
Abstract
Selective breeding of dogs over hundreds of years has inadvertently resulted in breed-specific propensities to particular diseases. Furthermore, it has likely induced more subtle affects on the physiology of certain breeds and moved them from their evolutionary optima. In the absence of obvious disease phenotypes such subtle changes could have yet unrecognised breed-specific implications for health and well-being. Here we have applied NMR metabolomics as a discovery-driven approach to identify the impact of breed on the urinary profile of dog and to determine if non-disease-related breed differences can be identified. Multiple urines were collected non-invasively over a two-week period from seven neutered male Labrador retrievers and miniature Schnauzers. Following NMR analyses by 1-dimensional 1H and 2-dimensional 1H J-resolved (JRES) spectroscopy, principal component analysis revealed that the metabolic variability within each individual is relatively small compared to inter-individual variability, and that some separation between breeds was evident. A supervised model, using partial least squares discriminant analysis (PLS-DA) with class based upon breed, was trained using the JRES data. The model predicted correctly the breed of seven additional urines, yielding a model sensitivity and specificity of 100%. Several significant metabolic differences between the breeds were identified. A second model was developed using PLS-DA with class based upon individual dogs, which again achieved high classification accuracy for the test set. Overall, this confirms that canine urine is information-rich and that breed is a major determinant of urinary metabolic fingerprints. In the future this may enable a more accurate development of specific nutritional care for an individual or breed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Natural selection results in the accumulation of traits for optimal success, which include improved health and fitness. Humans have undertaken artificial selection of dog, arguably for 15,000–40,000 years (Savolainen et al. 2002). Evidence of breeding for specific roles, e.g., hunting and guard dogs, exists from Egyptian times and the other main current types, including lap and herd dogs, since Roman times. Initially, the selection criteria would have been behavioural but also included diminution (through hormonal and dietary changes) and resulted in early maturation, larger litters and shortened generation time (Clutton-Brock 1999). Selection for more specific roles has lead to a diverse range of morphologies, making the dog being more variable than any other mammalian species. Indeed, many of the recognised breeds are a consequence of a restricted breeding pool over the last 400–600 years. There has been a change in selection from specific function to aesthetic aspects as a consequence of the development of kennel clubs 150 years ago, which may have further reduced the physiological fitness of many pure-bred dogs through the creation of breed barriers and the drive toward a breed standard. The selective breeding within breed type has inadvertently resulted in the development of breed-specific propensities to specific diseases. Indeed, it is on this basis that dog is such a useful genetic model for mapping disease traits that are common to both dog and humans (Patterson 2000). However, as well as presenting distinct susceptibilities to disease states, the development of breed classes is likely to have also had more subtle effects on the physiology of certain breeds and moved them from their evolutionary optima. These changes may have some, as yet unrecognised, welfare effects upon some breeds and without obvious disease phenotypes appropriate care or treatment may not be offered. The consequence of these breed-specific effects may be identified through the use of -omics technologies. Methods to measure a vast range of factors at the DNA, RNA, protein and metabolite level exist and may shed light on breed-specific differences, which may be compensated by appropriate care and diet.
Urine is a practical diagnostic sample because it is relatively safe, non-invasive and simple to collect, and contains systemic information about the individual in a concentrated form. However, beyond diagnostic markers to diagnose pregnancy and specific diseases, urine can also provide other relevant information about the individual, its health, physiological function and response to xenobiotics (Strasinger and Di Lorenzo 2001). The extent to which urine can act as a surrogate sample to more invasive samples is being extended through the development of metabolomics as a discovery tool. Metabolic fingerprinting has been used to identify markers associated with a number of phenotypes, such as drug safety and toxicity (Nicholson et al. 2002; Pelczer 2005), disease (Brindle et al. 2002; Coen et al. 2005; Rosenblum et al. 2006), gender (Plumb et al. 2005; Stanley et al. 2005), ethnicity (Dumas et al. 2006), ageing (Plumb et al. 2005), diet and the impact of caloric restriction (Bijlsma et al. 2006; Shi et al. 2002a, b; Vigneau-Callahan et al. 2001; Walsh et al. 2006), and the environment (Hines et al. 2007; Lin et al. 2006; Viant et al. 2003).
