Working Group (CAWG) discussed and developed qualitative reporting standards to use when reporting metabolite data (Fiehn et al. 2007). These standards have been considered the primary minimum reporting standards over the past 10 years (Creek et al. 2014; Salek et al. 2013; Spicer et al. 2017).
Notwithstanding, looking back on more than a decade of development, there remains a lack of conformity in adhering to minimum reporting standards in many metabolomics studies published after the 2007 MSI paper outlining reporting standards, with reporting standards poorly implemented or largely ignored by both publishers and researchers (Salek et al. 2013). This problem has been further exacerbated by the lack of consensus and consistency, or complete omission, with respect to reporting confidence levels in metabolite assignments and the lack of practical options for ranking and reporting confidence levels in metabolite assignments.
Qualitative, quantitative, and integrated methods have been suggested for metabolite identification in metabonomics studies (Bingol et al. 2016; Creek et al. 2014; Dona et al. 2016; Everett 2015; Posma et al. 2017; Rohnisch et al. 2018; Salek et al. 2013; Sanchon-Lopez and Everett 2016; Spicer et al. 2017; Tardivel et al. 2017; van der Hooft and Rankin 2016). Qualitative methods largely rely on the original guidelines set out by the MSI and CAWG, which involve categorizing putative metabolites by a variety of parameters and methods (Creek et al. 2014; Spicer et al. 2017). Quantitative methods vary, but typically are based on an exhaustive chemical analysis to generate a ‘score’ for each possible metabolite using a combination of chemical parameters and database comparison from multiple experiments (Bingol et al. 2016; Everett 2015; Sanchon-Lopez and Everett 2016). As of this writing there is not a widely accepted method that has been effectively developed and adopted to facilitate assignment and reporting of confidence levels in metabolite identifications from experimental NMR data (Spicer et al. 2017).
The MSI standards for classifying metabolite assignments (Sumner et al. 2007) are listed below:
-
Identified compounds (there is a significant amount of additional information that is recommended by the CAWG/MSI for this level of significance).
-
Putatively annotated compounds (e.g. assignments made without reference to chemical standards, based upon physicochemical properties and/or spectral similarity with public/commercial spectral libraries).
-
Putatively characterized compound classes (e.g. based upon characteristic physicochemical properties of a chemical class of compounds, or by spectral similarity to known compounds of a chemical class).
-
Unknown compounds—although unidentified or unclassified these metabolites can still be differentiated and quantified based upon spectral data.
For a metabolite to be “identified”, at least two orthogonal properties of an authentic reference standard measured in the researcher’s laboratory must be matched to features measured in the sample spectrum. However, most laboratories do not maintain a comprehensive validated library for making such metabolite identifications. Rather, most laboratories rely on reference to, and comparison with, standard compound reference spectra available in large commercial databases (e.g. ChenomX) or federally funded or public databases (HMDB, COLMAR) for making metabolite identifications. Consequently, in the context of the MSI guidelines, most NMR-based metabolomics researchers make “putatively annotated compound” assignments based on spectral similarity to commercial or public databases. The problem herein lies with the significant and inherent ambiguity in making the assignment of a “putatively annotated compound”. For example, the ambiguity associated with a “putatively annotated compound” assignment can range from a high degree of uncertainty, e.g. in the case where the assignment is based on a single matching singlet without any further corroboration from the database spectra, to an assignment that has essentially no uncertainty, e.g. in the case of an assignment being based on multiple spectral features, including both singlets and multiplets with matching intensities, and corroborated by more than one 2D NMR dataset, but not confirmed by an authentic reference standard in the researcher’s own laboratory. In these two extreme examples, both assignments would be indicated as a “putatively annotated compound” in a publication, and researchers in other laboratories or fields would have no way of knowing the relative magnitude of the uncertainty, ambiguity, or reliability in classifying the assignment as a “putatively annotated compound”.
Here, we introduce the Rank and AssigN Confidence to Metabolites (RANCM) algorithm, pronounced “Rank ‘Em”, to augment and refine the “putatively annotated compound” classification category suggested in the MSI recommendations. Our goal is to minimize the ambiguity associated with the “putatively annotated compound” assignment classification, and to provide a platform for which all assignments to this category can be documented in a common format that can be submitted to the data repository sites alongside the raw data, thus providing a resource that can be reviewed by other investigators. The significance of preserving such documentation in public data repositories can be appreciated when considering that most researchers who conduct NMR-based metabolomics studies draw biochemical and biological conclusions based on “putatively annotated compound” assignments, and therefore providing and preserving the documentation supporting the “putatively annotated compound” assignments will provide critically valuable information to future investigators considering the published work. In the new RANCM algorithm, we introduce a 3-point ordinal ranking scale for assigning confidence to putative metabolite annotations. The 3-point ordinal ranking scale makes use of experimentally acquired 1D and/or 2D NMR spectra in combination with the widely and commonly used reference databases such as ChenomX, the HMDB and COLMAR. The 3-point ordinal scale is supported by a detailed decision tree to facilitate easy assignment to a specific rank. We recommend that the ranking scores be reported in some manner in NMR-based metabolomics publications, either in a table or in supplementary material. Further, a standardized Excel spread sheet template is provided to aid in implementation of the RANCM algorithm, which helps in deciding the rank assignment for each metabolite, and to document the data used to make the rank assignments, which can be submitted to data repository sites along with raw NMR datasets.
2 Materials and methods
2.1 Samples
Urine samples from mice from the NOD-ShiLtJ mouse model of type 1 diabetes (unpublished data) were used to demonstrate the ranking scheme introduced in this study. The NOD-ShiLtJ urine samples were obtained by placing the mouse into a metabolism cage for 12 h and collecting urine into a beaker containing light mineral oil to prevent evaporation and sodium azide to prevent bacterial growth. The urine under the light mineral oil layer was collected by pipette and centrifuged at 10,000xg for 5 min at 4 °C, and liquid supernatant was collected. The samples were prepared for NMR analysis by mixing 540 µL of urine with 66 µL of a pH 7.4 phosphate buffer containing 10 mM trimethylsilylpropanoic acid (TSP) as a chemical shift reference. The samples were pH adjusted to 7.4 by adding HCl and NaOH as necessary, and finally added to 60 µL of D2O. The resulting 666 µL solutions were frozen at -80 °C until NMR experiments could be conducted. 600 µL of each sample was transferred into a 5 mm NMR tube for NMR analysis. The animal research protocol for this project (protocol number 898) was approval by the Miami University Institutional Animal Care and Use Committee.
