
Abstract
Producing a comprehensive overview of the chemical content of biologically-derived material is a major challenge. Apart from ensuring adequate metabolome coverage and issues of instrument dynamic range, mass resolution and sensitivity, there are major technical difficulties associated with data pre-processing and signal identification when attempting large scale, high-throughput experimentation. To address these factors direct infusion or flow infusion electrospray mass spectrometry has been finding utility as a high throughput metabolite fingerprinting tool. With little sample pre-treatment, no chromatography and instrument cycle times of less than 5 min it is feasible to analyse more than 1,000 samples per week. Data pre-processing is limited to aligning extracted mass spectra and mass-intensity matrices are generally ready in a working day for a month’s worth of data mining and hypothesis generation. ESI-MS fingerprinting has remained rather qualitative by nature and as such ion suppression does not generally compromise data information content as originally suggested when the methodology was first introduced. This review will describe how the quality of data has improved through use of nano-flow infusion and mass-windowing approaches, particularly when using high resolution instruments. The increasingly wider availability of robust high accurate mass instruments actually promotes ESI-MS from a merely fingerprinting tool to the ranks of metabolite profiling and combined with MS/MS capabilities of hybrid instruments improved structural information is available concurrently. We summarise current applications in a wide range of fields where ESI-MS fingerprinting has proved to be an excellent tool for “first pass” metabolome analysis of complex biological samples. The final part of the review describes a typical workflow with reference to recently published data to emphasise key aspects of overall experimental design.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Although, no single analytical platform can offer a fully comprehensive survey of the chemical diversity representing the metabolome, continuous improvements in mass spectrometry (MS) instrumentation have allowed development of relatively standardised metabolite profiling or fingerprinting procedures (e.g. Dunn 2008; Saito and Matsuda 2010; Koek et al. 2011). A fundamental principle of MS is the representation of metabolite features in any biological matrix by measurement of the spectrum of signals reflecting the mass to charge ratios (m/z) of their ionisation products. Traditional approaches for assaying metabolite content by mass spectrometry involve the use of liquid (LC–MS) or gas (GC–MS) chromatography to first attempt to separate metabolites before detection, thus generating quantitative or semi-quantitative information on individual metabolites (e.g. Lisec et al. 2006; De Vos et al. 2007). These metabolite ‘profiling’ methods demand careful control over the chromatographic process to ensure reproducibility and require significant time, effort and expertise for data pre-processing in order to deconvolve, align and annotate peaks correctly. Unfortunately, any chromatography column matrix will undergo gradual detoriation with repetative use, resulting in significant changes in data characteristics after a period of constant operation in larger (>200 samples) profiling experiments. An alternative approach to capture information relating to total metabolite content is to develop a spectrometric ‘fingerprint’ without recourse to any chromatographic separation (e.g. Smedsgaard and Frisvad 1996; Goodacre et al. 2002; Aharoni et al. 2002; Dunn et al. 2005a; Southam et al. 2007; Koulman et al. 2007; Beckmann et al. 2008; Madalinski et al. 2008; Fuhrer et al. 2011). The lack of a chromatography step before sample ionisation has a great influence on both experimental design and outcomes. For example, the absence of a ‘peak’ finding or ‘feature’ alignment step greatly simplifies data pre-processing approaches when compared to the complexity of converting raw data from hyphenated mass spectrometry experiments (e.g. LC–MS and GC–MS) into useful output.
Electrospray ionisation (ESI) is the most common method used for ionisation in the generation of mass spectrometric metabolite fingerprints (ESI-MS). During the electrospray process ionization is accomplished by the loss or gain of a proton, or other adducts, and charged analyte molecules can carry either single or multiple charges. Although ideally suited to more polar chemicals, even molecules that do not have acidic or basic groups can be charged through the formation of adducts with various ions such as Cl− ions in negative ion mode or commonly K+ or Na+ in positive ion mode. The formation of such adducts is highly dependent on the salt content of the crude sample matrix. As such minimal (usually no) fragmentation is produced during electrospray and a protonated (+ve ion data) or de-protonated (−ve ion data) analyte is often referred to as the “pseudo molecular ion” of the parent molecule (Gorlach and Richmond 1999). This property means that LC–MS analytical approaches using electrospray ionisation are able to directly predict m/z signal putative identity based on measured mass. The lack of extensive molecular fragmentation during electrospray ionisation is considered to result in reproducible ionisation patterns when extracts representing similar biological matrices are analysed and offers an experimental robustness suitable for larger scale investigations (e.g. Smedsgaard and Frisvad 1996; Catchpole et al. 2005; Dunn et al. 2005b; Parker et al. 2009; Beckmann et al. 2010; Scott et al. 2010; Ward et al. 2010; Fuhrer et al. 2011). ESI-MS fingerprinting methodology can be adapted to analyse both polar and lipophilic extracts in both positive and negative ionisation modes to provide comprehensive coverage of a wide range of chemistries. Relatively recently the use of ambient ionisation techniques based on variants of desorption electrospray ionisation (DESI) or extractive electrospray ionisation (EESI) have extended the utility of mass spectrometry fingerprinting to direct analysis of complex solid materials or liquid aerosols without the need for sample pre-treatment or extraction (e.g. Chen Wortmann and Zenobi 2007; Li et al. 2009, 2011; Wu et al. 2010; Chen Hu and Zhang 2010; Hu et al. 2011; Wang et al. 2011; Gu et al. 2010). A detailed description of these methods is out of the scope of the present article.
ESI-MS fingerprint data generated at high mass resolution can be analysed directly to both discover AND annotate metabolite signals explanatory of a biological difference between specific sample classes. Alternatively, many applications have used a hierarchical approach in which ESI-MS fingerprinting at nominal mass is followed by the targeted analysis of specific m/z signals by higher resolution methods. Different instrument configurations are more suited to these different analytical approaches. Nominal mass metabolite fingerprinting is often utilised in situations where there is a need for a high-throughput screening method with comprehensive coverage of metabolite diversity that allows sample classification or discrimination according to their origin or biological relevance, without actually structurally identifying and quantifying individual metabolites (for overview see Beckmann et al. 2008; Enot et al. 2008). With an analytical cycle time of typically 3–5 min per sample, coupled with the relative simplicity of data pre-processing and robustness of the data, it is considered that nominal mass ESI-MS fingerprinting in particular is an ideal choice for high throughput ‘first pass’ analysis.
A survey of publications reporting the use of non-targeted flow infusion ESI-MS fingerprinting (see Table 1 later) indicate clearly that this technology has been used as a credible high throughput alternative to much more laborious and time consuming analytical procedures employing prior chromatographic separation of analytes. In several cases in which a direct comparison was made, flow infusion ESI-MS fingerprinting has proved equally as informative as for example HPLC–MS analysis (Zahn et al. 2001; Han et al. 2008a; Lin et al. 2010) or GC–MS profiling (Catchpole et al. 2005; Beckmann et al. 2007; Mas et al. 2007; Pope et al. 2007; Lloyd et al. 2011c) or NMR fingerprinting (Scott et al. 2010; Ward et al. 2010).
The purpose of the present article is to firstly review the development of ESI-MS fingerprinting technology and summarise applications to date. Key technical and experimental design considerations which impact on the utility of ESI-MS fingerprinting will be explained with reference to recent data generated on a specific instrument following a typical workflow.
2 Choice of instrumentation for ESI-MS fingerprinting
2.1 General principles of ESI-MS fingerprint data generation
A key aspect of any non-targeted electrospray ionisation mass spectrometry (ESI-MS) fingerprinting method is the need to obtain a stable spray resulting in a sustained ion current throughout data acquisition. Early reports of metabolite fingerprinting used a ‘direct injection’ (DI-MS) approach utilising a syringe pump, or similar device, to constantly introduce the entire sample into the mass spectrometer (e.g. Goodacre et al. 2002). The majority of more recent reports have adopted the use of a ‘flow injection’ or ‘flow infusion’ strategy (FIE-MS) in which the sample is either introduced in an LC–MS running solvent as a ‘plug flow’ (Fig. 1a) in an short (2–3 min) HPLC run or introduced continuously using bespoke nano-infusion devices, such as the chip-based NanoMate™ (e.g. Boernsen et al. 2005; Southam et al. 2007; Beckmann et al. 2008; Fuhrer et al. 2011; Zamfir et al. 2005). Since atmospheric pressure chemical ionisation (APCI) has been used only in a few cases (e.g. Gray and Heath 2005) we will continue using the generic term ‘ESI-MS’ fingerprinting in this article to describe electrospray ionisation experiments using either a syringe pump (DI-MS), HPLC/nano-LC systems (flow infusion experiments or analysis, FIE-MS, FIA-MS) or chip-based infusion approaches.

ESI-MS fingerprint data generation. a Typical plug flow injection chromatogram of an ESI-MS experiment with a single peak containing all metabolites in the absence of a chromatographic column. In the resulting mass spectrum of human urine b the magnification of ion intensities by factor of 10 at m/z >500 shows a continuous data stream and therefore mass bin occupancy with positive intensity values, which is essential for most data analysis procedures. c Acquisition of ESI-MS data in four mass ranges and ion intensities in positive ESI-MS ionisation mode generated in a Thermo LTQ linear ion trap
Data are often collected as a full width spectrum in centroid mode and during data pre-processing an average chromatogram is calculated from scans within the central portion of the infusion/injection peak. A delay of a couple of minutes before the next sample is infused into the solvent stream can avoid any ‘carry over’ between consecutive injections and provides a region of signal ‘noise’ that can be used for background subtraction (Windig et al. 1996). Generally there is scope to use both positive and negative ionisation modes either independently or using alternate scans, depending on instrumentation and solvent or mobile phase preferences. As a default all instrumentation is generally capable of acquiring data to allow binning ions into nominal mass bins differing by 1 atomic mass unit (amu) within the range m/z 50 to m/z 1,000 (although many instruments have higher mass ranges) and thus a typical ‘full spectrum’ metabolite fingerprint will contain approaching 1,000 data features. In the absence of analyte pre-separation by chromatography processed ESI-MS fingerprint data can be considered essentially 2 dimensional (mass × intensity), differing only in the way in which the sample is introduced to the ionisation chamber (Fig. 1b). With two ionisation modes (+ve and −ve ion) data dimensionality in nominal mass ESI-MS fingerprinting often approaches 2,000 mass features.
2.2 Instrumentation and mass resolution
Researchers using LC–MS analytical methods will always seek to optimise the balance between mass resolution and sensitivity to maximise the information content of ESI-MS data without compromising reproducibility. Equipment used for ESI-MS fingerprinting combine single or hybrid quadrupole, time-of-flight, ion trap, Orbitrap and FT-ICR-MS technology, all of which offer different capabilities in terms of mass resolution and accuracy, dynamic range, sensitivity and general robustness (see Gross 2004). The choice of instrument has a major impact on experimental design.
Single quadrupole instruments and ion traps with quadrupole (Q) detectors are extremely robust and offer rapid scanning over large m/z ranges (see Gross 2004 for an overview). Ion traps, with an inherent capacity for ion storage, have the advantage that further successive fragmentation of selected ions (MSn experiments) could provide additional structural information. Although mass resolution is a valuable characteristic of instruments used to acquire nominal mass ESI-MS fingerprint data, the ability to capture large numbers of ions in constant sized ‘packages’ in each scan is also sometimes considered to be an advantage and linear ion trap instruments with automatic gain control (AGC) functions are particularly well suited to address this issue (March 1997). In both single quadrupole and ion trap instruments ESI-MS fingerprints can be regarded as simplified images of total sample composition in that the measured variables [mass to charge (m/z) ratios] are compiled by integrating the levels of more than one metabolite at a mass resolution of 1 amu. In some types of extract it will not be uncommon for certain mass bins to contain upwards of 10–15 signals derived from near-isobaric or actually isobaric (e.g. hexose sugars) molecular species. Nominal mass data matrices are generally both extremely reproducible and very quick to generate. From a pragmatic perspective, non-targeted, nominal mass ESI-MS fingerprinting also has the advantage that any instrument in any laboratory should be able to replicate any measurements thus possibly providing extended scope for future data integration (Beckmann et al. 2007; Enot et al. 2007). To accomplish this objective the data analysis strategy is based on the measure of relative ratios of m/z-signals in each fingerprint generated from large numbers of samples in a single analytical batch.
In contrast to nominal mass fingerprinting the use of instruments fitted with a single time of flight (TOF) detector capable of higher mass resolution theoretically offers a route to the direct annotation of signals based on mass estimates. In addition this approach should be capable of resolving signals from near-isobaric metabolites that would normally be integrated into a single mass bin in nominal mass fingerprinting, offering the possibility to produce more information rich spectra. However, with such instruments it becomes increasingly difficult to achieve reproducible data generation as mass resolution demands are increased. The advent of relatively cheaper and more robust tandem mass spectrometers, and especially hybrid Quadrupole-time of flight (Q-TOF) and Quadrupole-ion trap (Q-trap) instruments (Gross 2004) offered the opportunity to acquire ESI-MS data more routinely at higher mass resolutions (see Bino et al. 2004; Dunn et al. 2005a, 2008; Dettmer et al. 2007; Villas-Boas et al. 2005 for reviews). At m/z 500 a typical performance would be an average of 10,000 (FWHM) with mass errors between 5 and 10 ppm. Such hybrid instruments are generally constructed with quadrupole and TOF mass analysers separated by a higher pressure collision cell which can be used to perform collision induced dissociation (CID) of selected ions, offering further scope for metabolite identification through MS/MS experiments. The reason behind the desire to use accurate mass approaches is clearly an expectation that the measured mass recorded will be close to the expected mass of a specific metabolite recorded in a publically accessible database, and thus mass information could potentially lead directly to putative annotation of a signal. Data generation is carried out as for single TOF instruments but with increasing resolution there is a need for even more powerful software for data pre-processing to ensure experimental reproducibility. The rewards in terms of data dimensionality and reproducibility for continually improving data-processing approaches are significant. For example in a recent report using a Q-TOF instrument, Fuhrer et al. (2011) collecting data in profile mode were able to detect over 1,500 ions reproducibly at a resolution averaging 10,000 (FWHM) in the mass range m/z 100–1,000 with an average mass accuracy of 0.001. However, to achieve this result required the inclusion of a computationally intensive data processing step based on a wavelet transformation algorithm in order to exhaustively identify reproducibly very low intensity ion peaks in mass spectra. In parallel with the refinement of data pre-processing software has been an explosion of software tools and databases to improve the annotation of ESI-MS signals based on accurate mass information (e.g. Kind and Fiehn 2006, 2007, 2010; Draper et al. 2009; Ohta et al. 2007; Iijima et al. 2008; Brown et al. 2009, 2011; Wishart 2009, 2011; Weber and Viant 2010; Rojas-Cherto et al. 2011).
