
Abstract
Metabolomic technologies produce complex multivariate datasets and researchers are faced with the daunting task of extracting information from these data. Principal component analysis (PCA) has been widely applied in the field of metabolomics to reduce data dimensionality and for visualising trends within the complex data. Although PCA is very useful, it cannot handle multi-factorial experimental designs and, often, clear trends of biological interest are not observed when plotting various PC combinations. Even if patterns are observed, PCA provides no measure of their significance. Multivariate analysis of variance (MANOVA) applied to these PCs enables the statistical evaluation of main treatments and, more importantly, their interactions within the experimental design. The power and scope of MANOVA is demonstrated through two different factorially designed metabolomic investigations using Arabidopsis ethylene signalling mutants and their wild-type. One investigation has multiple experimental factors including challenge with the economically important pathogen Botrytis cinerea and also replicate experiments, while the second has different sample preparation methods and one level of replication ‘nested’ within the design. In both investigations there are specific factors of biological interest and there are also factors incorporated within the experimental design, which affect the data. The versatility of MANOVA is displayed by using data from two different metabolomic techniques; profiling using direct injection mass spectroscopy (DIMS) and fingerprinting using fourier transform infra-red (FT-IR) spectroscopy. MANOVA found significant main effects and interactions in both experiments, allowing a more complete and comprehensive interpretation of the variation within each investigation, than with PCA alone. Canonical variate analysis (CVA) was applied to investigate these effects and their biological significance. In conclusion, the application of MANOVA followed by CVA provided extra information than PCA alone and proved to be a valuable statistical addition in the overwhelming task of analysing metabolomic data.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Plant metabolomics is the study of predominantly low molecular weight metabolites within cells, tissues or organisms and is a widely applied approach for the elucidation of gene function in a wide range of plant species. Due to the biochemical complexity of plant systems and the high diversity of chemical species to be analysed, there is currently no single technology, which enables measurement of the global metabolome (Sumner et al., 2003). As a consequence there are a wealth of approaches and technologies being applied to study plant metabolomics (Holtorf et al., 2002; Fernie, 2003; Sumner et al., 2003; Bino et al., 2004). One of the many goals of researchers in the field of metabolomics is high-throughput screening of crude samples with little or no sample preparation time. The methods of fourier transform infra-red spectroscopy (FT-IR), nuclear magnetic resonance (NMR) and direct injection electrospray mass spectrometry (DIMS) have emerged with this in mind (Goodacre et al., 2004; Kim et al., 2005), all producing high dimensional, biochemically discriminatory data. FT-IR is a ‘fingerprinting’ technique, which produces a biochemical signature of the sample to generate biologically relevant data without identifying individual metabolites (Fiehn, 2001; Johnson et al., 2003). DIMS can be classified as both a fingerprinting technique and ‘profiling’ technique, as terminologies used in this field often overlap. This article refers to DIMS as a profiling technique due to phase separation of the extracts and tentative identification of contributory mass ions (Dunn et al., 2005). Irrespective of the analytical method selected, the aim is to differentiate between samples and/or the classification of samples to their origin or in response to external stimuli (Fiehn, 2001).
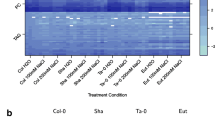
Both of these techniques yield huge multidimensional datasets which are virtually uninterpretable by visual analysis (figure 1A, B). Hence, chemometric methods are used to examine the shape of the data and to identify trends within it, so summarising the data. Principal component analysis (PCA) is a method commonly applied which enables the visualisation of sources of natural variability within the data in the form of 2-D or 3-D plots of different principal component (PC) combinations (Taylor et al., 2002; Johnson et al., 2003; Schulz et al., 2004). The method has several advantages: primarily that the data do not have to be multivariate normally distributed and that no a priori knowledge of the data set structure is required as it is an ‘unsupervised’ technique. As a result the user does not have to make any assumptions about significant effects or interactions within the data.

(A) Classical FT-IR spectra indicating very little obvious spectral variation between several replicates of the Arabidopsis genotypes Col-0, etr1-1 and ctr1-1 after sample preparation method 5, as described in Table 8. (B) An example of a DIMS spectrum from the pathogen challenge investigation. The spectra from each treatment combination share a high degree of similarity; hence, chemometric analysis is needed to allow differentiation between different treatments and controls.
In addition to data visualisation, PCA is also used to reduce data dimensionality (Martens and Næs, 1989). Metabolic data sets typically contain hundreds, or even thousands of variables, many of which are often highly correlated. Such a large number of variables relative to a much smaller number of observations can lead to problems of matrix singularity when trying to analyse data using methods such as canonical variate analysis (CVA) (Chatfield and Collins, 1980). This has been pointed out previously by Langsrud (2002), Jansen et al. (2005), Smilde et al. (2005) and Coombes (accessed 15/05/2007). Hence, PCA is applied to reduce the dimensionality of the data prior to further analysis (Goodacre et al., 1998; Johnson et al., 2004). CVA, or discriminant function analysis (DFA) as it can also be termed, is a clustering method commonly applied to metabolic data and is a ‘supervised’ method requiring a priori knowledge of the data (Alsberg et al., 1998).
Although PCA and CVA are very useful techniques for data visualisation, and dimensionality reduction in the case of PCA, often when dealing with complex, high dimensional data clear trends of biological interest are not observed when plotted. Even if patterns are observed, PCA provides no measure of their significance. Multivariate analysis of variance (MANOVA) is a statistical method which enables assessment of the statistical significance of the factors and their interactions within the experimental design, as demonstrated by Daoyu and Lawes (2000) in a study using phenotypic data for parental selection in kiwifruit breeding. In principle, MANOVA parallels the more familiar univariate analysis of variance (ANOVA), in that the form of the technique corresponds to the experimental design.
Analysis of variance has two main purposes: these being the summarisation of data and the drawing of inferences from data. When using multivariate methods, the emphasis is often on data summarisation rather than drawing inferences as these data sets can contain hundreds or thousands of variables and there is a need to find structure in the data. However, many studies are also multi-factorial in their design, containing more than one factor. If the experimental design contains only a single factor then CVA may be sufficient.