2.2 NMR data collection
All NMR experiments on the NOD-ShiLtJ samples were conducted on a Bruker Avance Spectrometer at 298°K and 850.104 MHz. One-dimensional 1H CPMG or NOESY NMR experiments and two-dimensional 1H-1H TOCSY and 1H-13H HSQC NMR experiments were performed as described in our previous publications (Chihanga et al. 2018a, b, c; Romick-Rosendale et al. 2009, 20122014; Schmahl et al. 2018; Wang et al. 2015; Watanabe et al. 2012a, b, 2011). The CPMG experiment was generally preferred as it consistently produced superior baselines free from distortion due to incompletely suppressed water signal and residual protein and lipid in the samples, although it can introduce minor peak intensity distortions away from the water signal due to the T2 filtering scheme. Phase correction, baseline correction, and other necessary processing of spectra were performed manually using TopSpin 3.5. All NMR data used to demonstrate the use of the ranking algorithm have been deposited to the figshare data repository (https://figshare.com/s/1b2fcd14658f32e65fcb) and the MetaboLights data repository (accession ID: MTBLS781). The complete description of the files included in the data repositories in included in Supplementary material file ESM_3.
2.3 Software used for metabolite annotations
One-dimensional NMR spectra were investigated using the ChenomX Profiler software (https://www.chenomx.com/). Putative metabolite identifications made using the ChenomX software were further analyzed using the HMDB (http://www.hmdb.ca/) (Wishart et al. 2007, 2009, 2013, 2018 ). Analysis of 2D NMR data was performed using the COLMAR software (http://spin.ccic.ohio-state.edu/) (Bingol et al. 2014, 2016). Ranking score assignments were determined using the decision tree described below.
2.4 Construction of the ranking decision tree
As we are suggesting and introducing the use of a new ranking scheme to define confidence levels in “putative metabolite assignments”, we provide some context for the philosophy underlying the construction of the decision tree. The overall construction was intended to reflect our own heuristic experience encountered when going through the process of metabolite identification. The decision tree is meant to parallel the very process and decisions that the investigator follows during the process. With the advent of the widely used public databases and tools that aid in metabolite identification, we naturally screen the databases in order to identify putative compounds that might explain peaks in the experimental spectra. This naturally leads to metabolites that have only one or more singlets and metabolites that have multiple peaks including multiplets. We therefore designed an algorithm that contains these two major branches. Depending on which branch the metabolite belongs to, there are different kinds of data that can be used to support the putative assignment. This includes varying levels of confirmation by 2D HSQC data in the cases of metabolites containing only singlets and a combination of 2D TOCSY and 2D HSQC experiments in the case that a metabolite contains multiple peaks including multiplets. In constructing the algorithm, we created three tiers of questions resulting in assignments to guide ranking of the confidence in the putative metabolite assignments. The 3-point ordinal scale and decision tree are constructed as follows:
2.4.1 Rank 1S/1
Rank 1 or Rank 1S corresponds to the lowest confidence level assignment of a “putatively annotated compound”. A “putatively annotated compound” assigned at this rank is uncertain but possible. Researchers should be cautious about drawing biochemical or biological conclusions from Rank 1 “putatively annotated compound” assignments. A “putatively annotated compound” is assigned a Rank = 1 or Rank 1S when one of the following scenarios is true.
Rank 1S: The putative metabolite has one or more singlets in the 1D 1H spectrum, and
-
a.
The intensity and chemical shift of the singlets in the sample spectrum match that of the putative metabolite in a 1D reference spectrum database (e.g. ChenomX).
-
b.
The singlet assignment is not supported by 2D data, i.e. either a cross peak in a 2D 1H-13C HSQC or in a 1H-15N HSQC, (with the sign of a 1H-13C cross-peak being consistent with that expected from multiplicity editing (Willker et al. 1993)).
Rank 1: The putative metabolite has multiple peaks, including multiplets, and
-
a.
The intensities and chemical shifts of at least two multiplets matches that of the putative metabolite in a 1D reference spectrum database (e.g. ChenomX).
-
b.
None of the matching peak assignments are supported by 2D homonuclear 1H-1H TOCSY data.
-
c.
None of the matching peaks are supported by 2D heteronuclear 1H-13C or 1H-15N HSQC NMR data (with the sign of a 1H-13C cross-peak being consistent with that expected from multiplicity editing (Willker et al. 1993)).
2.4.2 Rank 2S/2
Rank 2 or Rank 2S indicates an intermediate level of confidence in identification of a “putatively annotated compound”. Researchers can be reasonably confident when drawing biochemical or biological conclusions based on measured changes in the concentration of Rank 2 or Rank 2S “putatively annotated compounds”. The “putatively annotated compound” is assigned to Rank 2 or Rank 2S when one of the three following scenarios is true.
Rank 2S: The putative metabolite has one or more singlets in the 1D 1H spectrum, and
-
a.
The intensity and chemical shift of one or more singlets in the sample spectrum matches that of the putative metabolite in a 1D reference spectrum database (e.g. ChenomX).
-
b.
At least one singlet assignment based on the 1D database is supported by 2D data, i.e. either a cross peak in a 2D 1H-13C HSQC or 2D 1H-15N HSQC (with the sign of a 1H-13C cross-peak being consistent with that expected from multiplicity editing (Willker et al. 1993)).
Rank 2: The putative metabolite has multiple peaks, including multiplets, and
-
a.
The intensities and chemical shifts of at least two multiplets match that of the putative metabolite in a 1D reference spectrum database (e.g. ChenomX).
-
b.
At least two matching multiplet assignments from the 1D data are supported by 2D homonuclear 1H-1H TOCSY data.
-
c.