Traditional time-of-flight (TOF) and hybrid Q-TOF mass analysers could potentially achieve (with internal calibration) a resolving power as high as 50,000. This level of resolution however is not sufficient to distinguish between a large proportion of metabolites of different elemental composition which remain effectively isobaric at the maximum mass resolution achievable (Castrillo et al. 2003; Dunn et al. 2005; Overy et al. 2005; Davey et al. 2008; Yang et al. 2009; Sun and Chen 2011). Unfortunately an increase in resolution also is generally accompanied by a loss in sensitivity and often precision in mass accuracy which can lead to problems with data stability and alignment (for review see Dunn 2008). Although much data is yet to be published, many of these problems have recently been resolved in a new generation of LC–MS instruments which can operate efficiently at 100,000 mass resolution routinely and provide sub-ppm mass accuracies.
With the advent of ultra-high accuracy mass resolving analyzers such as the Orbitrap (maximum resolution ~240,000) and Fourier transform ion cyclotron resonance (FT-ICR) analyzer (resolution potentially in excess of 1,600,000 with 18 T magnets) the problem of precision in mass accuracy is reduced to sub-ppm errors allowing efficient prediction of ion elemental formulas (for reviews see Brown Kruppa and Dasseux 2005; Marshall and Hendrickson 2008; Junot et al. 2010; Ohta et al. 2010b). Data collection at high mass resolution effectively improves the number of metabolites to be detected when compared with lower resolution instruments. In such cases the data may be considered more to be metabolite ‘profiles’ in which each peak represents a single chemical entity (Junot et al. 2010; Ohta et al. 2010). It has however been demonstrated that applying accurate isotopic pattern calculation on data generated at 3 ppm mass accuracy could be advantageous to routinely generating 0.1 ppm mass resolution data over the full mass range on a currently still hypothetical instrument (Kind and Fiehn 2006; Miura et al. 2010).
In addition to syringe pumps and HPLC delivery systems all of the above instruments have been operated in conjunction with chip-based nanospray infusion devices with great effect to generate ESI-MS fingerprints. One advantage of such sample delivery systems is the ability to infuse samples with great stability over long periods of time (Schultz et al. 2000). With long data acquisition times, analytical sensitivity and dynamic range can be increased, making it possible to simultaneously detect hundreds of compounds in excess of what can be measured in LC–MS systems (Boernsen et al. 2005; Wickremsinhe et al. 2006). In most general aspects the structure of processed spectra generated by FT-MS technology is very similar to that produced by tandem mass spectrometry using new generation Q-TOF or Q-Trap instruments. However, the ultra-high resolution offers additional scope to annotate individual m/z signals by comparisons of signals across the spectrum as the mass accuracy is such that pseudo-parental ions, isotopes and salt adducts can be recognised by mass differences with much more confidence (e.g. Aharoni et al. 2002; Stoll et al. 2006; Tautenhahn et al. 2007; Southam et al. 2007; Giavalisco et al. 2008; Han et al. 2008a; Madalinski et al. 2008; Takahashi et al. 2008; Draper et al. 2009; Payne et al. 2009; Taylor et al. 2009; Favé et al. 2011; Lloyd et al. 2011c), although care is needed with interpretation of Fourier artefact peaks (Brown et al. 2009).
3 Technical aspects of ESI-MS data generation
3.1 Dynamic range and sensitivity
In a complex biological extract analytes will be present at a wide range of concentrations, differing by several orders of magnitude (Fiehn 2002; Sumner et al. 2003; Bino et al. 2004; Dunn 2008; Scalbert et al. 2009). The dynamic range of any instrument configuration (i.e. the ratio of the highest to lowest concentration metabolites detected) needs to be increased to allow the detection of lower abundance ions. Compared to single quadrupole instruments linear ion traps provide an enhanced dynamic range with a reduced low mass cutoff due to the spatial distribution of the ion cloud on the linear axis, resulting in an improved sensitivity (March 1997; Gross 2004). FT-ICR-MS and Orbitrap-MS instruments potentially offer ultra-high mass accuracy and resolution, however, a dynamic response across a wide mass range can only be achieved at the expense of mass accuracy, since the large numbers of ions entering the ICR detector induce adverse space-charge effects (Junot et al. 2010; Ohta et al. 2010). Such charge repulsive effects particularly cause mass calibration problems at higher m/z values and a reduction in peak resolution efficiency (Zhang et al. 2005; Oikawa et al. 2006).
Similar approaches have been used to address the issue of dynamic range limitations in both high mass accuracy and nominal mass fingerprinting in which the data has been collected as a series of overlapping spectra comprising relatively narrow mass windows followed by ‘stitching’ the windows together after acquisition (Southam et al. 2007; Giavalisco et al. 2008; Beckmann et al. 2008, 2010; Parker et al. 2008, 2009; Scott et al. 2010; Ward et al. 2010; Favé et al. 2011; Lloyd et al. 2011b, 2011c). For example, in an optimised approach described by Southam et al. (2007) FT-ICR-MS data was generated using the selected ion monitoring (SIM) mode from m/z 70 to m/z 500, using 21 adjacent SIM windows of 30 m/z each overlapping by 10 m/z. Data gathered in such ‘windows’ can subsequently by combined (or stitched) together using novel algorithms (Payne et al. 2009). Using fish liver samples Southam et al. (2007) showed that compared to wide scan mode the ‘SIM stitching’ method increased peak detection by a factor of 5 and the mass error decreased by a factor of 4, leading to a maximum mass error <0.5 ppm. Beckmann et al. (2008) similarly reported the collection of nominal mass ESI-MS data in two scan ranges (low: 15–200 m/z; and high: 110–2,000 m/z) to represent the composition of domestic dog urine (Beckmann et al. 2010) and extracts from pathogen-challenged plant leaves (Parker et al. 2008, 2009). This method was recently extended to collect data over four scan ranges (m/z 15–110; 100–220; 210–510 m/z; 500–1,200 m/z) in profile mode (Fig. 1c) and has been used to great effect in a range of studies (Scott et al. 2010; Ward et al. 2010; Favé et al. 2011; Lloyd et al. 2011b, c). The data collected in the overlapping mass windows could either be combined to give a full spectrum from m/z 15 to 1,200 (e.g. see Fig. 1b), or each mass range could be analysed independently to reduce data dimensionality and for use in correlation analyses. In most experimental situations, collecting the data in smaller mass windows generally improved the modelling characteristics (e.g. classification sensitivity and specificity metrics) of nominal mass ESI-MS fingerprint data.
3.2 Signal quantitation using accurate mass profiling ESI-MS
Measurement of the true concentration of a metabolite in crude biological samples requires the signal intensities of the analytes concerned be compared to a calibration curve made using chemical standards. In any non-targeted metabolomics approach quantitation is normally limited to expression of signal intensities as relative ratios in arbitrary units (e.g. in relation to total ion current of each sample). Thus generally ESI-MS fingerprint data have rarely been considered anything but semi-quantitative with the exception of a few studies using high accurate mass data generated using sophisticated signal acquisition procedures in combination with powerful data pre-processing algorithms (e.g. Southam et al. 2007; Han et al. 2008; Madalinski et al. 2008; Fuhrer et al. 2011). As outlined in the previous section great care is required to optimise the dynamic range whilst maintaining high levels of both sensitivity and mass accuracy. As such many signals will not be represented in the final data after stringent data-pre-processing. For example using a 12 T FT-ICR mass spectrometer Han et al. (2008b) detected 570 ions in an aqueous plasma extract of human plasma, and an elemental formula was assigned to 250 of them solely on the basis of accurate mass measurements with sub-ppm mass errors. Using an LTQ-Orbitrap instrument although mass spectra in the range m/z 50–m/z 1,000 derived from yeast cell extracts contained a few thousand m/z signals only 400 were considered to be quantifiable (Madalinski et al. 2008). To qualify as ‘analytically relevant’ a signal had to occur in at least 60 % of acquisition profiles under identical experimental conditions and have an intensity 3-fold higher than that of the background noise. More recently, in an attempt to exploit fully the high resolution and mass accuracy of TOF-MS Fuhrer et al. (2011) described a high throughput platform capable of detecting >1,500 reproducible signals (i.e. >2-fold high than background noise) in both ionisation modes under conditions where >90 % of chemical standards showed a linear response when ions where recorded in profile mode from m/z 50 to m/z 1,000. Interestingly, between 400 and 800 signals were assigned to ionisation products of around 200 Escherichia coli metabolites. This recently reported experiment used samples comprising a hot water extraction of intact E. coli cells and as such preferentially measured polar chemicals associated with central metabolism. It remains to be seen if a similar performance can be demonstrated with more complex extraction procedures (e.g. solvent mixtures or sample partitioning through phase separation), particularly with crude sample types likely to contain complex membrane components or fragmented biological polymers.
3.3 ESI-MS fingerprint data processing and quality control
Concepts in quality control (QC) in LC–MS experiments are discussed extensively by Dunn et al. (2011) and will not be considered in detail in the present review. Randomizing sample classes in the run sequence is mandatory to avoid bias due to the position of samples within the injection order. The inclusion of a ‘mastermix’ QC solution made by combining an aliquot of all (or a representative sub-set of) samples used in the experiment provides a standardised quality control sample relevant to the matrix under study (e.g. Beckmann et al. 2007). In LC–MS procedures generating quantitative data these QC samples can be used to monitor instrument response throughout the analysis of a large batch of samples to calculate technical precision within each block, and these QCs can also be used to perform local corrections thereby compensating for instrument drift (e.g. Dunn et al. 2011). In instances where ESI-MS fingerprinting is used to generate a comprehensive metabolite data set then such QC measures are essential for data normalisation, especially when samples are analysed in large batches (e.g. Fuhrer et al. 2011). In all types of ESI-MS fingerprinting experiments, the infusion of several QC samples at the beginning of a batch of samples is advisable to ‘condition’ the LC–MS system after any system maintenance following the analysis of a large sample batch (see Dunn et al. 2011). Signal variance appears apparently much greater for m/z typically exhibiting higher intensities when all signals are measured on the same scale in the raw data. The logarithmic transformation of signal intensity can help alleviate the dependency of the variance with the intensity because it makes multiplicative models additive (van den Berg et al. 2006) and is often a consensus step in many metabolomics applications.
Further inter-spectrum sources of variability are often present in ESI-MS fingerprint data that come mainly from different sample concentrations, loss of sensitivity of the detector over time or degradation of certain samples. A range of methods are routinely applied in mass spectrometry to normalize fingerprint data based on the use of one or more internal standards of ‘known’ concentration. This is not often possible in direct infusion ESI-MS fingerprint data which are generally not quantitative and so the most widely used solution consists of a so-called global normalization by rescaling each measurement within a spectrum by a constant factor, such as the sum of all the spectra intensities (TIC). Like any LC–MS experimental strategy ESI-MS fingerprint data quality can also be effected by increasing contamination of the ionisation source that gradually reduces ionisation efficiency; this may be a particular problem with crude extracts from biological matrices containing large amount of proteins or complex carbohydrates. Additional cumulative effects on data quality many result from the loss of ion transmission efficiency and a gradual deterioration of detector sensitivity at set values or the absence of regular calibration and tuning which is a necessity in high and ultra-high resolution mass spectrometry. For such reasons ESI-MS fingerprinting experiments are generally carried out in batches of carefully randomized samples during periods of adequate instrument performance and specific ‘corrections’ applied post-data acquisition to align batches of data from the same experiment.
3.4 Ion suppression and ESI-MS fingerprinting
Ion suppression is a well described and often unpredictable effect described in many experimental contexts when quantitative LC–MS data are generated (e.g. King et al. 2000; Annesley 2003; Liu et al. 2010). It can be thought of as a ‘matrix effect’ resulting from the presence of co-eluting compounds which change the efficiency of ESI-MS droplet formation/evaporation, cause precipitation in the LC mobile phase, or cause charge neutralisation of an effected analyte in the gas phase (King et al. 2000). Ion suppression is manifestly a potential problem in LC–MS as it can reduce signal intensity from a sub-set of metabolites eluting at specific retention times in the chromatogram, thus invalidating any strictly quantitative measurement (Antignac et al. 2005). There were occasional suggestions early in the development of metabolomics technology which perhaps gave the impression that the phenomenon of ion suppression might be more of a problem with flow infusion ESI-MS fingerprinting (e.g. Hall et al. 2002). However it is difficult to find any evidence that ion suppression has more of a detrimental effect on flow infusion fingerprinting than in LC–MS methods using chromatography to separate metabolites. Indeed, when examined in detail, studies by several groups all showed that the relationship between signal intensity and metabolite concentration was generally linear at higher sample dilutions (Dunn et al. 2005b; Southam et al. 2007; Madalinski et al. 2008; Han et al. 2008; Fuhrer et al. 2011). In addition, the use of long infusion times and slower sample delivery rates, achievable using NanoMate technology (Schultz et al. 2000), in combination with more dilute sample extracts, has been reported to reduce ion suppression effects greatly. When direct infusion results were compared with LC–MS analyses of the same extract reproducibility of the observed metabolic fingerprint was significantly improved (Boernsen et al. 2005; Southam et al. 2007). From a practical perspective the impact of ion suppression effects are generally minimised in ESI-MS fingerprinting by running dilutions of any new biological matrix to optimise data quality by ensuring that there is a good spread of signal intensities across the whole spectrum (Dunn et al. 2008; Beckmann et al. 2008; Madalinski et al. 2008; Fuhrer et al. 2011). However, even after such optimisation it is not uncommon for an ESI-MS fingerprint to still be dominated by signals derived from an abundant metabolite class which easily acquires charge. However, as all analytes within a flow infusion electrospray sample experience the ionisation process in an identical chemical background then any such matrix effects should at least be imposed uniformly in all samples derived from similar types of tissues (Fiehn 2002). Furthermore, high through-put of many biological replicates actually allows discrimination of classes based on very small but nevertheless statistically significant changes. Therefore, subtle, but consistent changes in the abundance of even potentially suppressed ions can be extracted by powerful data mining strategies.