It is the presence of many sources of variation and the significance of this variation which are of key importance in the development of metabolomics technologies/methodologies. There is both biological and experimental variation, with the latter phrase used to encompass sample generation, harvesting, preparation and analysis. In order to preserve the biological variability of interest, that is to preserve the biochemical differences between the sample classes such as those due to genotype, abiotic or biotic treatments, one must aim to maximise the range and amounts of metabolites extracted, whilst minimising the experimental variance (Gullberg et al., 2004). The term biological variability can be slightly ambiguous; so far we have used the term to refer to those differences between samples which are expected, as the samples are genetically different or have been exposed to different experimental treatments. However, there is also the biological variation which occurs between plants which are genetically identical or between those which are replicates from the same treatment. Roessener et al. (2000) and Fiehn et al. (2000) found that the biological variation between genetically identical samples (plant variability) was greater than that associated with the analytical technique of GC-MS (machine variability). These observed degrees of variability reflect the intrinsic metabolic flexibility of plants, and hence metabolomics must cope with this (Holtorf et al., 2002). Overall these findings highlight the need for biological replication (the analysis of samples from the same statistical population), appropriate experimental design and the standardisation of methods (Chen et al., 2004).
In this article we will show how MANOVA can be applied to metabolic data from different metabolomic techniques; profiling using direct injection mass spectroscopy (DIMS) and fingerprinting using fourier transform infra-red (FT-IR) spectroscopy, to study the significance of all experimental factors and their interactions. Langsrud (2002) also used MANOVA on multivariate data of reduced dimensionality by PCA, but merely noted where significant interactions occurred. Here we investigate the nature of significant interactions arising in a MANOVA by CVA. In all experiments there are specific factors of biological interest and there are also factors incorporated within the experimental design, which affect the data. The data shown here are all collected from Arabidopsis thaliana plant material from two separate investigations. The first investigation involves response to pathogenic challenge with the economically important necrotroph Botrytis cinerea (the cause of grey mould in soft fruits) at 12 and 48 h post infection. Resistance against necrotrophic pathogens is dependent upon the synergistic action of jasmonic acid and ethylene signalling cascades (Penninckx et al., 1998; Thomma et al., 1998). The model plant Arabidopsis exhibits disease symptoms following challenge with B. cinerea and offers a wide range of well-characterised mutants with altered ethylene perception and transduction (Guzman and Ecker, 1990), which may exhibit enhanced or compromised defence. Three different genotypes were used in this work, the wild type Columbia (Col-0), an ethylene insensitive mutant etr1-1, which has a dominant mutation in a gene that encodes an ethylene receptor and a constitutive signalling ethylene mutant ctr1-1 that is mutated in a MAP3kinase CTR1 gene (Bleecker et al., 1988; Kieber et al., 1993). The aim was to obtain snapshots of the metabolome of the mutants and wild-type during metabolic reprogramming of the host upon infection with B. cinerea, using DIMS, and then to use these data to discriminate by genotype, infection and time within two replicate experiments. The second investigation uses FT-IR data from an experiment to study the effect of sample preparation method, namely freezing or freeze-drying, on the metabolome of these same three genotypes, but without infection, providing a system within which comparisons could be made between the different sampling methods.
Materials and methods
Plant material
Plants of mutants etr1-1 and ctr1-1 and their wild-type, Col-0, were cultivated on Levington Universal compost in black trays with 24 compartment inserts in a 6 × 4 arrangement, with one plant per module. Plants were grown in Conviron (Controlled Environments Ltd) growth rooms at 24 °C providing a light intensity of 70 μmol m−2 s−1 at shelf height and with an 8 h photoperiod (i.e. short day) light cycle for 5 weeks. For ease of treatment, plants were transferred at 5 weeks to Fotron 600-H growth cabinets (Fisons Environmental Equipment) set at the same conditions and kept here for the duration of the investigation. Aerial plant parts were harvested at the fully expanded rosette stage (stage 3.7 as defined by Boyes et al., 2001).
Pathogen Challenge Investigation
Botrytis cinerea, strain IMI 169558, was cultured on potato dextrose agar plates for 4 weeks (PDA; Oxoid) at 20 °C with a 12 h photoperiod in a Gallenkamp illuminated cooled incubator (Sanyo Biomedical Europe BV). Conidia were harvested from the surface of the plates by flooding with sterile potato dextrose broth (PDB; Oxoid) and dislodging conidia with an L-shaped glass rod, filtering to remove mycelium and then diluted to 1 × 105 spores mL−1 in PDB. Plants of mutants etr1-1 and ctr1-1 and Col-0 were inoculated by spraying using a mini airbrush (Amtech) attached to a compressor (35/20 Bambi air Ltd), subsequently covered with propagator hoods and placed back into the Fotron 600-H growth cabinets for the duration of the experiment. Non-infected control plants were ‘mock inoculated’ by spraying with PDB. The aerial parts of the plant were harvested at 12 and 48 h post infection, with three individual plants of each genotype and infection combination being bulked together [40 mg of fresh weight (FW)] for each biological replicate, and immediately frozen as whole rosettes in liquid nitrogen and stored at −80 °C. Two replicate experiments were performed (hereafter referred to as experiment 1 and experiment 2) within 2 months of each other. These plant samples were analysed using DIMS.
Sample preparation and storage investigation
Three trays of each genotype (Col-0, etr1-1 and ctr1-1) were grown as described above and three plants of each genotype were bulked together, a plant randomly selected from each of the three trays, to give a sample. Three samples were prepared for each genotype and preparation combination. The plants were processed immediately after harvesting. The samples were treated in the following ways. (1) Whole rosettes, frozen in liquid nitrogen, stored at −80 °C. (2) Whole rosettes, frozen in liquid nitrogen, ground by pestle and mortar, stored at −80 °C. (3) Whole rosettes, frozen in liquid nitrogen, freeze-dried for 24 h, stored at −80 °C. (4) Whole rosettes, frozen in liquid nitrogen, freeze-dried for 24 h, stored at room temperature in desiccators. (5) Whole rosettes, frozen in liquid nitrogen, ground by pestle and mortar and analysed on day of harvest. Samples prepared using methods 1–4 were all stored for 8 weeks prior to analysis by FT-IR. A new batch of plants was grown as described above and these provided material for preparation method 5, which was not stored but analysed immediately.