The matching multiplets supported by the 1D data and 2D 1H-1H TOCSY data are not supported by 2D heteronuclear 1H-13C or 1H-15N HSQC NMR data (with the sign of a 1H-13C cross-peak being consistent with that expected from multiplicity editing (Willker et al. 1993)).
2.4.3 Rank 3S/3
Rank 3 or Rank 3S indicates the highest level of confidence in assignment of a “putatively annotated compound”. The researcher can be highly confident when making biochemical or biological conclusions regarding changes in the concentration of the “putatively annotated compound”. The “putatively annotated compound” is assigned a Rank 3 or Rank 3S when one of the following scenarios is true.
Rank 3S: The putative metabolite has two or more singlets in the 1D 1H spectrum, and
-
a.
The intensity and chemical shift of the majority of at least two singlets in the sample spectrum matches that of the putative metabolite in a 1D reference spectrum database (e.g. ChenomX).
-
b.
The majority of at least two singlet assignments are supported by 2D data, i.e. either a cross peak in a 2D 1H-13C HSQC or in a 1H-15N HSQC, (with the sign of a 1H-13C cross-peak being consistent with that expected from multiplicity editing (Willker et al. 1993)).
Rank 3: The putative metabolite has multiple peaks, including multiplets, and
-
a.
The intensities and chemical shifts of the majority of at least two multiplets match that of the putative metabolite in a 1D reference spectrum database (e.g. ChenomX).
-
b.
The majority of at least two matching multiplet assignments are supported by 2D homonuclear 1H-1H TOCSY data.
-
c.
The majority of at least two matching multiplet assignments are supported by 2D heteronuclear 1H-13C or 1H-15N HSQC NMR data (with the sign of a 1H-13C cross-peak being consistent with that expected from multiplicity editing (Willker et al. 1993)).
In order to facilitate rank assignments using RANCM, a decision tree flow chart has been constructed as shown in Fig. 1. One utilizes the decision tree by first identifying a putative metabolite present in the reference spectrum database. For example, one can “filter” the database for reference compounds that have a peak at or near the chemical shift of the peak you are attempting to identify. Once you have identified a putative compound, the next step is to determine which of the two branches in the decision tree to take in making your rank assignment, i.e. does the putative metabolite have only “one or more singlets” (left branch in Fig. 1), or does the metabolite have “multiple peaks, including multiplets” (right branch in Fig. 1). One you have determined the correct branch, simply start at the top of the branch, answering the tier 1, tier 2 and tier 3 questions in the boxes (Fig. 1). At the first instance where you answer “NO” to the question, you stop and assign the “putatively annotated compound” the rank indicated at the right of the question. An Excel worksheet template is provided in supplementary material that can be used as a guide for making rank assignments and to document every “putatively annotated compound” rank assignment, by adding a sheet for each metabolite. It is intended that the Excel sheet should be used at the time the researcher is making “putatively annotated compound” assignments. At the completion of the study, it is recommended that the investigator prepare a table indicating the rank assignment for each “putatively annotated compound” and that the table be included either in the manuscript or as supplementary material, and that the completed Excel sheet be submitted together with the raw data to document the data used to make the “putatively annotated compound” assignments. The Excel sheet template that can be used to curate and document metabolite and rank assignments has been deposited to the figshare data repository (https://figshare.com/s/1b2fcd14658f32e65fcb).

Decision tree flowchart for deciding rank assignments for “putatively annotated compounds”. The decision tree contains two branches. The branch selection depends on the number and nature of the peaks expected in a 1D 1H NMR spectrum according to the following: a metabolite having one or more singlet peaks (left, red branch) or metabolite having multiple peaks including multiplets (right, blue branch). To determine which branch to use, one first identifies a putative matching compound and determines the number and complexity of expected peaks for the putative compound from a reference database. Once the appropriate branch has been selected, one proceeds to answer the questions at each tier, starting at Tier 1. Each tier is separated into a distinct row within the decision tree. If one answers “YES” to the question for a given tier, one proceeds to the question in the next tier. In the first instance for which one answers “NO” to a question, the investigator would not proceed further, and would assign the rank indicated beside the boxed question to the “putatively annotated compound”
3 Results and discussion
3.1 Rank 1S example—3,7-dimethyluric acid
Here, we demonstrate a Rank 1S assignment of 3,7-dimethyluric acid (strongest acidic pKa = 8.13). Based on filtering for the observed chemical shifts of ~ 3.37 ppm and ~ 3.42 ppm in ChenomX, we determined that 3,7-dimethyluric acid was a potential candidate (Fig S1A). Since 3,7-dimethyluric acid had only two predicted singlets, this determined that we should use the “one or more singlets” branch in the decision tree (Fig. 1). We were able to match chemical shifts of the two singlets as well as the intensity of the singlet at ~ 3.37 ppm in ChenomX (Fig S1A), thus satisfying the tier 1 criterion in the decision tree (Fig. 1). This ensured assignment of the metabolite as 3,7-dimethyluric acid with at least a Rank 1S confidence level. In order to advance to a Rank 2S level, at least one of the peaks would need to be supported by 2D 1H-13C HSQC data. Inspection of the 2D 1H-13C HSQC in the HMDB reference data base for 3,7-dimethyluric acid (HMDB01982) revealed the chemical shifts at which we should expect cross peaks (Fig S1B). However, inspection of the experimental 2D 1H-13C HSQC spectrum in Fig S1C revealed that the expected peaks were either absent or had insufficient signal to noise to be observed. Therefore, the tier 2 criterion could not be satisfied and the metabolite could only be assigned as 3,7-dimethyluric acid with a Rank 1S confidence level in this sample.