4 Applications of ESI-MS fingerprinting
ESI-MS fingerprinting has gained popularity using a range of instrumentation applied to an ever increasing number of biological problems (Table 1) since Smedsgaard and Frisvad (1996, 1997) first described the use of direct infusion ESI-MS to carry out taxonomic studies in crude extracts of filamentous fungi producing potential bioactive metabolites. Over the past decade a major application of ESI-MS fingerprinting methodology has centred on classification experiments using multivariate data analysis tools (e.g. Broadhurst and Kell 2006; Enot et al. 2008). These include the pre-screening of samples to confirm the ‘discreteness’ of sample classification to ensure, for example, that data are not artificially partitioned as a result of analytical batches, sample collection sites or unforeseen environmental or genotypic influences (Beckmann et al. 2008). This can be of particular benefit in epidemiological studies involving ‘free living’ humans where metabolome differences derived from unrecorded experimental factors orthogonal to the biological question under consideration (e.g. drug intake, inflammation/disease status, exercise, genotype, colonic microflora differences, diet) can confound data analysis (e.g. Scalbert et al. 2009; Favé et al. 2011). Similarly, discovery of sample classification or sample outlier problems by ‘first pass’ metabolite fingerprinting approaches at an early stage of an analytical process can avoid confounding problems later in an experimental ‘pipeline’, particularly when modelling will ultimately depend on data with very high dimensionality derived from much more expensive and time consuming quantitative LC–MS or GC–MS profiling methods. In addition to sample classification ESI-MS fingerprint data has been increasingly interrogated by a range of powerful feature selection methods (e.g. Broadhurst and Kell 2006; Enot et al. 2008) to provide initial clues to the potential identity of metabolites ‘explanatory’ of chemical differences between biological classes. A typical use is to highlight masses that can be targeted for structural identification and accurate quantification in follow up analyses using either hyphenated mass spectrometry (typically UHPLC-MS) or high mass accuracy fingerprinting methods. In general, most applications can be distinguished firstly by the mass resolution capability of the equipment used and the anticipated utility of ESI-MS data generated in any metabolomics ‘pipeline’. The following examples will be used to outline the range of applications reported to date.
4.1 Nominal mass and low resolution ESI-MS fingerprinting
Using simple mass spectrometers with single quadrupole detectors, Pietta and co-workers used direct infusion ESI-MS to characterise medicinal plants and plant resins (Mauri et al. 1999; Mauri and Pietta 2000a, b; Pietta et al. 2002). With only very modest resolution and limited sensitivity the nominal mass spectra generated in such experiments were useful to survey the diversity and relative concentrations of major potentially bioactive secondary metabolites found in crude extracts. Flow infusion ESI-MS fingerprinting using ion trap devices have similarly been used to analyse medicinal plants (Piovan et al. 1998, 2004; Favretto and Flamini 2000; Favretto et al. 2001), fruit polyphenol composition (McDougall et al. 2008), to confirm the correct identification of medicinal plants (Chen Lin and Harnly 2010c), to analyse the composition of derived herbal medicines (Sun and Chen 2011), and to phenotype fungal pathogen isolates (Lowe et al. 2010). With the advent of ‘first generation’ commercially available time of flight (TOF) instruments at the end of the 20th century (Guilhaus et al. 1997) came the first reports of ‘global’ non-targeted, nominal mass ESI-MS fingerprinting for species and strain differentiation in bacteria and yeast (Goodacre et al. 1999; Vaidyanathan et al. 2001, 2002; Allen et al. 2003, 2004; Kaderbhai et al. 2003). These publications were followed by reports of the use of similar methodologies in plant biology, food and environmental science, largely for sample classification, diagnostic or quality control purposes. For example Goodacre et al. (2002) first used direct infusion ESI-MS to look for evidence of olive oil adulteration and such methods continue to be used for oil and liquid hydrocarbon fuel analysis (e.g. Catharino et al. 2007; Alves et al. 2010). Non-targeted, nominal mass direct infusion ESI-MS fingerprinting similarly has found utility for the characterisation of herbal dietary supplements (Mattoli et al. 2011), for assessment of agronomic practices on food chemical composition (Luthria et al. 2008; Chen et al. 2010c) and for an examination of the wine aging process (Sawaya et al. 2011). In more fundamental applications involving plants Goodacre et al. (2003) looked for evidence of chemical signals involved in photoperiod responses in the vascular tissue (phloem) of the photoperiod-sensitive plant Pharbitis nil. Furthermore Catchpole et al. (2005) used flow infusion ESI-MS to determine whether the metabolome of genetically modified plants could be considered ‘substantially equivalent’ to that of cultivars developed using traditional plant breeding methods. In addition the chemical basis of cultivar classification in potato tubers was found to be associated with metabolites considered to be precursors of food quality characteristics (Beckmann et al. 2007). Johnson et al. (2007) used similar approaches to investigate metabolome differences between Arabidopsis ethylene signalling mutants and their wild-type; more recently Lloyd et al. (2011a) investigated the metabolome changes in similar mutants following infection with the fungal pathogen Botrytis cinerea. Similar ESI-MS fingerprinting approaches revealed that phospholipids were major discriminatory metabolites in responses by Brachypodium distachyon to challenge by Magnaporthe grisea (Allwood et al. 2006). There are several reports of non-targeted direct infusion ESI-MS fingerprinting using instruments with a single TOF detector (mass resolution around 5,000 FWHM (at m/z 200) in which data has either been processed into bins of narrower mass windows (e.g. 0.2 amu; Davey et al. 2008) or spectra collected in centroid mode as individual peaks of accurate mass down to 4 decimal places (Dunn et al. 2005b; Overy et al. 2005). Using such approaches Dunn et al. (2005b) were able to identify approximately 50 metabolites in a crude extract of tomato fruit and it was demonstrated that 10 were present at significantly different levels in two species of tomato and in some instances explanatory signals were detected that would have been masked in data binned to 1 amu.
Linear ion trap technology is associated with a general robustness over long periods of time which has proved extremely valuable for large scale experiments using nominal mass fingerprinting (for overview see Beckmann et al. 2008). This ability to analyse a larger number of samples at a high throughput was conducive to the generation of sufficient replicates to allow the efficient use of powerful supervised machine learning methods (Broadhurst and Kell 2006; Enot et al. 2008) not only to classify samples but also to run robust correlation and feature selection procedures. Thus, an important advance in the field was the use of non-targeted nominal mass ESI-MS fingerprinting, not only to classify samples but also to derive preliminary evidence that specific m/z signals were explanatory of metabolome differences between different biological classes. For example, there have been several instances where it has been used to investigate the biochemical origin of mutant phenotypes in Arabidopsis thaliana in functional genomics studies (e.g. Enot et al. 2006; Scott et al. 2010), or to screen for ‘unexpected’ differences in the metabolome of large populations of genetically modified potato plants (Enot et al. 2007; Enot Beckmann and Draper 2007. The same analytical strategy has showed particular utility in situations where the biological systems display dynamic temporal changes, for example the rapid changes in the leaf metabolome following challenge of plants by pathogens (Allwood et al. 2006; Parker et al. 2008, 2009; Ward et al. 2010). The problem of large levels of uncontrolled variability in the human urine metabolome from both habitual diet and individual ‘metabotype’ was similarly overcome by the use of adequate sample replication using the same technology (Fave et al. 2011; Lloyd et al. 2011b, c).
4.2 Accurate mass ESI-MS fingerprinting using Q-TOF and Q-Trap instruments
Non-targeted, flow infusion ESI-MS fingerprinting using Q-TOF and Q-trap mass spectrometers has been applied to a range of biological and diagnostic problems over the past decade. Many reports centred on applications in microbial strain identification (Lafaye et al. 2005; Smedsgaard et al. 2004; Mas et al. 2007; Pope et al. 2007; Hojer-Pedersen et al. 2008; Fuhrer et al. 2011), herbal medicine evaluation (Yue et al. 2011), plant genotyping (Scholz et al. 2004) or analysis of humic acid extracts (Baigorri et al. 2008) similar to those undertaken previously by nominal mass fingerprinting. A key difference in some of these reports from those using nominal mass fingerprinting was the desire by researchers to achieve putative identification of de novo explanatory metabolites of interest to the biological problem under study directly from the fingerprint data. More recent publications are witness to the emergence of a new generation of more robust, higher accuracy TOF instruments that have allowed researchers to utilise direct infusion accurate mass ESI-MS fingerprinting for ‘proof of principle’ and diagnostic uses in clinical studies (e.g. Lin et al. 2010; Lokhov et al. 2012; Yang et al. 2009; Dunn et al. 2012) and for systems biology and functional genomics studies (e.g. Fuhrer et al. 2011).
4.3 ESI-MS fingerprinting at ultra-high mass accuracy using Fourier transform mass spectrometry
Following a flurry of ‘proof of principal’ papers (e.g. Cooper and Marshall 2001; Aharoni et al. 2002) there has been an increasing number of reports of the use of ultra-high accurate mass fingerprinting in a wide range of research fields as FT-ICR-MS and Orbitrap-MS instruments have become more robust and widely available. Aharoni et al. (2002) produced one of the earliest descriptions of the application of FT-ICR-MS fingerprinting to a biological problem when they characterised in great detail the chemical composition of the ripening process in strawberries. Tohge et al. (2005) used a similar approach to study the effect of exogenous myb gene expression in transgenic Arabidopsis plants. Metabolic phenotyping studies and mechanistic biochemical investigations using flow infusion FT-ICR-MS fingerprinting have also been reported, using the model functional genomics plant species, Arabidopsis thaliana particularly to probe its responses to environmental stimuli and stresses, including mineral nutrition (Hirai et al. 2004), cold acclimation (Gray and Heath 2005) and light/dark regulation of metabolism (Nakamura et al. 2007). Flow infusion FT-ICR-MS equally has found utility in the de novo investigation of both metabolic flux and biochemical function of enzymes in studies involving both plant and microorganisms (Schmidt et al. 2007; Zulak et al. 2007; Furuya et al. 2008; Kai et al. 2009; Oikawa et al. 2006). In an interesting study Nam et al. (2008) used direct infusion FT-ICR-MS fingerprinting to look for evidence of the excretion of allelopathic chemicals by the aquatic plant Myriophyllum spicatum during co-existence with the cyanobacterium Microcystis aeruginosa.
Data processing, signal annotation success and quantification are improved further in approaches utilising stable isotopes (Mougous et al. 2002; Han et al. 2008; Madalinski et al. 2008; Giavalisco et al. 2008). For example Giavalisco et al. (2008) used extracts from Arabidopsis grown under conditions that allowed whole metabolome labelling with 13C CO2. When the 13C metabolite-derived mass spectrum is compared to that from plants grown under a 12C atmosphere then all metabolites of true biological origin will appear in both, but the 13C-labelled metabolites shifted with respect to its molecular mass by the number of carbon atoms contained. One consequence of this is that all natural metabolites will be present as 12C/13C ‘ion pairs’ and all ‘system’ peaks will only be found in the 12C mass spectrum and can be removed from any subsequent data analysis. Madalinski et al. (2008) similarly spiked extracted yeast samples used for Orbitrap FT-MS analysis with several different concentrations of extracts of yeast cultures uniformly labelled with 15N to explore how to minimise the matrix effects of the direct infusion ESI-MS process. With optimal conditions established non-targeted Orbitrap FT-MS fingerprinting was used to investigate the induction of the glutathione biosynthesis pathway in yeast culture challenged with the cadmium. Their data provided several new insights into the modulation of the sulphur amino acid pathway in yeast that were not evident in a prior targeted analysis using chromatographic procedures (LC–MS).
A pioneering publication by Cooper and Marshall (2001) first demonstrated the power of direct infusion FT-ICR-MS for the analysis of complex dietary components by discriminating five wines without any prior purification step. Gougeon et al. (2009) used FT-ICR-MS fingerprinting coupled with multivariate analysis to show that barrel-aged wines have a ‘metabologeographic signature’ relating to the forest location from which the wood used to manufacture the barrels was originally derived. Around the same time Liger-Belair et al. (2009) used FT-ICR-MS fingerprinting to reveal large numbers of compounds in aerosols derived from champagne bubbles and putatively assigned many signals to compounds with organoleptic properties or potential to be aroma precursors. Proschogo et al. (2012) suggested that FT-ICR-MS fingerprinting outperformed other analytical methods for the investigation of aging of oils extracted from macadamia nuts. The results were consistent with current industry measures of rancidity and showed that different methods of oil extraction and storage regimes prior to analysis resulted in different levels of peroxides in the oil that are associated with rancidity.