Direct injection electrospray mass spectrometry (DIMS)
The samples were frozen in liquid nitrogen, then ground using a ball mill (MM200, Retsch) and metabolites were extracted essentially following a procedure from Fiehn et al. (2000) for metabolite extraction. A chloroform, methanol and water-based solvent mixture (at a ratio of 1:2.5:1) was used as the extraction buffer. The samples were then mixed using a shaking table (Janke and Kunkel, VX 2E) in a cold room at 3 °C for 15 min. The aqueous layer was removed and 0.5 mL of sterile ultra pure dH2O was added. The extracts were mixed, centrifuged at 3 °C at 9000 × g (14000 rpm; Hettich, EBA 12R) for 3 min and then dried down in a Savant AES2000 (Thermo Electron Corp) automatic environmental speed vacuum concentrator and stored at −80 °C. Prior to analysis the stored extracts were centrifuged as before to remove any small particulates and 75 μL were transferred into a 250 μL glass insert placed in a glass autosampling vial. Finally, extracts were reconstituted in 100 μL 80% [v/v] methanol (running solvent) and mixed gently. Metabolomic profiling of the extracts was carried out using flow injection (FI)-ESI-MS on a micromass LCT electrospray ionisation time-of-flight mass spectrometer. All extracts were introduced into the electrospray source via a Waters 2695 Alliance Separations Module (Waters Ltd) under the following conditions in negative ion mode (ES-): capillary voltage 2000V, sample cone voltage 30V, source temperature 120 °C, desolvation temperature 250 °C, RF lens 125V. Mass spectra were collected in the mass-to-charge (m/z) range 100–1400 Th.
The data were exported from Masslynx version 3.5 software (Micromass Ltd, Manchester, UK), used to run the LCT, and converted into an ASCII format using a databridge program written by Dr D Broadhurst (University of Manchester, UK). The ASCII data were then loaded into Matlab (version 6.5; Mathworks Inc, Natwick, MA, USA) and, prior to data analyses, normalised to total ion count, so each m/z measurement became a proportion of the total ion count for each sample.
FT-IR spectroscopy
Double distilled water was added to each freeze-dried sample in a ratio 1 mg freeze-dried weight: 40 μL water and to each frozen (not freeze-dried) sample in a ratio of 1 mg fresh weight: 5 μL water. The sample was then mixed gently and 5 μL of sample were loaded onto a 400-well bespoke aluminium plate. The plates were then dried at 50 °C for 45 min. After drying, the plate was loaded onto a motorised stage of an adapted reflectance TLC (thin layer chromatography) accessory connected to a Bruker IFS28 FT-IR spectrometer (Bruker Spectrospin, Coventry, UK) equipped with an MCT (mercury-cadmium-telluride) detector cooled with liquid nitrogen (Goodacre et al., 1996) The spectra were collected over the wavenumber range 4000–600 cm−1. A total of 256 spectra were co-added and averaged per sample to improve the signal to noise ratio. Further instrument and methodological details are given in Winson et al. (1997) and Goodacre et al. (1998).
The spectrum for each sample contained 1764 data points, ranging from 4000 to 600 cm−1 wavenumbers, each representing absorbance values at a particular wavelength. These data were imported into Matlab. Prior to performing any data analyses the spectra were normalised by row using the prestd.m algorithm in the statistics toolbox of Matlab. PCA was carried out on the correlation matrix which autoscales the data as part of the process (Otto, 1999).
Principal component analysis
Principal component analysis is a transformation which produces a new set of uncorrelated variables from the original correlated variables. The derived variables, termed principal components (PCs), are linear combinations of the original variables and are arranged in decreasing order of importance with respect to the percentage of total variability accounted for (Joliffe, 1986). Often the first few PCs account for a high proportion of the total variance within the original data; as such, it is often assumed that these contain the important descriptive information about the experimental samples.
PCA was performed using a routine written in Matlab and which computed the PCs of the correlation matrix rather than those of the variance-covariance matrix.
Multivariate analysis of variance
Analysis of variance (ANOVA) provides a method for analysing and interpreting univariate data; and often in situations where multiple responses are measured, each is analysed separately by ANOVA (Smith et al., 1962). However, by analysing each separately, the investigator is failing to study correlations that may exist between the responses; and in a situation where there are many hundreds of variables, as in spectroscopy, such a strategy is totally impractical.
For ANOVA the total sum of squared deviations for the overall mean is partitioned into a series of sums of squares (SS) due to the experimental factors, their interactions and a residual (error) sum of squares. With each factor are associated degrees of freedom and these when divided into the corresponding sum of squares give the mean squares, the ratios of which constitute the F-statistics. The partitioning is set out in an ANOVA table as shown in Table 1 for the simplest situation – a single factor (one-way ANOVA). Multivariate analysis of variance uses exactly the same principle, except the data comprise p variates instead of a single variate. Hence, the SS are replaced by sums of squares and products matrices (SSPM) for the p variates. A comparison between ANOVA and MANOVA can be seen in Tables 1 and 2, with both showing a generalised form for a one-way situation.
In Table 1, all the quantities are, of course, single numbers, whereas in Table 2, H, R, T are all square matrices of dimensions p × p, where p is the number of variates involved.
The degrees of freedom are exactly the same for the two methods, but in Table 2 they have each been assigned a symbol of their own (corresponding to the symbol given to the sums of squares and product matrix) in order to simplify the formulation of equation (1). In MANOVA there are no quantities corresponding to the mean squares of ANOVA. The test statistic, Wilks’-Lambda (Λ) is given as the ratio of the determinant of matrix R divided by the determinant of the matrix which is the sum of both H and R. The next column in Table 2 is the F-statistic approximation which is calculated from the Wilks’-Lambda using equation (1). The F-statistic has degrees of freedom equal to ph and ab−c, as defined in equation (1). If the latter expression does not evaluate to a whole number then the degrees of freedom are rounded up or down in the usual way.