3.2 Rank 1 example—homovanillate
Based on filtering for the chemical shifts at ~ 6.88 ppm, we determined that homovanillate (carboxylic acid pKa = 3.74) was a potential match (Fig S2A). Five peaks, including multiplets were predicted based on the ChenomX data base (Fig S2A), indicating that we should use the “multiple peaks including multiplets” branch of the decision tree (Fig. 1). The chemical shifts could be matched for three of the five expected features and the intensities could be matched for two of the peaks (Fig S2A), thus satisfying the tier 1 level criterion in the decision tree. In order to advance to a Rank 2 assignment, the majority of the peaks tentatively assigned using the 1D ChenomX database needed to be supported by 2D 1H-1H TOCSY data. The expected cross peaks in the 2D 1H-1H TOCSY were identified from the spectrum included in the HMDB for record HMDB00118 (Fig S2B). Inspection of the experimental 2D 1H-1H TOCSY spectrum in the region where the cross peaks were expected (Fig S2B, red inset) did not allow definitive observation of the expected cross peaks given their proximity to the diagonal (Fig S2B, green inset), and therefore it was not possible to satisfy the tier 2 criterion in the list, thus limiting the confidence to a Rank 1 assignment. In order to advance to a Rank 3 assignment, the tier 3 criterion would have needed to be satisfied. This would have required that the majority of the peaks tentatively assigned in the 1D spectrum be supported by 2D 1H-13C HSQC data. The positions of the expected cross peaks for homovallinate were found from the reference spectrum provided in the HMDB (Fig S2C), however submission of the peak list of the experimental 2D 1H-13C HSQC spectrum matched only one of the three expected cross peaks (Fig S2C, inset), which would have failed to satisfy the tier 3 criterion and prevented advancement to a Rank 3 assignment.
3.3 Rank 2S example—allantoin
Based on filtering for the chemical shifts at ~ 5.4 ppm in the 1D ChenomX database, we determined that allantoin (strongest acidic pKa = 8.93) was a potential match for this metabolite (Fig S3A). As can be seen in Fig S3A, two peaks were expected in the 1D 1H spectrum of allantoin at 5.4 and 6.0 ppm. Since the two expected peaks in allantoin were singlets, this dictated that we should use the “one or more singlets” branch of the decision tree. Inspection of the 1D 1H spectrum indicated that the chemical shift and intensities of both predicted peaks could be adjusted to fit the experimental spectrum (Fig S3A) thus satisfying the tier 1 criterion in the decision tree (Fig. 1). This ensured that allantoin could be assigned with at least a Rank 1S confidence level. In order to advance to the Rank 2S level, at least one of the two peaks would need to be supported by 2D 1H-13C HSQC data. Inspection of the structure of allantoin (Fig S3B) indicated that it has only one non-exchangeable methine proton that would be observable in a 2D 1H-13C HSQC spectrum. Comparison of the experimental 2D 1H-13C HSQC spectrum with that of the reference spectrum included in the HMDB (HMDB0000462) indicated that expected peak was a potential match (Fig S3C), and this was further confirmed by submitting the data to the COLMAR software which identified allantoin as a unique match for the experimentally observed cross peak (Fig S3D). This ensured that the “putative metabolite” could be assigned as allantoin with at least a Rank 2S confidence level. In order to advance to a Rank 3S level, both of the expected peaks tentatively identified in the 1D spectrum would need to be supported by 2D HSQC data. However, because the second singlet corresponded to one or more of the exchangeable NH protons, it was impossible to support the assignment of this peak with 2D 1H-15N HSQC NMR data, and therefore it was impossible to satisfy the tier 3 criterion, thus limiting the assignment to a Rank 2S level of confidence.
3.4 Rank 2 example—hydroxyphenyllactate
Based on filtering the ChenomX 1D database for peaks around 6.9 ppm and 7.2 ppm, hydroxyphenyllactate (strongest acidic pKa = 3.58) was identified as a potential metabolite candidate. Hydroxyphenyllactate was found to have several peaks including multiplets, indicating that we should use the “multiple peaks, including multiplets” branch of the decision tree (Fig. 1). It was found that the chemical shifts and intensities of multiple peaks could be fit using the ChenomX database (Fig S4A), satisfying the tier 1 criterion indicating that the metabolite could be assigned at least a Rank 1 confidence level. In order to advance to the Rank 2 level, the majority of at least two peaks tentatively identified in the 1D spectrum would need to be supported by 2D 1H-1H TOCSY data. Submission of the experimental 2D 1H-1H TOCSY spectrum to the COLMAR database query returned matches for multiplets identified in the 1D ChenomX database (Fig S4B) satisfying the tier 2 criterion that ensured at least a Rank 2 assignment. In order to advance to the Rank 3 level, the majority of the peaks tentatively identified in the 1D ChenomX database needed to be supported by 2D 1H-13C HSQC data. Five cross peaks were expected in the 2D 1H-13C HSQC data based on the spectrum included in the HMDB for hydroxyphenyllactate (HMDB00755) (Fig S4C). Out of the five expected peaks, only two were detected: one for proton #2 (Fig S4C, red inset) and one for proton #4 (Fig S4C, blue inset) whereas peaks were missing for proton #1 (Fig S4C, green inset) and proton #3 (Fig S4C, blue inset). The lack of support for the assignment of the majority of peaks identified in the 1D ChenomX database by the 2D 1H-13C HSQC data failed to satisfy the tier 3 criterion and thus limit the confidence in the assignment of the metabolite to hydroxyphenyllactate to a Rank 2 confidence level.
3.5 Rank 3S example—creatine
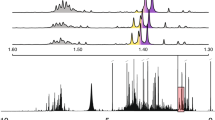
In this case, filtering the ChenomX database for peaks near 3.0 ppm and 3.9 ppm led to the identification of creatine (strongest acidic pKa = 3.5) as a potential metabolite. Inspection of the peak list indicated that we should expect two singlets for creatine, dictating that we should use the “one or more singlets” branch of the decision tree (Fig. 1). It was found that the chemical shifts and intensities of both peaks could be matched in the 1D ChenomX database (Fig. 2a), thus satisfying the tier 1 criterion and ensuring that the metabolite could be assigned as creatine with at least a Rank 1S confidence level. In order to advance to the Rank 2S level, at least one of the singlet peaks tentatively assigned in the 1D ChenomX database would need to be supported by 2D HSQC data. Reference to the HMDB indicated that two cross peaks should be expected in the 2D 1H-13C HSQC spectrum for creatine (HMDB00064) (Fig. 2b). Inspection of the experimental 2D 1H-13C HSQC spectrum for this sample indicated that two peaks were found in the expected region of the spectrum (Fig. 2b, inset), thus satisfying the tier 2 criterion for assignment to the Rank 2S level. Submission of the list of cross peaks picked from the experimental 2D 1H-13C HSQC spectrum to the COLMAR database indicated that both of the peaks found in the correct region of the spectrum matched the cross peaks expected for creatine in the COLMAR database (Fig. 2c, inset). Furthermore, the methyl cross peak (peak #2) and the methylene cross peak (peak #1) had the expected sign expected for their respective multiplicities (Fig. 2b, inset). These observations satisfied the tier 3 criterion indicating that creatine could be assigned with a Rank 3S confidence level. As a point of reference, two nearby peaks both in the 1D ChenomX database and in the 2D COLMAR database were identified as creatinine in the COLMAR database.