Flow infusion FT-ICR-MS fingerprinting has been found to have great utility for the analysis of complex environmental samples containing botanical residues. For example, dissolved organic matter (DOM) represents one the largest and probably the most heterogeneous pools of reactive carbon on earth. An understanding of specific mechanisms that add or remove compounds from the DOM pool has been held back by lack of detailed description of its molecular constituents. Flow infusion FT-ICR-MS has now been applied to this problem by characterising DOM in freshwater (e.g. Kim et al. 2006; Sleighter and Hatcher 2008; Minor et al. 2012) and marine (e.g. Tremblay et al. 2007; Dittmar and Koch 2006) systems providing enormous detail, on the composition of thousands of individual compounds (Kujawinski et al. 2004; Stenson et al. 2003 and Koch et al. 2005). Characteristic signatures of different environments have been developed and these findings are now starting to unravel the complex balance of abiotic and biotic processes which control DOM composition. Similarly, Ohno et al. (2010) were able to use direct infusion FT-ICR-MS data to discover molecular signals with scope for development as markers for different types of soil and plant-derived biomass. The field of environmental toxicology has also attracted interest in the use of ultrahigh mass accuracy FT-ICR-MS methods to discover biomarkers of pollution stress in indicator organism (Taylor et al. 2009; Rossello-Mora et al. 2008).
From a clinical perspective direct infusion ESI-MS fingerprinting has been use to improve knowledge of early molecular events occurring upon ischemia/reperfusion following liver transplantation (Hrydziuszko et al. 2010). Hasegawa et al. (2010) showed that urinary metabolic fingerprinting with FT-ICR-MS can be used to detect biomarkers representative of compounds with different mechanisms of toxicity which can have benefit at early stages of drug development. The results of Antunes et al. (2011a) using FT-ICR-MS fingerprinting suggested that phospholipids were an important source of carbon and energy for Salmonella during growth in the laboratory as well as during gallbladder infections of mice. In other reports the labelling of cells in culture with 34S has been used to differentiate sulphated from reduced sulphated compounds which led to the discovery of several new sulphated molecules in Mycobacterium species (Mougous et al. 2002). In a different strategy Han et al. (2008) used isotopically labelled internal standards to confirm the identity and quantification of choline in plasma and serum samples. Further utility of direct infusion FT-ICR-MS fingerprinting for diagnosis of chronic illnesses as diverse as liver injury from alcohol (Bradford et al. 2008), drug-induced phospholipidosis (Hasegawa et al. 2007, insulin insensitivity Lucio et al. 2010 and Crohn’s disease Jansson et al. 2009 have also been reported.
5 Recommendations on integration of ‘first pass’ non-targeted ESI-MS fingerprinting with further targeted analysis to develop hypotheses in metabolomics research
To date many published projects have employed an experimental design using a ‘hierarchical’ approach to generate metabolomics data that initiates with ESI-MS fingerprinting either to generate initial hypotheses, or to validate or optimise experimental design (Catchpole et al. 2005; Grata et al. 2007; Beckmann et al. 2007, 2010; Madalinski et al. 2008; Parker et al. 2009; Scott et al. 2010; Ward et al. 2010; Lin et al. 2010; Fuhrer et al. 2011; Favé et al. 2011; Lloyd et al. 2011b, c). For example, Fuhrer et al. (2011) used direct infusion accurate mass fingerprinting with a Q-TOF to investigate metabolome differences in E. coli deletion mutants and then performed targeted collision induced fragmentation on several abundant ions of interest and compared spectra to those in an in house library and the Massbank database. Madalinski et al. (2008) similarly used an LTQ-Orbitrap to generate direct infusion, accurate mass ESI-MS fingerprints to first discover metabolite signals explanatory of cadmium exposure in yeast. Explanatory signals were followed up by targeted MSn experiments using the linear ion trap to develop fragmentation ‘trees’.
Demonstrating that any non-targeted metabolomics procedure can develop good quality data whilst dealing with sources of extreme variability is a very important aspect of method development (Broadhurst and Kell 2006; Enot et al. 2008) and these mathematical processes should be adequately reported (Goodacre et al. 2007). It is equally important that the experimental strategy explicitly identifies potentially explanatory data features that can be easily investigated further for hypothesis development and biological validation. In the following sections we will outline the challenges and make recommendations for method development and data processing and modelling for procedures using direct infusion ESI-MS as the primary discovery tool.
5.1 Validating experimental design and sample overview using nominal mass fingerprinting
5.1.1 Defining an appropriate sample batch size
In several recent papers we have described the use of an optimised nominal mass, flow infusion ESI-MS fingerprinting workflow to generate information-rich spectra that are suitable for classification and feature selection in experiments containing large numbers (600 to >2,000) of samples (Fig. 2). This procedure initiates with the use of a linear ion trap to perform first pass ESI-MS fingerprinting for sample classification and feature selection (Beckmann et al. 2008). The collection of nominal mass spectra over four scan ranges (m/z 15–110; 100–220; 210–510 m/z; 500–1,200 m/z) in both negative ion and positive ion mode has been used to great effect using extracts derived from tubers, plant leaves, urine and blood (Scott et al. 2010; Ward et al. 2010; Favé et al. 2011; Lloyd et al. 2011b, c). A key part of the procedure is a generic routine to ‘mean-shift’ batches of data as part of our ESI-MS pre-processing workflow (Enot et al. 2008) which allows basically data alignment between many batches of hundreds of samples (Fig. 3). An appropriate number of samples per batch has to be explored empirically and can depend on experimental parameters such as ion source geometry and sample matrix (e.g. 20–30 mammalian serum samples or 240 plant extracts per batch can be reasonable). Batch size might differ between instruments of different make, but can be easily determined by principal component analysis (PCA) of quality control samples in pilot experiments. Aligned data batches can then be used to construct a large data matrix to allow further checks on data quality to be performed.

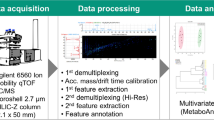
Experimental ‘pipeline’ for a hierarchical non-targeted metabolomics study initiating with nominal mass, flow infusion ESI-MS fingerprinting

Sample batch ‘mean intensity shifting’ to align ESI-MS fingerprint data. Plots show log10-transformed total ion intensity data points of each mass spectrum (e.g. Fig. 1b) in injection order with a solid line indicating the mean value for a batch of 24 samples: a before and b after mean shift of batch mean intensities
5.1.2 Pre-screening of fingerprint data set prior to feature selection
A useful first step in any metabolomics study is a pre-screen of either all, or a representative sub-set of samples, to determine whether the biological question under study is likely to be explainable by reproducible metabolome differences (Allwood et al. 2006; Enot et al. 2006, 2007, 2008; Parker et al. 2008). The data in Fig. 4 (adapted from Lloyd et al. 2011b) provide an example from a real-life study which examined the impact on human urine metabolite composition of both acute and habitual intake of citrus foods. Unsupervised multivariate data analysis methods such as PCA (Fig. 4a) are commonly used in metabolomics studies to screen for unexpected groupings or the presence of potential outlying samples in ESI-MS fingerprint data (e.g. Broadhurst and Kell 2006; Enot et al. 2008; Beckmann et al. 2008). Supervised multivariate data analysis techniques such as linear discriminant analysis (LDA) provide methods to discriminate labelled biological classes linked to the underlying signal behaviour within the ESI-MS fingerprints under comparison. When urine samples were labeled by volunteer ID and subjected to PC-LDA it can be seen clearly (Fig. 4b) that two individuals were separate from all other volunteers. Subsequent investigation revealed that both outlying individuals had paracetamol breakdown products in their urine which would have confounded efficient data modeling had these two individuals not been removed from the data set. Similar multivariate data analysis approaches can be used to look for evidence of experimental factors that might unknowingly partition data, for example gender differences (Fig. 4c). The use of such algorithms to provide an exploratory overview of the behavior of data representing a sample population, particularly after labeling by specific meta-data attributes, is an important step in confirming that the overall experimental statistical design is adequate.

Quality control and experimental design validation in a non-targeted, flow infusion, nominal mass ESI-MS fingerprinting experiment. a Analysis of spectra derived from urine samples by +ve ion mode ESI-MS (80–550 m/z) using principal component analysis (PCA). Classes are labelled with symbols related to time of collection: crosses pre-test day evening/night urine samples, circles fasting spot urine, triangles 3 h post-prandial urine samples. Total percentage explained variances for each PC are given in brackets. Circled in red are biological replicates from a potential volunteer outlier. Potential outlying technical/experimental replicates are highlighted in boxes; b Analysis of urine samples by +ve ion mode ESI-MS (100–550 m/z) using PC-linear discriminant analysis (PC-LDA), using ‘ID’ as the class structure, as a but with more samples per person. Eigenvalues (Tw values) are given in brackets; c Analysis of urine samples by +ve ion mode ESI-MS (100–550 m/z) using PC-LDA with ‘Gender’ as the class structure (F female and M male). Tw values are given in brackets; d PC-LDA of +ve ion mode ESI-MS fingerprints (100–220 m/z) of fasting urine using ‘citrus consumption’—high, medium and low as the class structure (adapted from Lloyd et al. 2011). Tw values are given in brackets; e pair-wise classification robustness comparisons between the citrus consumer classes. ACC classification accuracy; AUC area under the (ROC receiver operating characteristic) curve; RF Mar random forest (RF) classification margin (Color figure online)
5.1.3 Validating experimental design
To assess whether assigned sample classifications are not only distinctive, but also adequate to warrant deeper metabolome investigation it is desirable to have quantitative measures of the ‘goodness of separation’ of the biological classes under investigation, preferably using more than one data analysis approach (Enot and Draper 2007; Enot et al. 2006, 2007, 2008). Good discrimination of each citrus food consumption level class is evident in PC-LDA scores plots comparing ESI-MS fingerprints of fasting urine samples (Fig. 4d). The Eigenvalue (Tw) for separation between high and lower level citrus consumers in the first discriminant function dimension (DF1) was ~2 which indicated a likelihood of robust classification (Enot et al. 2008), providing confidence that further investigation of this biological problem using non-targeted flow infusion ESI-MS fingerprinting data was potentially worthwhile. As in any study using data with high dimensionality it is very important to validate that the differences between any biological classes are robust (Somorjai et al. 2003; Lyons-Weiler et al. 2005; Broadhurst and Kell 2006; Enot et al. 2008). It is therefore essential to confirm the robustness of the classification by using other data analysis tools, such as random forest agglomerative decision trees (RF; Breiman 2001) or partial least squares-discriminant analysis (PLS-DA; Szymanska et al. 2011) and to calculate informative measures of model sensitivity and specificity (Broadhurst and Kell 2006; Enot et al. 2006, 2007, 2008; Westerhuis et al. 2008). Validation of classification in the example study included firstly, assessing the area under the receiver operating characteristic (ROC) curve (AUC) which aggregates performance across the entire range of trade-offs between true positive rate and false positive rate and ranges from 0.5 (indicating random performance) to 1.0 (denoting perfect classification). Secondly, random forest (RF) was employed in the analysis of the urine fingerprint data, where the RF classification margin is defined as the proportion of votes for the correct class minus the maximum proportion of votes for the other classes—the larger the margin, the more confidence in the classification. The RF margin values of the pair-wise comparison between the high and low citrus groups was >0.25, and the AUC was >0.9, indicating excellent classification (Fig. 4e).
5.2 Feature selection in flow infusion ESI-MS fingerprint data models
Once adequate classification of samples has been demonstrated, feature selection techniques can be used to discover mass signals potentially responsible for ‘explaining’ metabolomic differences between each sample class. Random forest (RF) models cope well with high dimensional data sets and multiclass problems, are robust classifiers and, more importantly, also provide insight into the structure of the data under study by quantifying the confidence in classification voting and by quantifying the importance of each ‘explanatory’ variable for the classification task (Breiman 2001). The RF importance scores of the top 20 ranked signals responsible for discriminating between the high and low citrus exposure classes are shown in Fig. 5a. In any modelling experiment using data with high dimensionality it is important to be confident that feature selection methods are identifying attributes that not only are statistically significant but importantly make biological sense and lead to testable hypotheses (Enot et al. 2006). In our experience, in RF models comparing two biological classes displaying relatively discrete metabolome differences, it is not unusual for Importance Scores to drop sharply and then quickly level out as the signals become less significant as shown in Fig. 5b (Enot et al. 2006, 2007, 2008; Beckmann et al. 2007, 2010; Favé et al. 2011; Lloyd et al. 2011b). From the analysis of a wide range of data sets we have determined that features with an RF importance score >0.02 are likely to make a significant contribution to a binary (two class) classification model.