Where
and h, r and t are defined in Table 2 and p is the number of variates.
Test statistics for MANOVA have been proposed other than Wilks’-Lambda but this statistic is by far the commonest, and in our experience other test statistics such as Lawley-Hotelling and Pillai’s have given very similar results.
Unlike PCA, MANOVA basically assumes that within-group distribution is multivariate normal. However, like ANOVA, MANOVA is fairly robust to some departure of the data from normality. Data normality is important only when assessing the probability of the F-statistic, hence critical only when the significance is marginal with reference to the chosen probability level (usually 0.05). When, as is very often the case, probabilities turn out to be very different from 0.05, either higher or lower, then any deviations of the data from normality are of little consequence.
MANOVA was performed using Minitab (version 12.23).
Canonical variate analysis
If any factors or their interactions are found to be significant in the MANOVA, the precise details of the effects can be elucidated by CVA. This can be regarded as the multivariate equivalent to the univariate multiple range test (Chatfield and Collins, 1980).
CVA was calculated as part of the one-way MANOVA algorithm in the Matlab Statistics toolbox (MANOVA1.m). CVs are plotted against each other, allowing interrelationships between ‘sample groups’ to become apparent. On such a graph the mean of each sample class is plotted on each CV, and this can be surrounded by a confidence area. The latter is circular because each population, having been transformed to CVs, has a variance of unity and, like PCs, CVs are uncorrelated with one another. When significant factors or their interactions were found for the nested MANOVAs (two machine replicates were taken for each plant replicate to generate the FT-IR data, thus the former are said to be ‘nested’ within the latter), a mean of the machine replicates was calculated before performing CVA as ‘plant’ was the residual in these cases for assessment of the significance. The confidence area is an interval estimate for the population mean (Quinn and Keough, 2002) and is analogous to confidence intervals in the univariate situation; so that if, say, the 95% confidence circles are plotted around each mean this would provide a guide as to which sample groups are significantly different to each other. Confidence areas are obtained simply by equation (2) and for a 95% confidence circle z = 1.96, which is derived using the method as described by Seahl (1964) for a 90% confidence level.
where r = radius of the confidence circle.
z = the standard normal deviate for the degree of confidence required.
n = the sample size of each group.
Selection of the discriminatory metabolites
The selection of discriminatory metabolites between different factors was only applied to the first investigation (DIMS data) for demonstration purposes, but it can also be applied to FT-IR data where key regions of spectra can be identified in terms of groups of metabolites or compound classes, rather than individual metabolites.
If the plotted CV results show interrelationships between sample groups, interrogation of the loading vectors derived from the CVA can identify the most influential PCs. Examination of the most highly loaded PCs on the CV of interest will illustrate the mass-to-charge (m/z) ion contribution (DIMS) that leads to separation of the sample groups. The most positive and most negative m/z ion contributions which appear in the relevant PC are considered as being key m/z ions influencing the separation of the factors. The most highly discriminatory key m/z ions can be tentatively identified based upon comparisons with DIMS metabolite libraries and databases, ultimately leading to the elucidation of the key differences between sample groups, although confirmation is required from further MS analyses.
Results and discussion
Selection of the number of principal components
Prior to performing the MANOVA, the dimensionality of each dataset was reduced using PCA. The selection of the number of PCs to apply MANOVA proved to be a very difficult problem. In purely statistical terms, the aim is to retain a sufficient number of PCs to account for a high percentage of the total variability, e.g. 95%. However, the problem is that in data of this kind there is a very high correlation between the spectra from all the experimental units which results in a very large percentage of the total variability being accounted for by the first PC. In the first investigation reported in this article, more than 95% of the total variability was accounted for by just the first two PCs (Table 3).
On the other hand, in the course of analysing the work reported in this article we have found that in the main, PC1 reflects the general correspondence of the spectra between the individual samples, while the subsequent PCs are the potential sources of biological variation. Useful biological interpretations often lie in PCs which contain only a small percentage of the total variability. In general, we have found that the more PCs included for further analysis by MANOVA, the greater the number of significant main effects and interactions will be found (unpublished data). But against this can be stated that these extra PCs will account for only a minute amount of statistical variability, and so the significance of any biological information that such PCs may contain would be very questionable.
Ideally, the selection of the number of PCs to retain for the rest of the statistical analyses would be based on biological criteria alone. However, because the biological information in the various PCs derived from data of a new investigation is unknown, and because an objective solution to this problem is ideally required, recourse does have to be made to the statistical aspect. For the first investigation reported in this article, we set the variability threshold to 99%, which required eight PCs (Table 3). In the second investigation, because less of the total variability was accounted for by the first few PCs a threshold of 95% was chosen, which resulted again in eight PCs being used.
Pathogen challenge investigation
In this investigation the first factor is experiment, which has two levels, and the second factor is genotype with three levels corresponding to the wild-type Arabidopsis line and the two ethylene mutants. The third factor is infection, which has two levels corresponding to plus or minus B. cinerea infection as previously described. The fourth factor is time, which has two levels equivalent to 12 h post infection and 48 h post infection. Six replicate plants were used for each experiment × genotype × infection × time combination. Hence the variability of the individual plants constitutes the residual or error line in the MANOVA table and is used to assess the four main effects and their interactions.
The dimensionality of the DIMS data was reduced and the first eight PCs were used in the MANOVA, the results of which are shown in Table 4.
The MANOVA shows that there is a highly significant experiment effect, genotype effect, infection effect and time effect (all P < 0.001). More importantly, there are also highly significant two and three factor interactions. The aim of this investigation was discrimination between the three Arabidopsis genotypes upon infection over time and within two independent experiments. The presence of the significant experiment × infection × time effect (P < 0.001), means that it is not appropriate to study the single effects of genotype, time and infection alone or even the first order interactions among them. Due to all the interactions, it was not possible to visualise the relationships between the factors and the experiments; so it was decided to break down the data, considering the two time points and the two replicate experiments individually. Thus we carried out four separate analyses on three-factor data (experiment, genotype, infection in the first two analyses; genotype, infection, time in the latter two). It must be noted that these four separate analyses use different sub-sets of the data. Hence this can introduce apparently conflicting interpretations.