Data used for the Rank 3S assignment for creatine. a The 1D 1H experimental NMR spectrum (black) overlaid with two matching singlets from the 1D 1H reference spectrum of creatine (blue) determined by ChenomeX. Both peaks representative of creatine are present in the experimental spectrum at the expected intensities. b The 2D 1H-13C HSQC reference NMR spectrum for creatine included in the HMDB. The chemical structure of creatine and the chemical shifts for the cross peaks are included as insets. The experimental 2D 1H-13C HSQC NMR spectrum is included as an inset, with red circles denoting experimental cross peaks that match the database peaks in the reference spectrum. c The COLMAR output after submitting the cross peaks that were picked from the experimental spectrum to the COLMAR database. The green solid circles indicate cross peaks that match the expected peak for creatine within defined tolerances. Due to similarities in structure and chemical shifts, creatinine has also been indicated using red circles in this database query
3.6 Rank 3 example—lactate
Filtering of the 1D ChenomX database for peaks near 1.3 ppm and 4.1 ppm led to identification of lactate (strongest acidic pKa = 3.86) as a potential candidate (Fig. 3a). Inspection of the peak list for lactate indicated that two multiplets should be expected for the metabolite, dictating that the “multiple peaks, including multiplets” branch of the decision tree should be used (Fig. 1). It was found that the chemical shifts and intensities of the peaks in the experimental spectrum could be matched with the reference spectrum for lactate in the 1D ChenomX database, thus satisfying the tier 1 criterion and ensuring that the metabolite could be assigned as lactic acid with at least a Rank 1 confidence level. In order to advance to the Rank 2 level, it would be necessary that the two peaks tentatively assigned to lactate be supported by 2D 1H-1H TOCSY data. Submission of the experimental 2D 1H-1H TOCSY spectrum to the COLMAR database returned a match for the cross peak expected between the two tentatively assigned multiplets in the 1D ChenomX database (Fig. 3b). This satisfied the tier 2 criterion, ensuring that the metabolite could be assigned to lactate with at least a Rank 2 confidence level. In order to advance to the Rank 3 level, the two tentatively assigned multiplets in the 1D ChenomX database needed to be supported by 2D 1H-13C HSQC data. The cross peaks expected in the 2D 1H-13C HSQC spectrum for L-lactate were found in the reference spectrum included in the HMDB (HMDB0000190)Footnote 1 (Fig. 3c). Submission of the list of cross peaks picked from the experimental 2D 1H-13C HSQC spectrum to the COLMAR database returned unique matches for both of the expected cross peaks (Fig. 3d), thus satisfying the tier 3 criterion and justifying the identification of lactate with a Rank 3 confidence level. It should be noted that simple reference to the NMR databases did not enable distinction between the d- and l- stereoisomers of lactate.

Data used for the Rank 3 assignment for L-lactic acid. a The 1D 1H experimental NMR spectrum (black) overlaid with matching multiplets from the 1D 1H reference spectrum of l-lactic acid (blue) determined by ChenomX. The two peaks representative of l-lactic acid are present in the experimental spectrum at the expected chemical shifts, with one feature of a multiplet overlapping with other signals. b The COLMAR output after submitting the experimental 2D 1H-1H TOCSY spectrum to a COLMAR query. An inset shows that two peaks identified by COLMAR match the two peaks for l-lactic acid contained in the COLMAR database. c The 2D 1H-13C HSQC reference NMR spectrum for l-lactic acid included in the HMDB. The chemical structure of lactic acid and the chemical shifts for the cross peaks are included as insets. d Two peaks characteristic of lactic acid identified by a COLMAR query of the experimental 2D 1H-13C HSQC NMR detailing the expected peaks (in red) matching with the peaks identified by COLMAR(in blue) Other identified peaks are present, but are not circled. Further insets detail the matching ratio result from the COLMAR query, as well as detailed insets for both experimental cross peaks(in blue and red) that match the expected positions from the HMDB database within defined tolerances
3.7 Recommended use of RANCM in NMR-based metabolomics studies
The primary aim of the RANCM algorithm is to minimize ambiguity in assignments to the MSI “putatively annotated compound” category, which is by far the most common assignment category reported in modern NMR-based metabolic profiling studies. The persistent ambiguity in ubiquitous “putatively annotated compound” assignments significantly and negatively impacts the reliability and potential impact of NMR-based metabolic profiling studies. In many cases, the goal of a NMR-based metabolic profiling study is to establish a biochemical, biological and cellular understanding of a disease condition, or other parameter, being investigated based on the changes in concentrations of significant metabolites that are either “identified” or assigned as a “putatively annotated compounds”. Thus, the story that emerges from the inferences regarding the metabolic pathways based on the lists of metabolites identified in the metabolic profiling analysis depends on the confidence with which the “putatively annotated compound” assignments have been made. However, there is no current standard method or practice for assigning confidence levels to “putatively annotated compound” assignments, and consequently, there is also no mechanism for documenting, reporting and submitting such documentation to the national and international data repositories alongside the raw NMR data files. The new RANCM ranking system introduced here is intended to fill these gaps, and by doing so, hopefully to increase the reliability, impact and utility of NMR-based metabolomics investigations.