Feature selection, targeted FT-ICR-MS analysis and signal annotation in a flow infusion ESI-MS fingerprint model discriminating volunteers on the basis of citrus consumption levels. a The top 10 positive ion features (m/z 100–550) discriminating between high and low habitual citrus consumers; b random forest importance scores of the top ranked signals discriminating between ‘High’ and ‘Low’ citrus fruit consumption. Inset shows a dendrogram constructed from Pearson correlation analysis of top positive ion signals. c An example of a nominal mass bin containing several signals, in this case proline betaine [M + K]1+, within the nominal mass bin m/z 182, is masking a 4-hydroxyproline betaine [M + Na]1+; d structure of proline betaine, a compound highlighted several times following MZedDB annotation of signals explanatory of citrus exposure levels. e Electrospray ionization tandem mass spectrometry (ESI MS/MS) spectra of the nominal mass bin containing putative proline betaine [M + H] 1+ (m/z 144) from a pool of urine derived from mixing samples from 4 volunteers; f the ESI-MS/MS spectra of an authentic sample of synthetic proline betaine [M + H]1+; g Box plot of the top explanatory metabolite signal (m/z 182) discriminating between High and Low habitual citrus fruit consumers. Where: the box indicates the interquartile range; the red horizontal bar the median; vertical bars the maximum and minimum values up to 1.5× interquartile range; error bars represent the standard error of 12 volunteers. e–g Are adapted from Lloyd et al. (2011) (Color figure online)
Non-targeted ESI-MS fingerprint models additionally have several properties inherent to the data structure which can help to perform a ‘sanity’ check on any potentially explanatory variables. Firstly, the fact that most metabolites are represented by several signals (including salt adducts, isotopes, clusters and neutral losses) means that even if an single adduct is obscured by the presence of isobaric molecules found in the same mass bin then there is always the chance that other mass bins containing different adducts derived from the same parent molecule will not be effected (see Catchpole et al. 2005; Enot et al. 2007 for detailed examples). An additional factor resulting from the presence of multiple ions derived from the same metabolite is that many of the more abundant signals should co-vary and would have an equal chance of being selected as an explanatory signal after feature selection; such correlation behaviour can be used to guide signal annotations (Tautenhahn et al. 2007; Overy et al. 2008; Enot et al. 2008; Takahashi et al. 2008; Draper et al. 2009; Fuhrer et al. 2011; Brown et al. 2011; Lloyd et al. 2011c). Displayed as Pearson correlation coefficient data, the inset in Fig. 5b shows part of the results of a correlation analysis between the highly ranked ESI-MS positive ion m/z signals discriminating high-level from low-level citrus food consumers. It can be seen that several of the top ranked signals are tightly correlated which suggests they may be related to the same metabolite or metabolite pathway. A cursory examination of the nominal mass mathematical differences between tightly linked m/z signals reveals several differing by 1 amu which are potentially isotopes (e.g. 144:145, and 218:219) and several differing by 22 amu (e.g. 144:166) or 38 amu (e.g. 144:182) which could present an [M + H]+, [M + Na]+ and [M + K]+ series. Thus knowing that multiple ionised representations of the same molecule would be expected to be highly ranked in classification experiments provides a further useful internal check for the quality of data models (Overy et al. 2008). Importantly, in situations where accurate mass information is available then these relationships, if confirmed, can quickly indicate which of the ion species is in fact a pseudo-parental ion to guide the annotation process. There have been an increasing number of reports in which ‘in house’ algorithms and bespoke ESI-MS signal annotation tools have been developed specifically to identify ions with specific mathematical relationships using both nominal mass data (e.g. Overy et al. 2008; Parker et al. 2008; Ward et al. 2010) and accurate mass data (Gorlach and Richmond 1999; Tautenhahn et al. 2007; Draper et al. 2009; Payne et al. 2009; Brown et al. 2011; Favé et al. 2011; Lloyd et al. 2011b, c; Kuhl et al. 2012).
It is recommended that a thorough evaluation of relative statistical significance levels, correlation behaviour and mathematical relationships is undertaken before committing samples to further targeted analysis in order to structurally annotate and/or quantify metabolites potentially explanatory of the biological question under study.
5.3 Explanatory signal annotation and hypothesis generation
5.3.1 Targeted FT-ICR-MS analysis of selected nominal mass bins
As nominal mass bins can contain one or several isobaric mass peaks when analysed by targeted FT-ICR-MS it is important to identify signals that behave consistently to identify ion species that may be responsible for the selection of a potentially explanatory mass bin. Thus to reduce the impact of inter-class differences, it is recommended that targeted FT-ICR-MS analysis is performed on several pools of samples (at least 3 pools) of each class. Two methods of FT-ICR-MS analysis can be implemented depending on whether a particular matrix is to be used repeatedly in a range of investigations. Firstly, as demonstrated by the citrus food consumption experiment, ‘one off’ narrow mass window scans (3–10 amu) around the nominal mass regions of interest can be undertaken using longer flow infusions times in narrow SIM mode, particularly in an instance where the signal intensity is relatively weak. Alternatively, if the sample type is to be regularly used then committing time and resources to a detailed FT-ICR-MS ‘matrix’ analysis in narrow SIM mode (across whole mass range; 50–1,400 m/z) could be worthwhile in order to produce a ‘checklist’ of accurate mass signals to be routinely expected in any particular matrix. It is well known that the number of scans acquired for each spectrum greatly affects the reproducibility of the resulting spectrum (Southam et al. 2007; Han et al. 2008; Giavalisco et al. 2009; Junot et al. 2010; Ohta et al. 2010). Optimization in our lab has shown that approximately 50 scans (in overlapping 30 amu ‘sliding’ windows) are required for each mass range, addressing the balance between quantification accuracy and acquisition time (and thus sample throughput). In both methods (narrow mass window scanning and ‘matrix’ analysis) the output is a table of accurate masses (down to 5 decimal places) together with their relative intensities for signals seen to be reproducible within each biological sample type. These data are then available for attempts at structural annotation to identify the areas of metabolism highlighted by non-targeted, flow infusion ESI-MS fingerprinting.
5.3.2 Structural annotation of FT-ICR-MS peaks
The description and criticism of procedures for the structural annotation of accurate mass data developed using ESI-MS analytical platforms have been the subject of a range of excellent original papers over the past decade and the detailed functionalities of the algorithms used are beyond the scope of the present review (for examples see Stoll et al. 2006; Kind and Fiehn 2006, 2007, 2010; Tautenhahn et al. 2007; Iijima et al. 2008; Nakamura et al. 2008; Werner et al. 2008; Draper et al. 2009; Wishart 2009; Brown et al. 2009, 2011; Kuhl et al. 2012). In parallel with the rapid increase in the number of publications describing experiments using ESI-MS analytical technology (both UHPLC-MS and direct infusion methods) has been the emergence of new (and modification of existing) databases containing metabolite information in order to facilitate putative annotation of accurate mass signals (e.g. PubChem, http://pubchem.ncbi.nlm.nih.gov/; HMDB, http://www.hmdb.ca/; KEGG, http://www.genome.jp/kegg/; KNapSAcK, http://kanaya.naist.jp/KNApSAcK/; MZedDB, http://maltese.dbs.aber.ac.uk:8888/hrmet/index.html; and MMD, Brown et al. 2009).
As described in Sect. 5.2, a small number (12–15) of nominal mass bins in ESI-MS fingerprints of fasting urine were selected as explanatory of different exposure to citrus foods in free-living human volunteers. Five nominal mass signals within this group (m/z 198, 182, 166, 144, 145) were very strongly correlated (Fig. 5b) and were therefore subjected to targeted FT-ICR-MS analysis using narrow SIM windows. High resolution (100,000 FWHM) spectra of each targeted mass bin was examined in replicates of 4 independent sample pools representing urine from individual classes as either high or low citrus consumers (e.g. see Fig. 5c). After interrogation of MZedDB at 1 ppm mass accuracy we observed that proline betaine (Fig. 5d), an abundant component of citrus fruits was tentatively explanatory of habitual exposure to citrus-containing foods. Further MS/MS analysis confirmed this putative identification using a chemical standard as a reference (Fig. 5e, f). This result makes biological sense (Fig. 5g) and is a good indicator of the robustness of this metabolomics workflow used routinely in Aberystwyth. These results form a solid foundation for the recommendation of proline betaine as a potential biomarker for citrus food exposure (Lloyd et al. 2011b) which can now be validated using target analytical methods in other human populations.
6 Conclusions and final comments
There are many mass spectrometry-based approaches to metabolomics and this review has provided details of how flow infusion ESI-MS fingerprinting has been established in a plethora of experimental situations. This approach is employed without recourse to chromatographic separation and therefore its speed and minimal data pre-processing requirements make it particularly attractive when dealing with large sample sets. The lack of a deconvolution step means that neither artefacts nor apparent missing values are introduced into the metabolite data. Isomers are not separated and cannot be readily identified; however, if a pure metabolite is found within a particular accurate mass window its empirical formula can be generated and the metabolite’s fragmentation pattern can be used to generate structural information and hence its unequivocal identification.
ESI-MS offers a very powerful ‘first pass’ screening of large sample populations in pilot experiments using nominal mass measurements. At this stage an early indication of any differences between biological classes can be generated at the metabolite level. This screening stage can be readily validated as large sample numbers can be analysed. Thus robust statistical analysis may be conducted providing an objective indication of the discriminatory ability and the population sizes needed in the different biological classes. Following this stage more time consuming chromatographic separation by LC or CE can be used and specific metabolites identified if necessary using high mass resolution spectrometers with MS–MS or MSn capabilities.
It has long since been believed that ion suppression limits the use of ESI-MS fingerprinting for metabolomics investigations. This review has admirably demonstrated that this is a myth and comments like this should be ignored. The opportunity of rapidly obtaining useful information from a first pass ESI-MS fingerprinting experiment greatly outweighs the probability that ion suppression will render the data useless. Indeed it is pertinent to note that the suppression is largely removed by using nano-infusion with dilute sample extracts and many recent investigations have indeed used such devices as the interface to the ESI source. The majority of ESI-MS fingerprinting experiments specifically measure signal relative ratios and are not used to generate quantitative data. This being said, several recent publications have advocated the use of a commercial ‘Absolute IDQ’ kit (Biocrates Life Sciences AG) in order to routinely generate quantitative data from flow infusion experiments (e.g. Mittelstrass et al. 2011; Rubio-Aliaga et al. 2011). However it should be noted that such experiments are in fact targeted to a subset of more lipophilic metabolites (dominated by carnitines and phospholipids, but including some amino acids) and as such cannot offer comprehensive metabolite coverage and are in fact relevant mainly to the analysis of animal tissues and animal biofluids. It is likely that the trend of using commercially supplied sets of reference chemicals in combination with standardized data generation methods is likely to increase in the future to provide coverage of relatively abundant metabolite classes found in a wider range of sample types
More than a decade of ESI-MS fingerprinting has shown us not only its capabilities and its position in metabolomics along with NMR, GC–MS and LC–MS but also the need for high resolution and accurate mass measurements to push metabolite profiling to its limits. Researchers have used FT-ICR-MS technology since it became commercially available at the end of the last century and about one half of the, relatively comprehensive, list of applications in Table 1 has used ultra-high resolution instrumentation either alone or in hierarchical workflows following high throughput nominal mass analysis. The advantage of high resolution, accurate mass experiments is the direct generation of chemical sum formula which, combined with data base searches, allow immediate tentative annotation of signals. In-line or off-line fragmentation experiments together with isotopic pattern analysis can help in confirming potential identities and databases to aid in this process are likely to become increasing sophisticated and more comprehensive in their coverage.
The new developments are exciting. TOF instruments like Leco’s Citius™ LC-HRT, Brucker’s maXis™ UHR-TOF, Agilent’s 6550 iFunnel Q-TOF LC/MS, AB SCIEX TripleTOF™ 5600 System and Waters’ SYNAPT G2-S MS are approaching 100,000 mass resolution routinely and/or sub-ppm mass accuracies at a breathtaking 50 to 200 Hz sampling rate. Thermo Scientific’s Orbi-trap technology aims to replace FT-ICR-MS technology with hybrids like the Orbitrap Velos Pro or the Q Exactive LC–MS. Whereas the Thermo Scientific’s FT-Ultra achieves 1 million mass resolution Orbi-trap technology is currently limited to 140,000 with 1 ppm mass accuracy at a 1 Hz sampling rate. Bruker however has installed already some new 18 T SolariX instruments for those who need more accuracy and resolution. All these new generation instruments will replace older Q-TOF and ion-trap instrumentation like the Premier series, QSTAR, early Bruker/Agilent TOFs and Thermo LTQs. It becomes clear that for each application, interest or budget one can get a suitable high or even ultra-high resolution LC–MS instrument, but it is certain that the new Q-TOFs and Orbitraps are well suited for ESI-MS experiments.
With ever improving TOF and Orbi trap hybrid technologies for LC–MS applications linked to nano-flow infusion and improving data pre-processing tools we expect not only more applications reporting flow injection, direct or nano-flow infusion, but also a shift from ESI-MS fingerprinting to non-targeted high resolution, accurate mass ESI-MS profiling in the foreseeable future.
Abbreviations
- amu:
-
Atomic mass unit
- Da:
-
Dalton
- MS:
-
Mass spectrometry
- m/z :
-
Mass-to-charge ratio
- DIMS:
-
Direct infusion MS
- FIE-MS:
-
Flow injection electrospray-ionisation MS
- Q-TOF:
-
Quadrupole-time-of-flight
- FT-ICR-MS:
-
Fourier transform ion cyclotron resonance MS
- 7 T:
-
7 Tesla, strength of magnet
- LC:
-
Liquid chromatography
- UHPLC:
-
Ultra high pressure/performance liquid chromatography
- GC:
-
Gas chromatography
- SIM:
-
Single ion monitoring
- DF:
-
Discriminant function
- LDA:
-
Linear discriminant analysis
- PCA:
-
Principal components analysis
- AUC:
-
Area under the receiver/operator curve
- QC:
-
Quality control
- MEDE:
-
Metabolomics to characterize dietary exposure (FSA-funded project)
- MZedDB:
-
Database: tools for the annotation of high resolution MS metabolomics data
References
Aharoni, A., De Vos, C. H. R., Verhoeven, H. A., et al. (2002). Nontargeted metabolome analysis by use of Fourier transform ion cyclotron mass spectrometry. OMICS: A Journal of Integrative Biology, 6, 217–234.
Allen, J., Davey, H. M., Broadhurst, D., et al. (2003). High-throughput classification of yeast mutants for functional genomics using metabolic footprinting. Nature Biotechnology, 21, 692–696.
Allen, J., Davey, H. M., Broadhurst, D., et al. (2004). Discrimination of modes of action of antifungal substances by use of metabolic footprinting. Applied and Environmental Microbiology, 70, 6157–6165.
Allwood, J. W., Ellis, D. I., Heald, J. K., Goodacre, R., & Mur, L. A. J. (2006). Metabolomic approaches reveal that phosphatidic and phosphatidyl glycerol phospholipids are major discriminatory non-polar metabolites in responses by Brachypodium distachyon to challenge by Magnaporthe grisea. Plant Journal, 46, 351–368.
Alves, J. D. O., Neto, W. B., Mitsutake, H., Alves, P. S. P., & Augusti, R. (2010). Extra virgin (EV) and ordinary (ON) olive oils: Distinction and detection of adulteration (EV with ON) as determined by direct infusion electrospray ionization mass spectrometry and chemometric approaches. Rapid Communications in Mass Spectrometry: RCM, 24, 1875–1880.
Annesley, T. M. (2003). Ion suppression in mass spectrometry. Clinical Chemistry, 49, 1041–1044.