As described above, the domestic dog is a unique mammalian species in that it has over 400 well-defined genetic classes with a diverse range of physiological and behavioural phenotypes. It is reasonable to propose that such changes are likely to result in subtle differences in breed care requirements. However, without clear disease phenotypes, there has been little possibility to research this using hypothesis-led research. As such this area of research would benefit from taking a discovery-driven approach using -omic technologies. With urine being a valid and non-invasive sample, metabolomics studies on urine would appear to be a useful approach. Previously, metabolomics analyses in the domestic dog have included a study related to the plasma fingerprints of congenital and acquired liver disease (Whitfield et al. 2005), a GC-MS investigation that compared human urine with that from Dalmatian dogs and Shetland sheepdogs (Matsumoto et al. 1995), and most recently a study to monitor urinary metabolic profiles throughout the lifetimes of control-fed and diet-restricted dogs (Wang et al. 2007). Here we have undertaken a study to develop and apply an NMR analysis to identify the impact of breed on the urinary profile of dog and to test whether non disease-related breed differences could be identified. The first objective was to establish optimised sample preparation methods for the analysis of metabolites in dog urine. Subsequently we sought to measure the NMR fingerprints of multiple urine samples from two dog breeds, Labrador retriever and miniature Schnauzer. Samples were collected from several male dogs across a 2-week period, allowing us to assess intra-individual metabolic variability, inter-individual variability in the same breed, and inter-breed variability. The final objective was to construct and test a supervised multivariate model that could predict the breed of dog based upon its urinary metabolome.
2 Materials and methods
2.1 Ethical standards statement
All dogs were fed commercially available, complete diets throughout the study period at energy levels to maintain adult body weight. The dogs were housed at the WALTHAM Centre for Pet Nutrition (UK) where they were housed in purpose-built, environmentally enriched facilities and treated in accordance with the Centre’s research ethics and UK Home Office Regulations.
2.2 Collection of urine
Urine samples were obtained from three neutered male dogs of one breed (Labrador retriever, LR), and four neutered male dogs of a second breed (miniature Schnauzer, MS) (Table 1), all fed the same brand diet for at least 4 weeks prior to urine collection. For each animal, either 5 or 6 urine samples were collected on different days within a 2-week longitudinal study (Table 2). Samples were obtained using a “free catch” approach during the morning walk using an individually designated uripet urine collection pot. Samples were maintained at ambient temperature for up to 30 min, placed on ice and aliquots then frozen within 1 h of sampling. Urine was then maintained at −80°C until it was shipped to the University of Birmingham on dry ice where it was stored at −80°C. The total set comprised of 37 identified urine samples and a further 7 samples (randomly obtained from the same seven dogs) for which the identities were withheld from the metabolomics group at Birmingham (Table 2).
2.3 Preparation of urine for NMR analysis
Each urine sample was thawed to room temperature, vortexed and centrifuged (10,000g; 3 min). Then 540 μl of supernatant was mixed with 180 μl 0.4 M sodium phosphate buffer (pH 7.0; 0.75% w/v sodium azide) and 60 μl 13 mM sodium 3-trimethylsilyl-2,2,3,3-d4-propionate (TMSP) in D2O, producing a final solution of 0.1 M buffer, 0.2% NaN3, 1 mM TMSP and 8% D2O. The solution was left to stand for 20 min, centrifuged, and the supernatant removed. The pH was adjusted to 7.05 (±0.05) using 1 M HCl or NaOH, the solution left to stand for 20 min, centrifuged, the supernatant removed, and the pH was measured again. This pH adjustment protocol was repeated until the pH fell within 7.00–7.10. The solution was then transferred to a 5 mm NMR tube.