Time interactions
By applying MANOVA to the two individual time points, very similar conclusions can be drawn. Due to this, only the first time point (12 h post infection) was expanded further in this article.
From the MANOVA (Table 5) for 12 h post infection it can be concluded that no further breakdown was required due to a non-significant triple interaction of experiment × genotype × infection (P = 0.432). There are highly significant experiment, genotype and infection effects (P < 0.001, P < 0.001 and P = 0.001 respectively) but, more importantly, all the first order interactions of experiment × genotype, experiment × infection and genotype × infection are highly significant. By removing the fourth (time) factor, the aim of these breakdowns (time 1 and 2 separately) was to differentiate between Arabidopsis genotypes, with and without infection within two replicate experiments. It is not appropriate to study the single factor effects only due to the presence of the significant interactions. So therefore CVA was used to study the interrelationships between experiment, genotype and infection.
The experiment × genotype interaction. The low P-value of <0.001 of the experiment × genotype interaction shows that the relationships between the three genotypes are not the same for each experiment. The actual relationships can be observed using CVA, with a priori knowledge of the number of classes, in this case, six consisting of each experiment and genotype combination (figure 2A, B). The data, in the form of sample means (one for each population) are plotted on the first two CV axes. It should be noted that the two plots show exactly the same CVA results; it is merely that different means are joined together to aid interpretation. By joining together the means of the two genotypes within each experiment, as shown in figure 2A, a clear experimental difference can be seen. CV2 separates the three genotypes showing the same trends, i.e. with Col-0 having the highest scores on CV2, followed by etr1-1, and then ctr1-1. But CV1 also has some influence on the separation of the genotypes and it is here, where there are some subtle differences in orientation that the cause of the significant experiment × genotype interaction originates. As the trends of the three genotypes are very similar, the experiment × genotype interaction is clearly not biologically significant, only significant statistically. Even though there seems to be large metabolomic differences between the two experiments the significance with respect to genotype is minimal as the three genotypes are behaving similarly between the two experiments.

CVA for the experiment × genotype interaction, where experiment is the first number and genotype is the second number (where 1 = Col-0, 2 = etr1-1 and 3 = ctr1-1) (A) means of the three genotypes within each experiment joined, and (B) means of the two experiments for each genotype joined. Both plots show confidence circles which equal 0.5658 units where n = 12.
The experiment × infection interaction. The presence of an interaction, (P < 0.001), indicates that the relationships between the two treatments of B. cinerea infected and non-infected were not the same for each experiment. Again, the relationships can be elucidated using CVA, in which each experiment and infection combination is shown, yielding a class structure of two treatments plus two controls. Again, as before, there is a clear experimental split on CV1, with experiment 1 to the left-hand side and experiment 2 to the right (figure 3A, B). However, the interaction between experiment and infection is clearly shown in that the two infection regimes are significantly different in experiment 1, but not in experiment 2. By joining together the means of the two experiments for each infection regime (figure 3B), it can be seen that in non-infected plants, the differences between the two experiments are greater than are the differences between the two experiments in the infected plants.

CVA for the experiment × infection interaction, where experiment is the first number and infection is the second number (1 = B. cinerea infected and 2 = non-infected) (A) means of the two infection regimes within each experiment joined, and (B) means of the two experiments for each infection regime joined. Both plots show confidence circles which equal 0.462 units where n = 18.
Examination of the CV loading vectors will identify PCs used to derive the CVA model for the experiment × infection interaction. Figure 4 shows the m/z plot of PC2, which is the most highly loaded PC on CV1 (positively loaded at 0.721), illustrating the m/z ion contribution to the variation between experiments given in figure 3. Some of the top m/z intensities are 150, 156, 211 and 213 of which all are highly positively loaded. On the other hand 116, 147, 210, 224 and 437 m/z are all negatively loaded on this PC. On CV1, experiment 1 has a low score while experiment 2 has a high score. Hence experiment 1 tends to have low 150, 156, 211 and 213 mass ions compared with experiment 2, but higher 116, 147, 210, 224 and 437 m/z. Tentative identification of these mass ions can allow possible metabolite pathways in which they belong to be explored leading to the elucidation of the key pathway differences between experiments, although confirmation using further MS techniques would be required.

Principal component (PC) loading plot; the most highly loaded PC loading vector plotted against m/z revealing the major ions contributing variance in the DIMS spectra ultimately allowing the identification of key discriminatory m/z between the two replicate experiments.
The genotype × infection interaction. Each genotype and infection combination is produced by pooling the data from the two experiments resulting in a class structure of six groups. This highly significant genotype × infection (P = 0.002) interaction indicates that the relationships between the three genotypes are not the same upon infection. The ethylene insensitive mutant, etr1-1, has been shown to be more susceptible to B. cinerea due to premature symptom development compared with Col-0 and ctr1-1 (data not shown). On the other hand, ctr1-1 shows delayed symptom responses and increased resistance to B. cinerea. Thus it can be hypothesised that there would be large differences between the metabolic profiles of the three genotypes upon infection with B. cinerea. The CVA results (figure 5A, B) show a separation of the genotypes on CV1, but the differences in orientation of Col-0 and etr1-1 is the cause of the interaction here. The two infection regimes are separated along CV2 (figure 5B), with the infected plants together having a lower score than the non-infected ones, but again there are differences in orientation of Col-0 and etr1-1. This interaction suggests that etr1-1 does not respond in the same way to infection as do Col-0 and ctr1-1.

CVA for the genotype × infection interaction, where genotype is the first number (where 1 = Col-0, 2 = etr1-1 and 3 = ctr1-1) and infection is the second number (1 = B. cinerea infected and 2 = non-infected) (A) means of each genotype joined, and (B) means of each infection joined together. Both plots show confidence circles which equal 0.5658 units where n = 12.
Experiment interaction
The aim of these breakdowns (experiments 1 and 2 separately) was the discrimination between the genotypes, with and without infection over time, removing the effect of the replicate experiment. Both of the breakdown analyses yielded very similar conclusions in terms of significant main effects and interactions. So even though significant differences were evident between the two experiments in the previous breakdown analysis for Time, and key m/z ion intensities contributing to this variation have been identified, the trends within each experiment remain the same. This suggests that even with the highly significant experiment difference the model is still reproducible.