Antignac, J. P., De Wasch, K., Monteau, F., et al. (2005). The ion suppression phenomenon in liquid chromatography-mass spectrometry and its consequences in the field of residue. Analytica Chimica Acta, 529, 129–136.
Antunes, L. C. M., Andersen, S. K., Menendez, A., et al. (2011a). Metabolomics reveals phospholipids as important nutrient sources during salmonella growth in bile in vitro and in vivo. Journal of Bacteriology, 193, 4719–4725.
Antunes, L. C. M., Arena, E. T., Menendez, A., et al. (2011b). Impact of Salmonella infection on host hormone metabolism revealed by metabolomics. Infection and Immunity, 79, 1759–1769.
Baigorri, R., Mari Zamarreno, A., Fuentes, M., et al. (2008). Multivariate statistical analysis of mass spectra as a tool for the classification of the main humic substances according to their structural and conformational features. Journal of Agricultural and Food Chemistry, 56, 5480–5487.
Beckmann, M., Enot, D. P., Overy, D. P., & Draper, J. (2007). Representation, comparison, and interpretation of metabolome fingerprint data for total composition analysis and quality trait investigation in potato cultivars. Journal of Agricultural and Food Chemistry, 55, 3444–3451.
Beckmann, M., Parker, D., Enot, D. P., Duval, E., & Draper, J. (2008). High-throughput, nontargeted metabolite fingerprinting using nominal mass flow injection electrospray mass spectrometry. Nature Protocols, 3, 486–504.
Beckmann, M., Enot, D. P., Overy, D. P., et al. (2010). Metabolite fingerprinting of urine suggests breed-specific dietary metabolism differences in domestic dogs. British Journal of Nutrition, 103, 1127–1138.
Bino, R. J., Hall, R. D., Fiehn, O., et al. (2004). Potential of metabolomics as a functional genomics tool. Trends in Plant Science, 9, 418–425.
Boernsen, K. O., Gatzek, S., & Imbert, G. (2005). Controlled protein precipitation in combination with chip-based nanospray infusion mass spectrometry. An approach for metabolomics profiling of plasma. Analytical Chemistry, 77, 7255–7264.
Bradford, B. U., O’connell, T. M., Han, J., et al. (2008). Metabolomic profiling of a modified alcohol liquid diet model for liver injury in the mouse uncovers new markers of disease. Toxicology and Applied Pharmacology, 232, 236–243.
Breiman, L. (2001). Random forests. Machine Learning, 45, 5–32.
Broadhurst, D. I., & Kell, D. B. (2006). Statistical strategies for avoiding false discoveries in metabolomics and related experiments. Metabolomics, 2, 171–196.
Brown, S. C., Kruppa, G., & Dasseux, J. L. (2005). Metabolomics applications of FT-ICR mass spectrometry. Mass Spectrometry Reviews, 24, 223–231.
Brown, M., Dunn, W. B., Dobson, P., et al. (2009). Mass spectrometry tools and metabolite-specific databases for molecular identification in metabolomics. Analyst, 134, 1322–1332.
Brown, M., Wedge, D. C., Goodacre, R., et al. (2011). Automated workflows for accurate mass-based putative metabolite identification in LC/MS-derived metabolomic datasets. Bioinformatics, 27, 1108–1112.
Castrillo, J. I., Hayes, A., Mohammed, S., Gaskell, S. J., & Oliver, S. G. (2003). An optimized protocol for metabolome analysis in yeast using direct infusion electrospray mass spectrometry. Phytochemistry, 62, 929–937.
Catchpole, G. S., Beckmann, M., Enot, D. P., et al. (2005). Hierarchical metabolomics demonstrates substantial compositional similarity between genetically modified and conventional potato crops. Proceedings of the National academy of Sciences of the United States of America, 102, 14458–14462.
Catharino, R. R., Milagre, H. M. S., Saraiva, S. A., et al. (2007). Biodiesel typification and quality control by direct infusion electrospray ionization mass spectrometry fingerprinting. Energy & Fuels, 21, 3698–3701.
Chen, H., Wortmann, A., & Zenobi, R. (2007). Neutral desorption sampling coupled to extractive electrospray ionization mass spectrometry for rapid differentiation of biosamples by metabolomic fingerprinting. Journal of Mass Spectrometry, 42, 1123–1135.
Chen, H.-W., Hu, B., & Zhang, X. (2010a). Fundamental principles and practical applications of ambient ionization mass spectrometry for direct analysis of complex samples. Chinese Journal of Analytical Chemistry, 38, 1069–1088.
Chen, P., Harnly, J. M., & Lester, G. E. (2010b). Flow injection mass spectral fingerprints demonstrate chemical differences in rio red grapefruit with respect to year, harvest time, and conventional versus organic farming. Journal of Agricultural and Food Chemistry, 58, 4545–4553.
Chen, P., Lin, L.-Z., & Harnly, J. M. (2010c). Mass spectroscopic fingerprinting method for differentiation between Scutellaria lateriflora and the Germander (Teucrium canadense and T. chamaedrys) species. Journal of AOAC International, 93, 1148–1154.
Cooper, H. J., & Marshall, A. G. (2001). Electrospray ionization Fourier transform mass spectrometric analysis of wine. Journal of Agricultural and Food Chemistry, 49, 5710–5718.
Davey, M. P., Burrell, M. M., Woodward, F. I., & Quick, W. P. (2008). Population-specific metabolic phenotypes of Arabidopsis lyrata ssp. petraea. New Phytologist, 177, 380–388.
De Vos, R. C. H., Moco, S., Lommen, A., et al. (2007). Untargeted large-scale plant metabolomics using liquid chromatography coupled to mass spectrometry. Nature Protocols, 2, 778–791.
Dettmer, K., Aronov, P. A., & Hammock, B. D. (2007). Mass spectrometry-based metabolomics. Mass Spectrometry Reviews, 26, 51–78.
Dittmar, T., & Koch, B. P. (2006). Thermogenic organic matter dissolved in the abyssal ocean. Marine Chemistry, 102, 208–217.
Draper, J., Enot, D. P., Parker, D., et al. (2009). Metabolite signal identification in accurate mass metabolomics data with MZedDB, an interactive m/z annotation tool utilising predicted ionisation behaviour ‘rules’. BMC Bioinformatics, 10, 227.
Dunn, W. B. (2008). Current trends and future requirements for the mass spectrometric investigation of microbial, mammalian and plant metabolomes. Physical Biology, 5, 011001.
Dunn, W. B., Bailey, N. J. C., & Johnson, H. E. (2005a). Measuring the metabolome: Current analytical technologies. Analyst, 130, 606–625.
Dunn, W. B., Overy, S., & Quick, W. P. (2005b). Evaluation of automated electrospray-TOF mass spectrometryfor metabolic fingerprinting of the plant metabolome. Metabolomics, 1, 137–148.
Dunn, W. B., Broadhurst, D., Brown, M., et al. (2008). Metabolic profiling of serum using ultra performance liquid chromatography and the LTQ-orbitrap mass spectrometry system. Journal of Chromatography B-Analytical Technologies in the Biomedical and Life Sciences, 871, 288–298.
Dunn, W. B., Broadhurst, D., Begley, P., et al. (2011). Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nature Protocols, 6, 1060–1083.
Enot, D. P., & Draper, J. (2007). Statistical measures for validating plant genotype similarity assessments following multivariate analysis of metabolome fingerprint data. Metabolomics, 3, 349–355.
Enot, D. P., Beckmann, M., Overy, D., & Draper, J. (2006). Predicting interpretability of metabolome models based on behavior, putative identity, and biological relevance of explanatory signals. Proceedings of the National academy of Sciences of the United States of America, 103, 14865–14870.
Enot, D. P., Beckmann, M., & Draper, J. (2007). Detecting a difference—assessing generalisability when modelling metabolome fingerprint data in longer term studies of genetically modified plants. Metabolomics, 3, 335–347.
Enot, D. P., Lin, W., Beckmann, M., et al. (2008). Preprocessing, classification modeling and feature selection using flow injection electrospray mass spectrometry metabolite fingerprint data. Nature Protocols, 3, 446–470.
Fard, A. M., Turner, A. G., & Willett, G. D. (2003). High-resolution electrospray-ionization Fourier-transform ion cyclotron resonance and gas chromatography-mass spectrometry of macadamia nut oil. Australian Journal of Chemistry, 56, 499–508.
Favé, G., Beckmann, M., Lloyd, A., et al. (2011). Development and validation of a standardized protocol to monitor human dietary exposure by metabolite fingerprinting of urine samples. Metabolomics, 7, 1–16.
Favretto, D., & Flamini, R. (2000). Application of electrospray ionization mass spectrometry to the study of grape anthocyanins. American Journal of Enology and Viticulture, 51, 55–64.
Favretto, D., Piovan, A., Filippini, R., & Caniato, R. (2001). Monitoring the production yields of vincristine and vinblastine in Catharanthus roseus from somatic embryogenesis. Semiquantitative determination by flow-injection electrospray ionization mass spectrometry. Rapid Communications in Mass Spectrometry, 15, 364–369.
Fiehn, O. (2002). Metabolomics—the link between genotypes and phenotypes. Plant Molecular Biology, 48, 155–171.
Fuhrer, T., Heer, D., Begemann, B., & Zamboni, N. (2011). High-throughput, accurate mass metabolome profiling of cellular extracts by flow injection-time-of-flight mass spectrometry. Analytical Chemistry, 83, 7074–7080.
Furuya, T., Nishi, T., Shibata, D., et al. (2008). Characterization of orphan monooxygenases by rapid substrate screening using FT-ICR mass spectrometry. Chemistry & Biology, 15, 563–572.
Giavalisco, P., Hummel, J., Lisec, J., et al. (2008). High-resolution direct infusion-based mass spectrometry in combination with whole (13)C metabolome isotope labeling allows unambiguous assignment of chemical sum formulas. Analytical Chemistry, 80, 9417–9425.
Giavalisco, P., Koehl, K., Hummel, J., Seiwert, B., & Willmitzer, L. (2009). (13)C Isotope-labeled metabolomes allowing for improved compound annotation and relative quantification in liquid chromatography-mass spectrometry-based metabolomic research. Analytical Chemistry, 81, 6546–6551.
Goodacre, R., Heald, J. K., & Kell, D. B. (1999). Characterisation of intact microorganisms using electrospray ionisation mass spectrometry. FEMS Microbiology Letters, 176, 17–24.
Goodacre, R., Vaidyanathan, S., Bianchi, G., & Kell, D. B. (2002). Metabolic profiling using direct infusion electrospray ionisation mass spectrometry for the characterisation of olive oils. Analyst, 127, 1457–1462.
Goodacre, R., York, E. V., Heald, J. K., & Scott, I. M. (2003). Chemometric discrimination of unfractionated plant extracts analyzed by electrospray mass spectrometry. Phytochemistry, 62, 859–863.
Goodacre, R., Broadhurst, D., Smilde, A., Kristal, B. S., Baker, J. D., Beger, R., et al. (2007). Proposed minimum reporting standards for data analysis in metabolomics. Metabolomics, 3, 231–241.
Gorlach, E., & Richmond, R. (1999). Discovery of quasi-molecular ions in electrospray spectra by automated searching for simultaneous adduct mass differences. Analytical Chemistry, 71, 5557–5562.
Gougeon, R. D., Lucio, M., Frommberger, M., et al. (2009). The chemodiversity of wines can reveal a metabologeography expression of cooperage oak wood. Proceedings of the National academy of Sciences of the United States of America, 106, 9174–9179.
Grata, E., Boccard, J., Glauser, G., et al. (2007). Development of a two-step screening ESI-TOF-MS method for rapid determination of significant stress-induced metabolome modifications in plant leaf extracts: The wound response in Arabidopsis thaliana as a case study. Journal of Separation Science, 30, 2268–2278.
Gray, G. R., & Heath, D. (2005). A global reorganization of the metabolome in Arabidopsis during cold acclimation is revealed by metabolic fingerprinting. Physiologia Plantarum, 124, 236–248.
Gross, J. H. (2004). Mass spectrometry: A textbook. Heidelberg: Springer.
Gu, H., Hu, B., Li, J., et al. (2010). Rapid analysis of aerosol drugs using nano extractive electrospray ionization tandem mass spectrometry. Analyst, 135, 1259–1267.
Guilhaus, M., Mlynski, V., & Selby, D. (1997). Perfect timing: Time-of-flight mass spectrometry. Rapid Communications in Mass Spectrometry, 11, 951–962.
Hall, R., Beale, M., Fiehn, O., Hardy, N., Sumner, L., & Bino, R. (2002). Plant metabolomics: The missing link in functional genomics strategies. The Plant Cell, 14, 1437–1440.
Han, J., Danell, R. M., Patel, J. R., et al. (2008a). Towards high-throughput metabolomics using ultrahigh-field Fourier transform ion cyclotron resonance mass spectrometry. Metabolomics, 4, 128–140.
Han, X., Yang, K., & Gross, R. W. (2008b). Microfluidics-based electrospray ionization enhances the intrasource separation of lipid classes and extends identification of individual molecular species through multi-dimensional mass spectrometry: Development of an automated high-throughput platform for shotgun lipidomics. Rapid Communications in Mass Spectrometry, 22, 2115–2124.
Hasegawa, M., Takenaka, S., Kuwamura, M., Yamate, J., & Tsuyama, S. (2007). Urinary metabolic fingerprinting for amiodarone-induced phospholipidosis in rats using FT-ICR MS. Experimental and Toxicologic Pathology, 59, 115–120.
Hasegawa, M., Ide, M., Kuwamura, M., Yamate, J., & Takenaka, S. (2010). Metabolic fingerprinting in toxicological assessment using FT-ICR MS. Journal of Toxicologic Pathology, 23, 67–74.
Helm, R. F., Jervis, J., Ray, W. K., Willoughby, N., Irvin, B., Hastie, J., et al. (2010). Mass spectral analyses of corn stover prehydrolysates to assess conditioning processes. Agricultural and Food Chemistry, 58, 12642–12649.