2.4 1H NMR spectroscopy
Prepared urine samples were analyzed on a DRX-500 NMR spectrometer (Bruker), operating at 500.18 MHz proton frequency, equipped with a cryogenically cooled probe and a BACS-60 automatic sample changer. One-dimensional (1-D) 1H NMR spectra were obtained using a 60° read pulse flip angle, 5.3-kHz spectral width, 3-s relaxation delay, 128 transients collected into 16 k data points, and excitation sculpting to suppress the residual water resonance (Hwang and Shaka 1995). Datasets were zero-filled to 32 k points and exponential line-broadenings of 0.5-Hz applied before Fourier transformation. The resulting spectra were phased and baseline corrected manually, and their chemical shifts calibrated against TMSP (0.0 ppm) using TopSpin (version 1.3, Bruker). 2-D 1H J-resolved (JRES) NMR spectra were also obtained using excitation sculpting for water suppression (Hwang and Shaka 1995). Acquisition parameters included a 3-s relaxation delay, 90° read pulse flip angle, and 16 transients per increment for 16 increments collected into 16 k data points, using spectral widths of 5.3 kHz in F2 (chemical shift axis) and 50 Hz in F1 (spin-spin coupling axis). Datasets were zero-filled to 128 points in F1, and both dimensions multiplied by sine-bell window functions prior to Fourier transformation. JRES spectra were calibrated against TMSP (0.0 ppm), tilted by 45°, symmetrized about F1, and then 1-D skyline projections of the 2-D spectra were calculated (denoted pJRES), all using TopSpin.
2.5 Spectral pre-processing
The 1-D NMR spectra and 1-D projections of the 2-D J-resolved spectra were converted to an appropriate format for multivariate analysis using custom-written ProMetab software (Viant 2003) running within Matlab (version 7.1; The MathsWorks, Natick, MA, USA). First, the regions above 10 ppm, below 0.2 ppm, and between 4.50 and 6.45 ppm (residual water and urea) were excluded from all spectra, and the total area of each spectrum was normalized to one. All spectra were then generalized-log transformed (transformation parameter, λ = 3.0 × 10−8) to stabilise the variance across the spectral data points and to increase the weightings of the less intense peaks (Parsons et al. 2007; Purohit et al. 2004). Due to slight pH-induced chemical shift variability, all spectra were aligned using a beam search algorithm as described by Lee and Woodruff (2004). All spectra were then segmented into 0.005-ppm bins. For the 1-D spectra, bins between 2.405–2.425, 2.52–2.57, 2.66–2.71, 2.94–2.955, 3.105–3.13, 3.72–3.77, 3.955–3.99, 7.08–7.20, 8.00–8.18 and 8.45–8.48 ppm containing pH-sensitive resonances were each compressed into a single bin resulting in a matrix of 44 samples × 1855 bins. Similarly, for the 1-D projections, bins between 2.40–2.425, 2.52–2.57, 2.66–2.71, 2.935–2.955, 2.96–2.98, 3.105–3.13, 3.72–3.77, 3.955–3.99, 7.08–7.20 and 8.00–8.18 ppm were each compressed into a single bin resulting in a matrix of 44 samples × 1855 bins.
2.6 Statistical analyses
Pre-processed NMR data were mean-centred before principal component analysis (PCA) and partial least squares discriminant analysis (PLS-DA) using PLS_Toolbox (version 3.53, Eigenvector Research, Manson, WA, USA). PCA was used to identify any clustering of the 37 known urine samples in an unsupervised manner, to determine if this clustering was most evident in the 1-D or pJRES dataset, and to identify any outliers (Eriksson et al. 2001). PLS-DA was used to build two classification models, the first based upon breed (2 classes only) and the second based upon individual dogs (seven classes) (Eriksson et al. 2001). For both models, the 37 known urine samples were used as the training set (Tables 1 and 2), and cross-validation was employed to tune the model and to select the appropriate number of latent variables (LV). The seven remaining samples were used as an independent test set to assess the performance of the fully trained PLS-DA models; the identities of these seven samples remained unknown to the metabolomics group at Birmingham until after the completion of model building. NMR peaks corresponding to the largest weightings in the PLS analyses were quantified using Matlab and the significance of these differences in metabolite concentrations were examined using t-tests (Microsoft Excel). Methods to identify the peaks are described below.
2.7 Metabolite identification
A 2-D 1H, 1H TOCSY NMR experiment at 800 MHz was performed on a single 4-fold concentrated urine sample (from dog MS3) to aid the assignment of metabolites. The spectrum was acquired using a DIPSI3 isotropic mixing sequence (Shaka et al. 1988) for 120 ms at a field strength of 6 kHz. For analysis of the spectrum the data were processed using NMRLab (Gunther et al. 2000) and visualised using CcpNmr software (University of Cambridge). Peaks were then identified (or attempted to be identified) using the Chenomx NMR Suite and associated metabolite libraries (version 4.5; Chenomx Inc., Edmonton, Canada), the Madison Metabolomics Consortium Database (MMCD) (Markley et al. 2007), and the Human Metabolome Database (HMDB) (Wishart et al. 2007). Spiking of pure metabolite into urine was also used to attempt to confirm metabolite identity by 1-D NMR.