Experiment 1 breakdown was chosen to be explored further in this article. There are highly significant single factor effects of genotype, infection and time but, again, all the first order interactions are highly significant (Table 6). However, the non-significant three factor interaction of genotype × infection × time, (P = 0.336) in the MANOVA table for experiment 1 enables us to proceed straight to CVAs.
The infection × time interaction. The significant infection × time interaction (P < 0.001) indicates that the relationships between the B. cinerea infected and non-infected are not the same for each time point. The CVA results are shown in figure 6A and B, in which each infection and time combination is shown, yielding a class structure of four. Figure 6A indicates that the non-infected are not significantly different at the two time points but B. cinerea infected plants are significantly different at the two sampling time points. Figure 6B shows that at 12 h the two infection regimes are not significantly different, but at 48 h the two infection regimes differ more than at 12 h. These observations are biologically important as the metabolite profiles of the non-infected controls should differ less than those infected with B. cinerea, so therefore these results suggest a representative and robust experimental design.

CVA for the infection × time interaction, where infection is the first number (1 = B. cinerea infected and 2 = non-infected) and time is the second (1 = 12 h and 2 = 48 h) (A) means of each infection joined, and (B) means of each time joined. Both plots show confidence circles which equal 0.462 units where n = 18.
The genotype × time interaction. The significant genotype × time interaction (P < 0.001) suggests that the relationships between the three genotypes differ over time. In this investigation, ctr1-1 grows as if continuously exposed to ethylene, resulting in a profoundly dwarfed phenotype when compared to Col-0; while etr1-1, shows a greater resemblance to the Col-0 phenotype (Woester and Kieber, 1998; Bleecker and Kende, 2000). It might therefore be hypothesised that there would be larger differences between the metabolite profiles of ctr1-1 and Col-0 than etr1-1 and Col-0. Each genotype and time combination is produced by pooling the data from the two infection regimes resulting in a class structure of six groups. Figures 7A and B show a clear genotype split on CV1, with ctr1-1 having a lower score than Col-0 and etr1-1, thus supporting the hypothesis. CV2 separates the two time points showing that ctr1-1 is not significantly different at the two time points and both Col-0 and etr1-1 are significantly different. The subtle differences in orientation of etr1-1 and Col-0 have produced the significant genotype × time interaction.

CVA for the genotype × time interaction, where genotype is the first number (where 1 = Col-0, 2 = etr1-1 and 3 = ctr1-1) and time is the second (1 = 12 h and 2 = 48 h) (A) means of each genotype joined, and (B) means of each time joined. Both plots show confidence circles which equal 0.5658 units where n = 12.
The genotype × infection interaction. The CVA results for the significant genotype × infection interaction (figure 8A, B) imply very similar conclusions to the genotype × infection interaction seen in the first breakdown analysis of time 1 (figure 5A, B). But in this case each genotype and infection combination is produced by pooling the data from the two time points within experiment 1. Again CV1 shows the same genotypic split but ctr1-1 is more separated from Col-0 and etr1-1 (figure 8A). Again, CV2 shows the same split due to the two different infection regimes (figure 8B) except not so clearly as the genotype × infection interaction for the time breakdown.

CVA for the genotype × infection interaction, where genotype is the first number (where 1 = Col-0, 2 = etr1-1 and 3 = ctr1-1) and infection is the second number (1 = B. cinerea infected and 2 = non-infected) (A) means of each genotype joined, and (B) means of each infection joined together. Both plots show confidence circles which equal 0.5678 units where n = 12.
Sample preparation and storage investigation
The experimental design here resulted in a two factor nested MANOVA as there are three plant replicates per genotype × sample preparation combination, which constitute the residual for the genotype and treatment effects and their interaction. The two factors are genotype at three levels (Col-0 and mutants etr1-1 and ctr1-1), and sample preparation method which had five levels as previously described. Each plant sample was analysed in triplicate (machine replicates) the variability of which was used to assess plant-to-plant variation, as illustrated in the MANOVA (Table 7). The dimensionality of the FT-IR data was reduced prior to MANOVA and the first 8 PCs were retained as these accounted for 95.23% explained variance.
MANOVA shows a highly significant genotype and sample preparation method effect (both P < 0.001), along with a significant genotype × preparation method interaction (P = 0.001) (Table 7). There is also a highly significant plant effect, illustrating that there is much more variability between the replicate plants than there is between replicate machine runs within plants as was found for the pathogenic challenge investigation.
The relationship between the three genotypes is not the same within each sample preparation method as there is a highly significant interaction. Figure 9 shows the CVA plot for the genotype × sample preparation interaction, with means and confidence intervals, where figure 9A is labelled showing genotype and figure 9B is labelled showing sample preparation method. Table 8 describes the numerical key used to label the plots to aid interpretation.

CVA for the significant genotype × sample preparation interaction, with (A) annotated to indicate the three genotypes: Col-0, ctr1-1 and etr1-1, and (B) annotated to indicate the sample preparation method, where FR is treatment 5, FF is freezing whole (1) and after grinding (2) and FD is freeze-dried and stored at −80 °C (1) and at room temperature (2), as described in Table 8. Both plots show the treatment means and confidence circles which equal 0.65 units where n = 9.
Figure 9 shows discrimination between the sample preparation methods and the genotypes. CV1 predominantly separates the different sample preparation methods, where all samples stored for 8 weeks are distinct from the fresh (FR) samples and the freeze-dried samples (indicated by FD1 and FD2) being furthest away (figure 9B). Despite the sample preparation effect, etr1-1 is clearly discriminated from both Col-0 and ctr1-1 (figure 9A). All etr1-1 samples show a similar trend of tending towards the lower end of CV2 irrespective of the sample preparation method. These results indirectly indicate a closer similarity between the metabolic fingerprints of Col-0 and ctr1-1 despite the very distinct dwarf phenotype of ctr1-1. In contrast, etr1-1 shows a larger biochemical difference which is not reflected by its phenotype which is more comparable to that of the Col-0.