We recommend that NMR-based metabolomics researchers adopt the new RANCM ranking scheme by completing the Excel sheet template included in supplementary material for each “putatively annotated compound” and that the Excel sheet documenting the confidence in the identification of each “putatively annotated compound” be uploaded alongside the raw experimental data in the public data repositories. It is recommended that the investigator complete a sheet as each putative metabolite is assigned, treating the Excel sheet as an electronic notebook. By adopting this practice, the investigator essentially documents their curation of the putative metabolite assignment process, while at the same time assigning a confidence rank to each assignment. We have deposited a completed Excel sheet documenting the six rank assignments included in this manuscript as an example to the figshare data repository with the following accession number (https://figshare.com/s/1b2fcd14658f32e65fcb). Furthermore, we recommend that the rank assignments for all “putatively annotated compound” be reported in the manuscript either in a table or in supplementary material. By adopting these practices, not only will researchers be able to use a straight-forward instrument to assign confidence levels to their “putatively annotated compound” identifications, but submitting the associated documentation supporting those rank assignments alongside the data used to make the rank assignments will enable future investigators to always go back to the original data if there are any future queries regarding those metabolite identifications. Another strength of the RANCM ranking system is that it provides a detailed decision tree that is easily used with any combination of datasets, ranging from only 1D experimental 1H NMR spectra to a complete set of 1D and 2D homonuclear 1H spectra or heteronuclear 1H-13C or 1H-15N NMR spectra. Therefore, the RANCM ranking system is stable in the sense that the rigor of the rank assignment is independent of the number or type of datasets that are used to make the rank assignment. Finally, the ease with which the decision tree along with the Excel sheet template can be used to document “putatively annotated compound” assignments should ensure that the RANCM algorithm can be widely adopted across the field of NMR-based metabolomics investigations.
3.8 Discussion of limitations of the RANCM algorithm
The RANCM algorithm presented is deliberately simple for multiple reasons. One reason is that the algorithm was constructed with the intention that it could be used with any set of NMR data available to the investigator, ranging from 1D data only to extensive 2D data sets collected either at natural abundance or with the aid of isotopic labeling. While it is desirable to identify putative metabolites with the highest confidence possible, in practice there are always complications that limit confidence in assignments, including spectral overlap, limited signal-to-noise ratio, and incomplete reference databases, to mention a few. Some of these limitations can be overcome to some extent either using some means of sample enrichment or isotopic enrichment to overcome issues related to poor signal-to-noise, and these various approaches are well documented in the literature. Notwithstanding these limitations, and even if exceptional measures are taken to improve the quality of the NMR data, it is critical to document the confidence in metabolite assignments in the context of whatever NMR data was used to make the assignments. Besides the limiting factors just mentioned, there are other factors that could be considered when assigning the confidence level of a metabolite identification. For example, one could consider the likelihood that the putative metabolite will be present in the particular sample matrix under consideration based on information available in the various database, such as the HMDB. Using such information, a higher confidence in a metabolite assignment could be achieved if the measured concentration in the sample matrix is consistent with the average concentration, or within the range of the metabolite concentrations reported in the database for the given sample matrix (Canueto et al. 2018). The current RANCM algorithm does not take such information into consideration. Another technique that can be used in metabolite validation is STOCSY (Cloarec et al. 2005), however, our intentionally simple algorithm does not include reference to use of STOCSY. Once a putative metabolite assignment has been made, it is also possible to determine if the putative metabolite assignment and the inferred metabolic pathways makes biochemical sense and such information could be used as a means of cross-validation of putative metabolite assignments. Another potential issue that the investigator must be aware of, but is outside of the scope of the algorithm, is the importance and complexity of proper referencing of the experimental spectra so that accurate comparisons can be made with the reference database spectra. For example, it is common for the public databases to use DSS as a reference compound, whereas it has become common for investigators conducting NMR based metabolic profiling studies to use TSP, and one must either account for the chemical shift differences between the reference standards or allow for a slightly greater tolerance to accommodate lack of correction. Referencing issues are also a potential problem due to chemical shift variability across different sample matrices, e.g. urine, blood, etc., and the investigators must be cognizant of the potential problems that this can cause for accurate referencing and comparison to the spectra deposited in the public databases. A final limitation of the RANCM algorithm as presented is that it has not yet been independently validated by experts out in the field. It is our hope that the quality and utility of the RANCM algorithm can be confirmed and improved by further validation by experts who use NMR based metabolic profiling as a part of their research. The authors would welcome any suggestions for improvement of the RANCM algorithm.
4 Conclusions
In conclusion, we introduce the RANCM ranking system to assign confidence levels to “putatively annotated compound” metabolite identifications made from experimental NMR spectral data. This new ranking system is intended to augment the original MSI guidelines in a manner that minimizes the ambiguity associated with the “putatively annotated compound” assignment category. The RANCM ranking system is supported by an easy-to-use decision tree that can be implemented using any combination of experimental NMR spectra. The ranking system will add value to NMR based metabolic profiling studies by providing a consistent and rigorous ranking scheme can be used to report confidence levels in metabolite assignments regardless of the amount or type of experimental NMR data that is available for a given NMR based metabolomics study. It is recommended that the experimental data supporting the rank level assignments be documented in an Excel sheet for every metabolite identified in the course of the study, and that the supporting documentation be submitted along with the raw NMR data files associated with the metabolomics study. The rank confidence values obtained using the RANCM algorithm introduced here could easily be integrated with, combined with, or reported alongside other existing approaches for assignment confidence in metabolite identifications made in NMR based metabolic profiling studies.
Notes
Note that the spectrum included for l-lactic acid in the HMDB is labeled as d-lactic acid with record number (HMDB0001311).
References
Bingol, K., Bruschweiler-Li, L., Li, D. W., & Bruschweiler, R. (2014). Customized metabolomics database for the analysis of NMR H-1-H-1 TOCSY and C-13-H-1 HSQC-TOCSY spectra of complex mixtures. Analytical Chemistry, 86, 5494–5501. https://doi.org/10.1021/ac500979g.
Bingol, K., Li, D.-W., Zhang, B., & Bruschweiler, R. (2016). Comprehensive metabolite identification strategy using multiple 2D NMR spectra of a complex mixture implemented in the COLMARm web server. Analytical Chemistry, 88, 12411–12418.
Canueto, D., Gomez, J., Salek, R. M., Correig, X., & Canellas, N. (2018). rDolphin: a GUI R package for proficient automatic profiling of 1D 1H-NMR spectra of study datasets. Metabolomics, 14, 24.