3 Results and discussion
3.1 1-D and pJRES NMR fingerprints of dog urine
The initial pH of the urine samples, prior to buffering, varied significantly from animal to animal (from 6.44 to 7.66 for miniature Schnauzers and 6.60–7.27 for Labrador retrievers). Furthermore, the samples were somewhat resistant to buffering, and even after pH adjustments in the presence of buffer the pH values drifted over time. Therefore, drawing from a number of published methods (Keun et al. 2002; Lauridsen et al. 2007), a protocol was developed to produce and maintain a stable and reproducible urine pH that resulted in minimal pH-induced chemical shift variation in the NMR spectra; described in Materials and Methods above. A representative 1-D 1H NMR spectrum and a 1-D skyline projection of a 2-D 1H J-resolved NMR spectrum (pJRES) for dog urine are shown in Fig. 1a and b. The corresponding pre-processed NMR data are shown in Fig. 1c and d for the 1-D 1H and the pJRES spectra, respectively. The raw and pre-processed data highlight a number of interesting features. First, the quality of the water suppression using excitation sculpting is readily apparent from the raw spectra (Fig. 1a, residual water at ca. 4.7 ppm). This is one of few studies to have employed this approach in metabolomics (Aranibar et al. 2006). Second, and as expected, urea dominates the 1-D spectrum (Fig. 1a, ca. 5.8 ppm). However it is absent from the projection of the J-resolved spectrum (Fig. 1b) due to a combination of the spin-echo delay in the pulse sequence as well as the sine window function, which both minimise the contribution of protons that relax rapidly. Third, the dramatic effect of the generalised log transformation is clearly evident, with the pre-processed data (Fig. 1c and d) both exhibiting a suppression of the most intense peaks and an increase in the lower intensity peaks (Parsons et al. 2007; Purohit et al. 2004). Finally, the greatly reduced spectral congestion in the projection of the J-resolved spectrum is visible (Fig. 1d, versus the 1-D data in Fig. 1c), which greatly increases metabolite specificity and facilitates more accurate quantification via the integration of well-resolved resonances on a flat baseline (Viant 2003).

Representative raw and pre-processed NMR spectra of the dog urine samples: (a) raw 1-D 1H NMR spectrum, (b) raw 1-D skyline projection of a 2-D 1H J-resolved NMR spectrum (pJRES), (c) pre-processed 1-D 1H NMR data, and (d) pre-processed pJRES data. The primary advantage of the pJRES spectral projection method is a lower peak density resulting in improved spectral resolution
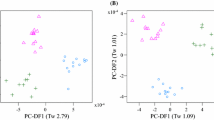
PCA was conducted on both the 1-D 1H and the pJRES pre-processed data from the 37 known urine samples. For the 1-D 1H dataset, two samples had large Q residuals (beyond 99% confidence limit) indicating that they do not fit the PCA model. Sample 7 (which showed chemical shift alterations compared with the other spectra) and sample 37 (no technical abnormality in NMR spectrum) were subsequently removed and the resulting PCA scores plot is shown in Fig. 2a. PCA also identified sample 37 as an outlier in the pJRES dataset, based upon a large Q residual, and the scores plot of the remaining 36 samples is shown in Fig. 2b. The scores plot of the pJRES data (derived from the 2-D J-resolved spectra) revealed improved clustering of each individual dog’s urine fingerprints and also an improved separation of the Labrador retrievers and miniature Schnauzers. This improvement in clustering using pJRES data is a result that we have documented previously (Viant 2003) and have observed for several other datasets. In addition, the PCA loadings plots and PLS weightings plots (discussed below) are significantly less congested for the pJRES dataset which greatly facilitates their interpretation. Consequently all subsequent multivariate analyses and interpretation were conducted only on the pJRES spectra.