Data from the two investigations suggests that the genotypes Col-0, etr1-1 and ctr1-1 have different metabolic fingerprints (FT-IR) and profiles (DIMS). However, the degree of discrimination of etr1-1 and ctr1-1 from Col-0 appears to be dependent on the analytical techniques employed.
When focusing on sample preparation method those samples which were analysed on the day of harvest (labelled as FR in figure 9B) are distinct from all those which had been stored for 8 weeks, irrespective of genotype. By joining together the means of each sample class, as shown in figure 9B, the relationships between genotypes with respect to sample preparation can be visualised more clearly. The shape of the triangles reflects the tendency for etr1-1 to cluster to the lower end of CV2. However the shapes of the five triangles corresponding to each sample preparation method differ, reflecting the complexity of the interaction. It is often not practical in highly replicated metabolomic experiments to analyse samples on the same day as harvest, so the material must be stored. Ideally storage of the plant material will preserve the metabolome as it was at the instant of harvest; however these results indicate that subtle changes may be occurring altering the biochemical composition. In this example the significance with respect to genotype is minimal as the same overall relationship between wild type and ctr1-1 on the one hand and etr1-1 on the other remains the same. Further work is required to assess the significance of sample preparation and storage duration on the metabolome and the consequences this might have.
Selection of the number of canonical variates
Although CVs have a similar property to PCs in that they both (sequentially) account for a decreasing percentage of the total variability, the criteria for selecting the number of CVs to retain in the interpretation of the results are quite different from considerations in respect of PCs where both are used sequentially to analyse multivariate data, as here.
In the first place, PCA is considering all the individual data points (individual plants or machine runs) envisaged as having come from a single statistical population, even though this is not true – every treatment is a different statistical population. The method is used simply to reduce the dimensionality of the data in order to make the data set more manageable. CVA, on the other hand, works on the means of samples derived from the different statistical populations.
Second, apart from a simple experimental situation where there is no factorial structure in the design and the results are first analysed by a one-way MANOVA and followed by a CVA to ascertain which treatments are significantly different from which others, a CVA following the MANOVA of a factorially designed investigation is using only a sub-set of the data to investigate a specified interaction. The amount of the total variability accounted for by the first CV will now depend only upon the differences between the sample means of the levels of the various factors included in the data sub-set being analysed. This point is brought out very well in the first investigation. Whenever the factor ‘experiment’ is part of the interaction being investigated, nearly all the variability is accounted for by the first CV, typically >90%. This is because the two experiments are so different from each other. However, in the case of a genotype × infection interaction, the first CV accounts for markedly less of the total variability (∼70%), and the second CV for ∼20%.
In our experience, it is rarely necessary to retain more than the first two CVs for an adequate interpretation of a CVA. But, like all questions of this type, every investigation is unique and a judgement has to be made according to circumstances. If most of the variability is accounted for by the first CV, then nothing will be gained by trying to interpret more than the first two CVs. On the other hand, in a situation like the genotype × infection interaction above where other CVs account for a substantial amount of the total variability (however that may be judged), then of course they should be included in the final assessment.
Conclusion
The value of statistical analyses as a test of analytical rigour of metabolomic data was highlighted in a review by Sumner et al. (2003), as was the daunting task of analysing large data sets. The application of MANOVA, a multivariate method, enables the simultaneous analysis of many variates; and therefore provides a powerful method by which the significance of multiple experimental factors and their interactions can be assessed. We have demonstrated how MANOVA can be applied to investigate the significance of all factors involved within an investigation from the main focus of genotype and pathogenic challenge to plant cultivation and preparation, plant reproducibility, analytical run and experimental reproducibility. CVA as a multiple range test permits the visualisation of the sample groups within the significant main effects or interactions of interest, and allows the rapid evaluation of inter-relationships. In the first investigation the experimental effect was not of biological interest. However, the inclusion of two replicate experiments was done specifically to assess the degree of reproducibility between identical experiments in the field of metabolomics. The identification of significant treatments is important for subsequent analysis, which may involve the application of other multivariate methods and perhaps evolutionary algorithms, many of which are of a supervised nature and therefore require a priori knowledge of the data set structure.
The application of MANOVA to metabolomic data incorporates the features of the experimental design in which all the effects of the different factors and their interactions are clearly revealed. This is a necessary statistical process in the overwhelming task of analysing complex multivariate datasets.