Higgs, R. E., Zahn, J. A., Gygi, J. D., & Hilton, M. D. (2001). Rapid method to estimate the presence of secondary metabolites in microbial extracts. Applied and Environmental Microbiology, 67, 371–376.
Hirai, M. Y., Yano, M., Goodenowe, D. B., et al. (2004). Integration of transcriptomics and metabolomics for understanding of global responses to nutritional stresses in Arabidopsis thaliana. Proceedings of the National academy of Sciences of the United States of America, 101, 10205–10210.
Hojer-Pedersen, J., Smedsgaard, J., & Nielsen, J. (2008). The yeast metabolome addressed by electrospray ionization mass spectrometry: Initiation of a mass spectral library and its applications for metabolic footprinting by direct infusion mass spectrometry. Metabolomics, 4, 393–405.
Hrydziuszko, O., Silva, M. A., Perera, M. T. P. R., et al. (2010). Application of metabolomics to investigate the process of human orthotopic liver transplantation: A proof-of-principle study. OMICS: A Journal of Integrative Biology, 14, 143–150.
Hu, B., So, P.-K., Chen, H., & Yao, Z.-P. (2011). Electrospray ionization using wooden tips. Analytical Chemistry, 83, 8201–8207.
Iijima, Y., Nakamura, Y., Ogata, Y., et al. (2008). Metabolite annotations based on the integration of mass spectral information. Plant Journal, 54, 949–962.
Jansson, J., Willing, B., Lucio, M., et al. (2009). Metabolomics reveals metabolic biomarkers of Crohn’s disease. PLoS One, 4, e6386.
Johnson, H. E., Lloyd, A. J., Mur, L. A. J., Smith, A. R., & Causton, D. R. (2007). The application of MANOVA to analyse Arabidopsis thaliana metabolomic data from factorially designed experiments. Metabolomics, 3, 517–530.
Junot, C., Madalinski, G., Tabet, J. C., & Ezan, E. (2010). Fourier transform mass spectrometry for metabolome analysis. Analyst, 135, 2203–2219.
Kaderbhai, N. N., Broadhurst, D. I., Ellis, D. I., Goodacre, R., & Kell, D. B. (2003). Functional genomics via metabolic footprinting: Monitoring metabolite secretion by Escherichia coli tryptophan metabolism mutants using FT-IR and direct injection electrospray mass spectrometry. Comparative and Functional Genomics, 4, 376–391.
Kai, K., Hashidzume, H., Yoshimura, K., et al. (2009). Metabolomics for the characterization of cytochromes P450-dependent fatty acid hydroxylation reactions in Arabidopsis. Plant Biotechnology, 26, 175–182.
Kim, S., Kaplan, L. A., & Hatcher, P. G. (2006). Biodegradable dissolved organic matter in a temperate and a tropical stream determined from ultra-high resolution mass spectrometry. Limnology and Oceanography, 51, 1054–1063.
Kind, T., & Fiehn, O. (2006). Metabolomic database annotations via query of elemental compositions: Mass accuracy is insufficient even at less than 1 ppm. BMC Bioinformatics, 7, 234.
Kind, T., & Fiehn, O. (2007). Seven golden rules for heuristic filtering of molecular formulas obtained by accurate mass spectrometry. BMC Bioinformatics, 8, 105.
Kind, T., & Fiehn, O. (2010). Advances in structure elucidation of small molecules using mass spectrometry. Bioanalytical Reviews, 2, 23–60.
King, R., Bonfiglio, R., Fernandez-Metzler, C., Miller-Stein, C., & Olah, T. (2000). Mechanistic investigation of ionization suppression in electrospray ionization. Journal of the American Society for Mass Spectrometry, 11, 942–950.
Koch, B. P., Witt, M. R., Engbrodt, R., Dittmar, T., & Kattner, G. (2005). Molecular formulae of marine and terrigenous dissolved organic matter detected by electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry. Geochimica et Cosmochimica Acta, 69, 3299–3308.
Koek, M. M., Jellema, R. H., Van Der Greef, J., Tas, A. C., & Hankemeier, T. (2011). Quantitative metabolomics based on gas chromatography mass spectrometry: Status and perspectives. Metabolomics, 7, 307–328.
Koulman, A., Tapper, B. A., Fraser, K., et al. (2007). High-throughput direct-infusion ion trap mass spectrometry: A new method for metabolomics. Rapid Communications in Mass Spectrometry, 21, 421–428.
Koulman, A., Woffendin, G., Narayana, V. K., et al. (2009). High-resolution extracted ion chromatography, a new tool for metabolomics and lipidomics using a second-generation orbitrap mass spectrometer. Rapid Communications in Mass Spectrometry, 23, 1411–1418.
Kuhl, C., Tautenhahn, R., Boettcher, C., Larson, T. R., & Neumann, S. (2012). CAMERA: An integrated strategy for compound spectra extraction and annotation of liquid chromatography/mass spectrometry data sets. Analytical Chemistry, 84, 283–289.
Kujawinski, E. B., Del Vecchio, R., Blough, N. V., Klein, G. C., & Marshall, A. G. (2004). Probing molecular-level transformations of dissolved organic matter: Insights on photochemical degradation and protozoan modification of DOM from electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry. Marine Chemistry, 92, 23–37.
Lafaye, A., Labarre, J., Tabet, J. C., Ezan, E., & Junot, C. (2005). Liquid chromatography-mass spectrometry and N-15 metabolic labeling for quantitative metabolic profiling. Analytical Chemistry, 77, 2026–2033.
Li, M., Hu, B., Li, J., et al. (2009). Extractive electrospray ionization mass spectrometry toward in situ analysis without sample pretreatment. Analytical Chemistry, 81, 7724–7731.
Liger-Belair, G., Cilindre, C., Gougeon, R. D., et al. (2009). Unraveling different chemical fingerprints between a champagne wine and its aerosols. Proceedings of the National academy of Sciences of the United States of America, 106, 16545–16549.
Lin, L., Yu, Q., Yan, X., et al. (2010). Direct infusion mass spectrometry or liquid chromatography mass spectrometry for human metabonomics? A serum metabonomic study of kidney cancer. Analyst, 135, 2970–2978.
Lisec, J., Schauer, N., Kopka, J., Willmitzer, L., & Fernie, A. R. (2006). Gas chromatography mass spectrometry-based metabolite profiling in plants. Nature Protocols, 1, 387–396.
Liu, G., Ji, Q. C., & Arnold, M. E. (2010). Identifying, evaluating, and controlling bioanalytical risks resulting from nonuniform matrix ion suppression/enhancement and nonlinear liquid chromatography-mass spectrometry assay response. Analytical Chemistry, 82, 9671–9677.
Liu, J., Wang, H., Cooks, R. G., & Ouyang, Z. (2011). Leaf spray: Direct chemical analysis of plant material and living plants by mass spectrometry. Analytical Chemistry, 83, 7608–7613.
Lloyd, A. J., Allwood, J. W., Winder, C. L., et al. (2011a). Metabolomic approaches reveal that cell wall modifications play a major role in ethylene-mediated resistance against Botrytis cinerea. Plant Journal, 67, 852–868.
Lloyd, A. J., Beckmann, M., Favé, G., Mathers, J. C., & Draper, J. (2011b). Proline betaine and its biotransformation products are potential biomarkers of habitual citrus fruit consumption in fasting urine samples. British Journal of Nutrition, 106, 812–824.
Lloyd, A. J., Favé, G., Beckmann, M., et al. (2011c). Use of mass spectrometry fingerprinting to identify urinary metabolites after consumption of specific foods. American Journal of Clinical Nutrition, 94, 981–991.
Lokhov, P. G., Kharybin, O. N., & Archakov, A. I. (2012). Diagnosis of lung cancer based on direct-infusion electrospray mass spectrometry of blood plasma metabolites. International Journal of Mass Spectrometry, 309, 200–205.
Lowe, R. G. T., Allwood, J. W., Galster, A. M., et al. (2010). A combined (1)H nuclear magnetic resonance and electrospray ionization mass spectrometry analysis to understand the basal metabolism of plant-pathogenic fusarium spp. Molecular Plant-Microbe Interactions, 23, 1605–1618.
Lucio, M., Fekete, A., Weigert, C., et al. (2010). Insulin sensitivity is reflected by characteristic metabolic fingerprints—a Fourier transform mass spectrometric non-targeted metabolomics approach. PLoS One, 5, e13317.
Luthria, D. L., Lin, L.-Z., Robbins, R. J., et al. (2008). Discriminating between cultivars and treatments of broccoli using mass spectral fingerprinting and analysis of variance-principal component analysis. Journal of Agricultural and Food Chemistry, 56, 9819–9827.
Lyons-Weiler, J., Pelikan, R., Zeh, H. J., et al. (2005). Assessing the statistical significance of the achieved classification error of classifiers constructed using serum peptide profiles, and a prescription for random sampling repeated studies for massive high-throughput genomic and proteomic studies. Cancer Informatics, 1, 53–77.
Madalinski, G., Godat, E., Alves, S., et al. (2008). Direct introduction of biological samples into a LTQ-Orbitrap hybrid mass spectrometer as a tool for fast metabolome analysis. Analytical Chemistry, 80, 3291–3303.
March, R. E. (1997). An introduction to quadrupole ion trap mass spectrometry. Journal of Mass Spectrometry, 32, 351–369.
Marshall, A. G., & Hendrickson, C. L. (2008). High-resolution mass spectrometers. Annual Review of Analytical Chemistry, 1, 579–599.
Mas, S., Villas-Boas, S. G., Hansen, M. E., Akesson, M., & Nielsen, J. (2007). A comparison of direct infusion MS and GC-MS for metabolic footprinting of yeast mutants. Biotechnology and Bioengineering, 96, 1014–1022.
Mattoli, L., Cangi, F., Ghiara, C., et al. (2011). A metabolite fingerprinting for the characterization of commercial botanical dietary supplements. Metabolomics, 7, 437–445.
Mauri, P., & Pietta, P. (2000a). Electrospray characterization of selected medicinal plant extracts. Journal of Pharmaceutical and Biomedical Analysis, 23, 61–68.
Mauri, P., & Pietta, P. (2000b). High performance liquid chromatography/electrospray mass spectrometry of Hypericum perforatum extracts. Rapid Communications in Mass Spectrometry, 14, 95–99.
Mauri, P., Migliazza, B., & Pietta, P. (1999). Liquid chromatography/electrospray mass spectrometry of bioactive terpenoids in Ginkgo biloba L. Journal of Mass Spectrometry, 34, 1361–1367.
McDougall, G., Martinussen, I., & Stewart, D. (2008). Towards fruitful metabolomics: High throughput analyses of polyphenol composition in berries using direct infusion mass spectrometry. Journal of Chromatography B, 871, 362–369.
Minor, E. C., Steinbring, C. J., Longnecker, K., & Kujawinski, E. B. (2012). Characterization of dissolved organic matter in Lake Superior and its watershed using ultrahigh resolution mass spectrometry. Organic Geochemistry, 43, 1–11.
Mittelstrass, K., Ried, J. S., Yu, Z., Krumsiek, J., Gieger, C., et al. (2011). Discovery of sexual dimorphisms in metabolic and genetic biomarkers. PLoS Genetics, 7(8), e1002215. doi:10.1371/journal.pgen.1002215.
Miura, D., Tsuji, Y., Takahashi, K., Wariishi, H., & Saito, K. (2010). A strategy for the determination of the elemental composition by Fourier transform ion cyclotron resonance mass spectrometry based on isotopic peak ratios. Analytical Chemistry, 82, 5887–5891.
Mougous, J. D., Leavell, M. D., Senaratne, R. H., et al. (2002). Discovery of sulfated metabolites in mycobacteria with a genetic and mass spectrometric approach. Proceedings of the National academy of Sciences of the United States of America, 99, 17037–17042.
Mungur, R., Glass, A. D. M., Goodenow, D. B., & Lightfoot, D. A. (2005). Metabolite fingerprinting in transgenic Nicotiana tabacum altered by the Escherichia coli glutamate dehydrogenase gene. Journal of Biomedicine and Biotechnology, 2, 198–214.
Nakamura, Y., Kimura, A., Saga, H., et al. (2007). Differential metabolomics unraveling light/dark regulation of metabolic activities in Arabidopsis cell culture. Planta, 227, 57–66.
Nakamura, Y., Kanaya, S., Sakurai, N., et al. (2008). A tool for high-throughput prediction of molecular formulas and identification of isotopic peaks from large-scale mass spectrometry data. Plant Biotechnology, 25, 377–380.
Nam, S., Joo, S., Kim, S., et al. (2008). Induced metabolite changes in Myriophyllum spicatum during co-existence experiment with the Cyanobacterium Microcystis aeruginosa. Journal of Plant Biology, 51, 373–378.
Ohno, T., He, Z., Sleighter, R. L., Honeycutt, C. W., & Hatcher, P. G. (2010). Ultrahigh resolution mass spectrometry and indicator species analysis to identify marker components of soil- and plant biomass-derived organic matter fractions. Environmental Science and Technology, 44, 8594–8600.
Ohta, D., Shibata, D., & Kanaya, S. (2007). Metabolic profiling using Fourier-transform ion-cyclotron-resonance mass spectrometry. Analytical and Bioanalytical Chemistry, 389, 1469–1475.
Ohta, D., Kanaya, S., & Suzuki, H. (2010). Application of Fourier-transform ion cyclotron resonance mass spectrometry to metabolic profiling and metabolite identification. Current Opinion in Biotechnology, 21, 35–44.
Oikawa, A., Nakamura, Y., Ogura, T., et al. (2006). Clarification of pathway-specific inhibition by Fourier transform ion cyclotron resonance/mass spectrometry-based metabolic phenotyping studies. Plant Physiology, 142, 398–413.
Overy, S. A., Walker, H. J., Malone, S., et al. (2005). Application of metabolite profiling to the identification of traits in a population of tomato introgression lines. Journal of Experimental Botany, 56, 287–296.