PCA scores plots from analyses of the 37 known urine samples using (a) the 1-D 1H NMR dataset (2 outliers removed), and (b) the pJRES NMR dataset (1 outlier removed). Dotted lines have been drawn around the clusters for each Labrador retriever (LR1 (△ ), LR2 (□), LR3 (○)) and solid lines around each miniature Schnauzer (MS1 (✱ ), MS2 (▴ ), MS3 (■), MS4 (•)). The pJRES dataset provides somewhat improved separation of the two breeds
Considering the PCA scores plot of the pJRES data (Fig. 2b), in general the metabolic variability within each individual is relatively small (except for LR2, samples 6–11) compared to the inter-individual variability. Furthermore, even using this unsupervised method of analysis, there is clearly some degree of separation between Labrador retrievers and miniature Schnauzers along the PC1-PC2 axis. The 14 urine samples from three of the four miniature Schnauzers are particularly tightly clustered with low PC1 and high PC2 scores. The fourth dog in this breed (MS3, samples 28–31) showed a somewhat different metabolic fingerprint, but health checks revealed no medical problems and no medication had been administered to this animal. The relatively low amount of variance explained by the first two principal components in Fig. 2b (43.3%) highlights the complexity of the metabolic variability within the NMR dataset; i.e., the dataset is not dominated by variance in just a few metabolites within multi-dimensional space. Overall, however, these unsupervised analyses clearly indicate that individuals and breeds occupy different regions of urinary metabolic space, and confirm the ability to obtain urine metabolic fingerprints in healthy dogs using a daily “free catch” sampling strategy. This sampling strategy is already widely used for dogs in both the clinic and domestic environments.
3.2 Classification model for dog breed
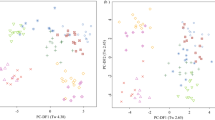
A PLS model was built using the pJRES pre-processed spectra of the 37 known urine samples (Tables 1 and 2), where each sample was identified as a Labrador retriever or miniature Schnauzer. The optimised model was constructed of 3 LVs based upon the minimization of the root mean square error of cross validation. None of the 37 samples exceeded the Q-residual or Hotelling T2 99% confidence limits, so all samples were retained in the training set for the model. The PLS scores plot is illustrated in Fig. 3a, which shows clear separation between the urinary NMR fingerprints of the Labrador retrievers and miniature Schnauzers. Subsequently, to investigate the classification accuracy of the PLS model, the breeds associated with each of the seven unknown urine samples were predicted using DA and a threshold determined using a Bayesian approach (as implemented in the PLS_Toolbox; Wise et al. 2004). The algorithm used the distribution of training set predictions obtained from the PLS model to determine a threshold value which best split the two classes with the least probability of false classifications for future predictions. The predicted breeds for the 37 known and 7 unknown samples are shown in Fig. 3b, and the predictions for the unknowns are summarised in Table 3. The model predicted that 4 of the unknown samples were from Labrador retrievers and 3 from miniature Schnauzers, which, when the identities of the samples were later revealed to the Birmingham metabolomics group, was determined to be the correct classification; i.e., the sensitivity and specificity for the PLS model based on breed are both 100%. The breed-specific nature of urines is clearly indicated by the degree of separation seen in the blind test set (Fig. 3b). There is therefore a high likelihood that metabolic differences representing the breed classification exist and may represent the different physiologies of the two breeds. Further examination of these discriminatory metabolites is important since it may provide a better understanding of which biological factors are the most influential, or sensitive, to breed physiology and genetics. In addition, these results suggest it may be possible to use metabolomics to identify small genetic differences in other species, including domesticated and wild animals.