Abbreviations
- MANOVA:
-
applied to metabolomic data
References
Alsberg B.K., Wade W.G., Goodacre R. (1998) Chemometric analysis of diffuse reflectance-absorbance Fourier transform infrared spectra using rule induction methods: application to the classification of Eubacterium species. Appl. Spectrosc. 52, 823–832
Bino R.J., Hall R.D., Fiehn O., et al (2004) Potential of metabolomics as a functional genomics tool. Trends Plant Sci. 9, 418–425
Bleecker A.B., Kende H. (2000) Ethylene: a gaseous signal molecule in plants. Annu. Rev. Cell. Dev. Biol. 16, 1–18
Bleecker A.B., Estelle M.A., Somerville C., Kende H. (1988) Insensitivity to ethylene conferred by a dominant mutation in Arabidopsis thaliana. Science 241, 1086–1089
Boyes D.C., Zayed A.M., Ascenzi R., et al (2001) Growth stage-based phenotypic analysis of Arabidopsis: a model for high throughput functional genomics in plants. Plant Cell 13, 1499–1510
Chatfield, C. and Collins, A.J. (1980) Introduction to Multivariate Analysis. Chapman and Hall
Chen J.J., Delongchamp R.R., Tsai C.A., et al (2004) Analysis of variance components in gene expression data. Bioinformatics 20(9), 1436–1446
Coombes, K.R. (2007). PCANOVA: combining principal components with analysis of variance to access group structure. Technical report available online: http://bioinformatics.mdanderson.org/TechReports/pca.pdf (Downloaded: 15/05/2007)
Daoyu Z., Lawes G.S. (2000) MANOVA and discriminant analyses of phenotypic data as a guide for parent selection in kiwifruit (Actinidia deliciosa) breeding. Euphytica 114, 151–157
Dunn W.B., Bailey N.J.C., Johnson H.E. (2005) Measuring the metabolome: current analytical technologies. Analyst 130, 606–625
Fernie A.R. (2003) Metabolome characterisation in plant system analysis. Funct. Plant Biol. 30, 111–120
Fiehn O., Kopka J., Dormann P., Altmann T., Tretheway R.N., Willmitzer L (2000) Metabolite profiling for plant functional genomics. Nat. Biotechnol. 18, 1157–1161
Goodacre R., Timmins E.M., Burton R., et al (1998) Rapid identification of urinary tract infection bacteria using hyperspectral whole-organism fingerprinting and artificial neural networks. Microbiology 144, 1157–1170
Goodacre R., Timmins E.M., Rooney P.J., Rowland J.J., Kell D.B. (1996) Rapid identification of Streptococcus and Enterococcus species using diffuse reflectance-absorbance Fourier transform infrared spectroscopy and artificial neural networks. FEMS Microbiol. Lett. 140, 233–239
Goodacre R., Vaidyanathan S., Dunn W.B., Harrigan G.C., Kell D.B. (2004) Metabolomics by numbers: acquiring and understanding global metabolite data. Trends Biotechnol. 22, 245–252
Gullberg J., Jonsson P., Nordstrom A., Sjostrom M., Moritz T. (2004) Design of experiments: an efficient strategy to identify factors influencing extraction and derivatization of Arabidopsis thaliana samples in metabolomic studies with gas chromatography/mass spectrometry. Anal. Biochem. 331, 283–295
Guzman P., Ecker J.R. (1990) Exploiting the triple response of Arabidopsis to identify ethylene-related mutants. Plant Cell 2, 513–523
Holtorf H., Guitton M., Reski R. (2002) Plant functional genomics. Naturwissenschaften 89, 235–249
Jansen J.J., Hoefsloot H.C.J., van der Greef J., Timmerman M.E., Westerhuis J.A., Smilde A.K. (2005) ASCA: analysis of multivariate data obtained from an experimental design. J. Chemometr. 19(9), 469–481
Johnson H.E., Broadhurst D., Goodacre R., Smith A.R. (2003) Metabolic fingerprinting of salt-stressed tomatoes. Phytochemistry 62, 919–928
Kieber J.J., Rothenberg M., Roman G., Feldmann K.A., Ecker J.R. (1993) CTR1, a negative regulator of the ethylene response pathway in Arabidopsis, encodes a member of the raf family of protein kinases. Cell 72, 427–441
Langsrud Ø. (2002) 50–50 multivariate analysis of variance for collinear responses. Statistician 51(3), 305–317
Otto M. (1999) Chemometrics: Statistics and Computer Application in Analytical Chemistry. Wiley-VCH Weinheim, Germany
Quinn G.P., Keough M.J. (2002) Experimental Design and Data Analysis for Biologists. Cambridge University Press, Cambridge, UK
Penninckx I.A.M.A., Thomma B.P.H.J., Buchala A., Métraux J.-P., Broekaert W (1998) Cooperative activation of jasmonate and ethylene response pathways in parallel is required for induction of a plant defensin gene in Arabidopsis. Plant Cell 10, 2103–2114
Roessner U., Wagner C., Kopka J., Trethewey R.N., Willmitzer L. (2000) Simultaneous analysis of metabolites in potato tuber by gas chromatography–mass spectrometry. Plant J. 23, 131–142
Seahl H. (1964) Multivariate Statistical Analysis for Biologists. Methuen, London, UK
Schulz H., Baranska M., Belz H.H., Rosch P., Strehle M.A., Popp J. (2004) Chemotaxonomic characterisation of essential oil plants by vibrational spectroscopy measurements. Vib. Spectrosc. 35, 81–86
Smilde A.K., Jansen J.J., Hoefsloot H.C., Lamers R.J., van der Greef J., Timmerman M.E. (2005) ANOVA-simultaneous component analysis (ASCA): a new tool for analyzing designed metabolomics data. Bioinformatics 21(13), 3043–3048
Smith H., Gnanadesikan R., Hughes J.B. (1962) Multivariate analysis of variance (MANOVA). Biometrics 18(1), 22–41
Sumner L.W., Mendes P., Dixon R.A. (2003) Plant metabolomics: large-scale phytochemistry in the functional genomics era. Phytochemistry 62, 817–836
Thomma B.P.H.J., Eggermont K., Penninckx I.A.M.A., et al (1998) Separate jasmonate-dependent and salicylated dependent defense-response pathways in Arabidopsis are essential for resistance to distinct microbial pathogens. Proc. Natl. Acad. Sci. U.S.A. 95, 15107–15111
Taylor J., King R.D., Altmann T., Fiehn O. (2002) Application of metabolomics to plant genotype discrimination using statistics and machine learning. Bioinformatics 18, S241–S248
Winson M.K., Goodacre R., Timmins E.M., et al (1997). Diffuse reflectance absorbance spectroscopy taking in chemometrics (DRASTIC). A hyperspectral FT-IR-based approach to rapid screening for metabolite overproduction. Anal. Chim. Acta 348, 273–282
Woeste K., Kieber J.J. (1998) The molecular basis of ethylene signalling in Arabidopsis. Philos. Trans. R. Soc. Lond. B Biol. Sci. 352, 1431–1438
Acknowledgements
The authors would like to thank the UK Biotechnology and Biological Sciences Research Council for part funding of this work through a studentship to AJL. Botrytis cinerea, strain IMI 169558, was the kind gift of Dr. Bart Thomma (Katholieke Universiteit Leuven, Belgium). Also thanks are given to Pat Causton, Tom Thomas and Ray Smith (Aberystwyth, UK), who grew and maintained the plant material. The authors would like to acknowledge the BBSRC for funding.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Johnson, H.E., Lloyd, A.J., Mur, L.A.J. et al. The application of MANOVA to analyse Arabidopsis thaliana metabolomic data from factorially designed experiments. Metabolomics 3, 517–530 (2007). https://doi.org/10.1007/s11306-007-0065-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11306-007-0065-3