Chihanga, T., Hausmann, S. M., Ni, S. S., & Kennedy, M. A. (2018a). Influence of media selection on NMR based metabolic profiling of human cell lines. Metabolomics, https://doi.org/10.1007/s11306-018-1323-2.
Chihanga, T., Ma, Q., Nicholson, J. D., Ruby, H. N., Edelmann, R. E., Devarajan, P., et al. (2018b). NMR spectroscopy and electron microscopy identification of metabolic and ultrastructural changes to the kidney following ischemia-reperfusion injury. American Journal of Physiology-Renal Physiology, 314, F154–F166. https://doi.org/10.1152/ajprenal.00363.2017.
Chihanga, T., Ruby, H. N., Ma, Q., Bashir, S., Devarajan, P., & Kennedy, M. A. (2018c). NMR-based urine metabolic profiling and immunohistochemistry analysis of nephron changes in a mouse model of hypoxia-induced acute kidney injury. American Journal of Physiology-Renal Physiology. https://doi.org/10.1152/ajprenal.00500.2017.
Cloarec, O., Dumas, M. E., Craig, A., Barton, R. H., Trygg, J., Hudson, J., et al. (2005). Statistical total correlation spectroscopy: An exploratory approach for latent biomarker identification from metabolic 1H NMR data sets. Analytical Chemistry, 77, 1282–1289. https://doi.org/10.1021/ac048630x.
Creek, D. J., Dunn, W. B., Fiehn, O., Griffin, J. L., Hall, R. D., Lei, Z., et al. (2014). Metabolite identification: Are you sure? And how do your peers gauge your confidence? Metabolomics 10, 350–353.
Dona, A. C., Kyriakides, M., Scott, F., Shephard, E. A., Varshavi, D., Veselkov, K., et al. (2016). A guide to the identification of metabolites in NMR-based metabonomics/metabolomics experiments. Computational and Structural Biotechnology Journal, 14, 135–153. https://doi.org/10.1016/j.csbj.2016.02.005.
Everett, J. R. (2015). A new paradigm for known metabolite identification in metabonomics/metabolomics: Metabolite identification efficiency. Computational and Structural Biotechnology Journal, 13, 131–144. https://doi.org/10.1016/j.csbj.2015.01.002.
Fiehn, O., Robertson, D., Griffin, J., van der Werf, M., Nikolau, B., Morrison, N., et al. (2007). The Metabolomics Standards Initiative (MSI). Metabolomics, 3, 175–178. https://doi.org/10.1007/s11306-007-0070-6.
Guijas, C., Montenegro-Burke, J. R., Domingo-Almenara, X., Palermo, A., Warth, B., Hermann, G., et al. (2018). METLIN: A technology platform for identifying knowns and unknowns. Analytical Chemistry, 90, 3156–3164. https://doi.org/10.1021/acs.analchem.7b04424.
Haug, K., Salek, R. M., Conesa, P., Hastings, J., de Matos, P., Rijnbeek, M., et al. (2013). MetaboLights—an open-access general-purpose repository for metabolomics studies and associated meta-data. Nucleic Acids Research, 41, D781–D786. https://doi.org/10.1093/nar/gks1004.
Nicholson, J. K., Lindon, J. C., & Holmes, E. (1999). ‘Metabonomics’: Understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological NMR spectroscopic data. Xenobiotica, 29, 1181–1189 doi. https://doi.org/10.1080/004982599238047.
Posma, J. M., Garcia-Perez, I., Heaton, J. C., Burdisso, P., Mathers, J. C., Draper, J., et al. (2017). Integrated analytical and statistical two-dimensional spectroscopy strategy for metabolite identification: Application to dietary biomarkers. Analytical Chemistry, 89, 3300–3309. https://doi.org/10.1021/acs.analchem.6b03324.
Rohnisch, H. E., Eriksson, J., Mullner, E., Agback, P., Sandstrom, C., & Moazzami, A. A. (2018). AQuA: An automated quantification algorithm for high-throughput nmr-based metabolomics and its application in human plasma. Analytical Chemistry, 90, 2095–2102. https://doi.org/10.1021/acs.analchem.7b04324.
Romick-Rosendale, L. E., Goodpaster, A. M., Hanwright, P. J., Patel, N. B., Wheeler, E. T., Chona, D. L., et al. (2009). NMR-based metabonomics analysis of mouse urine and fecal extracts following oral treatment with the broad-spectrum antibiotic enrofloxacin (Baytril). Magnetic Resonance in Chemistry, 47(Suppl 1), 36–46. https://doi.org/10.1002/mrc.2511.
Romick-Rosendale, L. E., Legomarcino, A., Patel, N. B., Morrow, A. L., & Kennedy, M. A. (2014). Prolonged antibiotic use induces intestinal injury in mice that is repaired after removing antibiotic pressure: implications for empiric antibiotic therapy. Metabolomics, 10, 8–20. https://doi.org/10.1007/s11306-013-0546-5.
Romick-Rosendale, L. E., Schibler, K. R., & Kennedy, M. A. (2012). A potential biomarker for acute kidney injury in preterm infants from metabolic profiling. Journal of Molecular Biomarkers & Diagnosis, https://doi.org/10.4172/2155-9929.S3-001.
Salek, R. M., Steinbeck, C., Viant, M. R., Goodacre, R., & Dunn, W. B. (2013). The role of reporting standards for metabolite annotation and identification in metabolomic studies. Gigascience, 2, 13. https://doi.org/10.1186/2047-217X-2-13.
Sanchon-Lopez, B., & Everett, J. R. (2016). New Methodology for known metabolite identification in metabonomics/metabolomics: Topological metabolite identification carbon efficiency (tMICE). Journal of Proteome Research, 15, 3405–3419. https://doi.org/10.1021/acs.jproteome.6b00631.
Schmahl, M. J., Regan, D. P., Rivers, A. C., Joesten, W. C., & Kennedy, M. A. (2018). NMR-based metabolic profiling of urine, serum, fecal, and pancreatic tissue samples from the Ptf1a-Cre; LSL-KrasG12D transgenic mouse model of pancreatic cancer. PLoS ONE, 13, e0200658. https://doi.org/10.1371/journal.pone.0200658.