Overy, D. P., Enot, D. P., Tailliart, K., et al. (2008). Explanatory signal interpretation and metabolite identification strategies for nominal mass FIE-MS metabolite fingerprints. Nature Protocols, 3, 471–485.
Parker, D., Beckmann, M., Enot, D. P., et al. (2008). Rice blast infection of Brachypodium distachyon as a model system to study dynamic host/pathogen interactions. Nature Protocols, 3, 435–445.
Parker, D., Beckmann, M., Zubair, H., et al. (2009). Metabolomic analysis reveals a common pattern of metabolic re-programming during invasion of three host plant species by Magnaporthe grisea. Plant Journal, 59, 723–737.
Payne, T. G., Southam, A. D., Arvanitis, T. N., & Viant, M. R. (2009). A signal filtering method for improved quantification and noise discrimination in Fourier transform ion cyclotron resonance mass spectrometry-based metabolomics data. Journal of the American Society for Mass Spectrometry, 20, 1087–1095.
Pietta, P. G., Gardana, C., & Pietta, A. M. (2002). Analytical methods for quality control of propolis. Fitoterapia, 73, S7–S20.
Piovan, A., Filippini, R., & Favretto, D. (1998). Characterization of the anthocyanins of Catharanthus roseus (L.) G. Don in vivo and in vitro by electrospray ionization ion trap mass spectrometry. Rapid Communications in Mass Spectrometry, 12, 361–367.
Piovan, A., Filippini, R., Caniato, R., et al. (2004). Detection of hypericins in the “red glands” of Hypericum elodes by ESI-MS/MS. Phytochemistry, 65, 411–414.
Pope, G. A., Mackenzie, D. A., Defemez, M., et al. (2007). Metabolic footprinting as a tool for discriminating between brewing yeasts. Yeast, 24, 667–679.
Proschogo, N. W., Albertson, P. L., Bursle, J., et al. (2012). Aging effects on macadamia nut oil studied by electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry. Journal of Agriculture and Food Chemistry, ahead of print.
Rojas-Cherto, M., Kasper, P. T., Willighagen, E. L., et al. (2011). Elemental composition determination based on MS(n). Bioinformatics, 27, 2376–2383.
Rossello-Mora, R., Lucio, M., Pena, A., et al. (2008). Metabolic evidence for biogeographic isolation of the extremophilic bacterium Salinibacter ruber. ISME Journal, 2, 242–253.
Rubio-Aliaga, I., de Roos, B., Sailer, M., McLoughlin, G. A., et al. (2011). Alterations in hepatic one-carbon metabolism and related pathways following a high-fat dietary intervention. Physiological Genomics, 43, 408–416.
Saito, K., & Matsuda, F. (2010). Metabolomics for functional genomics, systems biology, and biotechnology. In: S. B. W. R. O. D. Merchant (Ed.), Annual Review of Plant Biology, vol. 61.
Sana, T. R., Roark, J. C., Li, X., Waddell, K., & Fischer, S. M. (2008). Molecular formula and METLIN personal metabolite database matching applied to the identification of compounds generated by LC/TOF-MS. Journal of Biomolecular Techniques: JBT, 19, 258–266.
Sawaya, A., Catharino, R. R., Facco, E. M. P., et al. (2011). Monitoring of wine aging process by electrospray ionization mass spectrometry. Ciencia E Tecnologia De Alimentos, 31, 730–734.
Scalbert, A., Brennan, L., Fiehn, O., et al. (2009). Mass-spectrometry-based metabolomics: Limitations and recommendations for future progress with particular focus on nutrition research. Metabolomics, 5, 435–458.
Schmidt, J., Boettcher, C., Kuhnt, C., Kutchan, T. M., & Zenk, M. H. (2007). Poppy alkaloid profiling by electrospray tandem mass spectrometry and electrospray FT-ICR mass spectrometry after ring-13 C(6)-tyramine feeding. Phytochemistry, 68, 189–202.
Scholz, M., Gatzek, S., Sterling, A., Fiehn, O., & Selbig, J. (2004). Metabolite fingerprinting: Detecting biological features by independent component analysis. Bioinformatics, 20, 2447–2454.
Schultz, G. A., Corso, T. N., Prosser, S. J., & Zhang, S. (2000). A fully integrated monolithic microchip electrospray device for mass spectrometry. Analytical Chemistry, 72, 4058–4063.
Scott, I. M., Vermeer, C. P., Liakata, M., et al. (2010). Enhancement of plant metabolite fingerprinting by machine learning. Plant Physiology, 153, 1506–1520.
Sleighter, R. L., & Hatcher, P. G. (2008). Molecular characterization of dissolved organic matter (DOM) along a river to ocean transect of the lower Chesapeake Bay by ultrahigh resolution electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry. Marine Chemistry, 110, 140–152.
Smedsgaard, J., & Frisvad, J. C. (1996). Using direct electrospray mass spectrometry in taxonomy and secondary metabolite profiling of crude fungal extracts. Journal of Microbiological Methods, 25, 5–17.
Smedsgaard, J., & Frisvad, J. C. (1997). Terverticillate penicillia studied by direct electrospray mass spectrometric profiling of crude extracts. 1. Chemosystematics. Biochemical Systematics and Ecology, 25, 51–64.
Smedsgaard, J., Hansen, M. E., & Frisvad, J. C. (2004). Classification of terverticillate Penicillia by electrospray mass spectrometric profiling. Studies in Mycology, 49, 243–251.
Somorjai, R. L., Dolenko, B., & Baumgartner, R. (2003). Class prediction and discovery using gene microarray and proteomics mass spectroscopy data: Curses, caveats, cautions. Bioinformatics, 19, 1484–1491.
Soule, M. C. K., Longnecker, K., Giovannoni, S. J., & Kujawinski, E. B. (2010). Impact of instrument and experiment parameters on reproducibility of ultrahigh resolution ESI FT-ICR mass spectra of natural organic matter. Organic Geochemistry, 41, 725–733.
Southam, A. D., Payne, T. G., Cooper, H. J., Arvanitis, T. N., & Viant, M. R. (2007). Dynamic range and mass accuracy of wide-scan direct infusion nanoelectrospray Fourier transform ion cyclotron resonance mass spectrometry-based metabolomics increased by the spectral stitching method. Analytical Chemistry, 79, 4595–4602.
Stenson, A. C., Marshall, A. G., & Cooper, W. T. (2003). Exact masses and chemical formulas of individual Suwannee River fulvic acids from ultrahigh resolution electrospray ionization Fourier transform ion cyclotron resonance mass spectra. Analytical Chemistry, 75, 1275–1284.
Stoll, N., Schmidt, E., & Thurow, K. (2006). Isotope pattern evaluation for the reduction of elemental compositions assigned to high-resolution mass spectral data from electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry. Journal of the American Society for Mass Spectrometry, 17, 1692–1699.
Sumner, L. W., Mendes, P., & Dixon, R. A. (2003). Plant metabolomics: Large-scale phytochemistry in the functional genomics era. Phytochemistry, 62, 817–836.
Sun, J., & Chen, P. (2011). A flow-injection mass spectrometry fingerprinting method for authentication and quality assessment of Scutellaria lateriflora-based dietary supplements. Analytical and Bioanalytical Chemistry, 401, 1577–1584.
Szymańska, E., Saccenti, E., Smilde, A. K., & Westerhuis, J. A. (2011). Double-check: Validation of diagnostic statistics for PLS-DA models in metabolomics studies. Metabolomics,. doi:10.1007/s11306-011-0330-3.
Takahashi, H., Kai, K., Shinbo, Y., et al. (2008). Metabolomics approach for determining growth-specific metabolites based on Fourier transform ion cyclotron resonance mass spectrometry. Analytical and Bioanalytical Chemistry, 391, 2769–2782.
Tautenhahn, R., Boettcher, C., Neumann, S. (2007). Annotation of LC/ESI-MS mass signals. In: S. W. R. Hochreiter (Ed.), Bioinformatics Research and Development, Proceedings.
Taylor, N. S., Weber, R. J. M., Southam, A. D., et al. (2009). A new approach to toxicity testing in Daphnia magna: Application of high throughput FT-ICR mass spectrometry metabolomics. Metabolomics, 5, 44–58.
Tohge, T., Nishiyama, Y., Hirai, M. Y., et al. (2005). Functional genomics by integrated analysis of metabolome and transcriptome of Arabidopsis plants over-expressing an MYB transcription factor. Plant Journal, 42, 218–235.
Tremblay, L. B., Dittmar, T., Marshall, A. G., Cooper, W. J., & Cooper, W. T. (2007). Molecular characterization of dissolved organic matter in a North Brazilian mangrove porewater and mangrove-fringed estuaries by ultrahigh resolution Fourier transform-ion cyclotron resonance mass spectrometry and excitation/emission spectroscopy. Marine Chemistry, 105, 15–29.
Vaidyanathan, S., Rowland, J. J., Kell, D. B., & Goodacre, R. (2001). Discrimination of aerobic endospore-forming bacteria via electrospray-ionization mass spectrometry of whole cell suspensions. Analytical Chemistry, 73, 4134–4144.
Vaidyanathan, S., Kell, D. B., & Goodacre, R. (2002). Flow-injection electrospray ionization mass spectrometry of crude cell extracts for high-throughput bacterial identification. Journal of the American Society for Mass Spectrometry, 13, 118–128.
van den Berg, R. A., Hoefsloot, H. C. J., Westerhuis, J. A., Smilde, A. K., & van der Werf, M. J. (2006). Centering, scaling, and transformations: Improving the biological information content of metabolomics data. BMC Genomics, 7, 142.
Villagra, E., Santos, L. S., Gontijo Vaz, B., Eberlin, M. N., & Felipe Laurie, V. (2012). Varietal discrimination of Chilean wines by direct injection mass spectrometry analysis combined with multivariate statistics. Food Chemistry, 131, 692–697.
Villas-Boas, S. G., Mas, S., Akesson, M., Smedsgaard, J., & Nielsen, J. (2005). Mass spectrometry in metabolome analysis. Mass Spectrometry Reviews, 24, 613–646.
Wang, H., Manicke, N. E., Yang, Q., et al. (2011). Direct analysis of biological tissue by paper spray mass spectrometry. Analytical Chemistry, 83, 1197–1201.
Ward, J. L., Forcat, S., Beckmann, M., et al. (2010). The metabolic transition during disease following infection of Arabidopsis thaliana by Pseudomonas syringae pv. tomato. Plant Journal, 63, 443–457.
Weber, R. J. M., & Viant, M. R. (2010). MI-Pack: Increased confidence of metabolite identification in mass spectra by integrating accurate masses and metabolic pathways. Chemometrics and Intelligent Laboratory Systems, 104, 75–82.
Werner, E., Heilier, J.-F., Ducruix, C., et al. (2008). Mass spectrometry for the identification of the discriminating signals from metabolomics: Current status and future trends. Journal of Chromatography B-Analytical Technologies in the Biomedical and Life Sciences, 871, 143–163.
Westerhuis, J. A., Hoefsloot, H. C. J., Smit, S., Vis, D. J., Smilde, A. K., van Velzen, E. J. J., et al. (2008). Assessment of PLSDA cross validation. Metabolomics, 4, 81–89.
Wickremsinhe, E. R., Singh, G., Ackermann, B. L., Gillespie, T. A., & Chaudhary, A. K. (2006). A review of nanoelectrospray ionization applications for drug metabolism and pharmacokinetics. Current Drug Metabolism, 7, 913–928.
Windig, W., Phalp, J. M., & Payne, A. W. (1996). A noise and background reduction method for component detection in liquid chromatography mass spectrometry. Analytical Chemistry, 68, 3602–3606.
Wishart, D. S. (2009). Computational strategies for metabolite identification in metabolomics. Bioanalysis, 1, 1579–1596.
Wishart, D. S. (2011). Advances in metabolite identification. Bioanalysis, 3, 1769–1782.
Wu, Z., Chingin, K., Chen, H., et al. (2010). Sampling analytes from cheese products for fast detection using neutral desorption extractive electrospray ionization mass spectrometry. Analytical and Bioanalytical Chemistry, 397, 1549–1556.
Yang, L., Bennett, R., Strum, J., et al. (2009). Screening phosphatidylcholine biomarkers in mouse liver extracts from a hypercholesterolemia study using ESI-MS and chemometrics. Analytical and Bioanalytical Chemistry, 393, 643–654.
Yue, M.-E., Xu, J., Li, Q.-Q., & Hou, W.-G. (2011). Identification of coumarins in traditional Chinese medicine by direct-injection electrospray ionisation tandem mass spectrometry and high-performance liquid chromatography-mass spectrometry. Journal of Analytical Chemistry, 66, 290–295.
Zahn, J. A., Higgs, R. E., & Hilton, M. D. (2001). Use of direct-infusion electrospray mass spectrometry to guide empirical development of improved conditions for expression of secondary metabolites from actinomycetes. Applied and Environmental Microbiology, 67, 377–386.
Zamfir, A. D., Lion, N., Vukelic, Z., et al. (2005). Thin chip microsprayer system coupled to quadrupole time-of-flight mass spectrometer for glycoconjugate analysis. Lab on a Chip, 5, 298–307.
Zhang, L. K., Rempel, D., Pramanik, B. N., & Gross, M. L. (2005). Accurate mass measurements by Fourier transform mass spectrometry. Mass Spectrometry Reviews, 24, 286–309.
Zulak, K. G., Cornish, A., Daskalchuk, T. E., et al. (2007). Gene transcript and metabolite profiling of elicitor-induced opium poppy cell cultures reveals the coordinate regulation of primary and secondary metabolism. Planta, 225, 1085–1106.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Draper, J., Lloyd, A.J., Goodacre, R. et al. Flow infusion electrospray ionisation mass spectrometry for high throughput, non-targeted metabolite fingerprinting: a review. Metabolomics 9 (Suppl 1), 4–29 (2013). https://doi.org/10.1007/s11306-012-0449-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11306-012-0449-x