Smith, C. A., O’Maille, G., Want, E. J., Qin, C., Trauger, S. A., Brandon, T. R., et al. (2005). METLIN—a metabolite mass spectral database. Therapeutic Drug Monitoring, 27, 747–751 doi. https://doi.org/10.1097/01.ftd.0000179845.53213.39.
Spicer, R. A., Salek, R., & Steinbeck, C. (2017). Compliance with minimum information guidelines in public metabolomics repositories. Scientific Data, 4, 170137. https://doi.org/10.1038/sdata.2017.137.
Sud, M., Fahy, E., Cotter, D., Azam, K., Vadivelu, I., Burant, C., et al. (2016). Metabolomics Workbench: An international repository for metabolomics data and metadata, metabolite standards, protocols, tutorials and training, and analysis tools. Nucleic Acids Research, 44, D463–D470. https://doi.org/10.1093/nar/gkv1042.
Sumner, L. W., Amberg, A., Barrett, D., Beale, M. H., Beger, R., Daykin, C. A., et al. (2007). Proposed minimum reporting standards for chemical analysis Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics, 3, 211–221. https://doi.org/10.1007/s11306-007-0082-2.
Tardivel, P. J. C., Canlet, C., LeFort, G., Tremblay-Franco, M., Debrauwer, L., Concordet, D., et al. (2017). ASICS: An automatic method for identification and quantification of metabolites in complex 1D 1H NMR spectra. Metabolomics, 13, 109.
Ulrich, E. L., Akutsu, H., Doreleijers, J. F., Harano, Y., Ioannidis, Y. E., Lin, J., et al. (2008). BioMagResBank. Nucleic Acids Research, 36, D402–D408. https://doi.org/10.1093/nar/gkm957.
van der Hooft, J. J. J., & Rankin, N. (2016). Metabolite identification in complex mixtures using nuclear magnetic resonance spectroscopy. In G. A. Webb (Ed.), Modern magnetic resonance. Glascow: Springer.
Wang, B., Sheriff, S., Balasubramaniam, A., & Kennedy, M. A. (2015). NMR based metabolomics study of Y2 receptor activation by neuropeptide Y in the SK-N-BE2 human neuroblastoma cell line. Metabolomics, 11, 1243–1252. https://doi.org/10.1007/s11306-015-0782-y.
Watanabe, M., Sheriff, S., Kadeer, N., Cho, J., Lewis, K. B., Balasubramaniam, A., et al. (2012a). NMR based metabonomics study of NPY Y5 receptor activation in BT-549, a human breast carcinoma cell line. Metabolomics, 8, 854–868. https://doi.org/10.1007/s11306-011-0380-6.
Watanabe, M., Sheriff, S., Lewis, K. B., Cho, J., Tinch, S. L., Balasubramaniam, A., et al. (2012b). Metabolic profiling comparison of human pancreatic ductal epithelial cells and three pancreatic cancer cell lines using NMR based metabonomics. Journal of Molecular Biomarkers and Diagnosis, S3, 1–17.
Watanabe, M., Sheriff, S., Ramelot, T. A., Kadeer, N., Cho, J., Lewis, K. B., et al. (2011). NMR Based metabonomics study of DAG treatment in a C2C12 mouse skeletal muscle cell line myotube model of burn-injury. International Journal of Peptide Research and Therapeutics, 17, 281–299. https://doi.org/10.1007/s10989-011-9264-x.
Willker, W., Leibfritz, D., Kerssebaum, R., & Bermel, W. (1993). Gradient selection in inverse heteronuclear correlation spectroscopy. Magnetic Resonance in Chemistry, 31, 287–292 doi. https://doi.org/10.1002/mrc.1260310315.
Wishart, D. S., Feunang, Y. D., Marcu, A., Guo, A. C., Liang, K., Vazquez-Fresno, R., et al. (2018). HMDB 4.0: the human metabolome database for 2018. Nucleic Acids Research, 46, D608–D617. https://doi.org/10.1093/nar/gkx1089.
Wishart, D. S., Jewison, T., Guo, A. C., Wilson, M., Knox, C., Liu, Y., et al. (2013). HMDB 3.0—The human metabolome database in 2013. Nucleic Acids Research, 41, D801–D807. https://doi.org/10.1093/nar/gks1065.
Wishart, D. S., Knox, C., Guo, A. C., Eisner, R., Young, N., Gautam, B., et al. (2009). HMDB: a knowledgebase for the human metabolome. Nucleic Acids Research, 37, D603–D610. https://doi.org/10.1093/nar/gkn810.
Wishart, D. S., Tzur, D., Knox, C., Eisner, R., Guo, A. C., Young, N., et al. (2007). HMDB: the human metabolome database. Nucleic Acids Research, 35, D521–D526. https://doi.org/10.1093/nar/gkl923.
Acknowledgements
The research was conducted with the support of Miami University. The instrumentation used in this work was obtained with the support of Miami University and the Ohio Board of Regents with funds used to establish the Ohio Eminent Scholar Laboratory where the work was performed.
Author information
Authors and Affiliations
Contributions
MAK conceived the ranking scheme. WCJ tested and contributed to the development of the new ranking scheme. WJC and MAK wrote the manuscript. WCJ and MAK read and approved the manuscript.
Corresponding author
Ethics declarations
Conflict of interest Statements
William C Joesten declares that he has no conflict of interest. Michael A. Kennedy declares that he has no conflict of interest.
Ethical approval
Research involving Human Participants and/or Animals. This study did not involve the use of human participants. All procedures involving mice were approved by both the ethics committee and the Institutional Animal Care and Use Committee at Miami University (Animal Welfare Assurance Number: D16-00100). The protocol approved by the Miami University IACUC was assigned Project Number 898.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Joesten, W.C., Kennedy, M.A. RANCM: a new ranking scheme for assigning confidence levels to metabolite assignments in NMR-based metabolomics studies. Metabolomics 15, 5 (2019). https://doi.org/10.1007/s11306-018-1465-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11306-018-1465-2