(a) PLS scores plot from analysis of the pre-processed pJRES NMR spectra of the 37 known urine samples, where breed was used for the class descriptors. A dotted line has been drawn around all known Labrador retriever samples (■) and a solid line around the known miniature Schnauzer samples (•). The seven unknown samples, which were not included in the training of the PLS model, are indicated as open squares or open circles for Labrador retrievers and miniature Schnauzers, respectively. (b) Predictions from the 3-LV PLS model as to whether urine samples belong to the Labrador retriever class; labelling as above. Unknown samples 38, 40, 41 and 43 all lie significantly above the threshold (dashed line just above zero) indicating that they arise from Labrador retrievers, and unknowns 39, 42 and 44 lie below the threshold and are predicted to arise from miniature Schnauzers
3.3 Metabolic discrimination between dog breeds
The weightings plot associated with the LV1 + LV2 axis in the scores plot, which provides the greatest discrimination between the breeds, is shown in Fig. 4. Positive peaks correspond to metabolites that are at elevated concentration in miniature Schnauzers and negative peaks to metabolites that are elevated in Labrador retrievers. NMR peaks with the larger PLS weightings were quantified in the original pJRES NMR dataset, and fold differences in the metabolite levels between the two breeds were calculated along with the significance of those differences (Table 4). We then attempted to identify these peaks using a range of methods. A few metabolites were readily identified from their characteristic chemical shifts and spin-spin couplings, in conjunction with metabolite databases. Note that many more metabolites could be identified in the urine spectra than listed in Table 4, but these metabolites did not correspond to large PLS weightings and so are of minimal interest to this study. Next, peaks arising from unidentified metabolites that could discriminate between the two breeds and that exhibited spin multiplicity were analysed further, specifically a triplet at 0.95 ppm and a triplet at 1.09 ppm. An 800 MHz TOCSY spectrum of a concentrated urine sample (from dog MS3, who was known to have high levels of these metabolites) was recorded and analysed, providing a more complete description of the spin systems. The triplet at 0.95 ppm was coupled to NMR resonances at 1.70 ppm (multiplet of at least 6 peaks) and 4.04 ppm (triplet), suggesting a CH–CH2–CH3 spin system. The second triplet at 1.09 ppm was coupled to NMR resonances at 1.86 ppm (multiplet), 3.30 ppm (multiplet) and 3.41 ppm (multiplet). Extensive searching of databases using this additional information revealed no obvious candidate metabolites, although the spin system associated with the 0.95 ppm triplet was somewhat consistent with 2-hydroxybutyrate. We therefore spiked pure 2-hydroxybutyrate into the urine sample, but the peaks arising from the spiked compound were not coincident with the unknown metabolite. Unfortunately these two metabolites remained unidentified.

PLS weightings plot from analysis of the pre-processed pJRES NMR spectra of the 37 known urine samples. The weightings correspond to the LV1 + LV2 axis, which provides the greatest degree of discrimination between the Labrador retriever and miniature Schnauzer urine samples. Positive peaks correspond to metabolites that are at higher concentration in the miniature Schnauzers relative to the Labrador retrievers, and vice versa. Peak assignments are as listed in Table 4
Next, NMR peaks that appeared as singlets (no cross peaks in a TOCSY) were analysed further by examining the degree of correlation between the intensities of these peaks across multiple spectra. It was discovered that the peaks at 2.09 ppm and 2.95 ppm likely arose from a common metabolite, since their intensities were highly correlated (r 2 = 0.989). This conclusion was further supported by the similar ratio in peak intensities between miniature Schnauzers and Labrador retrievers, as well as the similar P-values (Table 4). However, even when considering the intensity ratio of these peaks together with their chemical shifts, none of the available metabolite databases were able to provide an assignment. Taken together, these results dramatically highlight the need for greatly expanded metabolite databases that can provide more extensive coverage of the mammalian metabolome. Clearly there are a number of highly significant metabolic differences between miniature Schnauzers and Labrador retrievers; for example a 4.35-fold higher concentration of an unidentified metabolite at 0.95 ppm in miniature Schnauzers (P = 0.0006) and a 2.58-fold higher concentration of an unidentified metabolite at 2.80 ppm in Labrador retrievers (P = 0.0004). Considering the relatively small number of dogs employed in this study, these highly significant metabolic discriminators between breeds indicate that the affect of breed may have significant impacts on their biology. Only when improved NMR databases of metabolites become available will a more in-depth interpretation of these biochemical differences between breeds become possible.
3.4 Classification model for individual dogs
A second PLS model was built using the pJRES pre-processed spectra of the 37 known urine samples (Tables 1 and 2), where class assignments were based on individual dogs. This dataset comprised of 5, 6, 6, 5, 5, 5 and 5 pJRES spectra from seven different animals. The optimised model was constructed of 6 LVs based upon the minimization of the root mean square error of cross validation. Only sample 37 in the training set exceeded the 99% confidence limit for the Q-residual and therefore was removed (this sample was determined to be an outlier from the earlier PCA results). The PLS scores plot for LV1 versus LV2 is shown in Fig. 5, which shows that the NMR fingerprints of each of the seven individual dogs tend to occupy different regions of metabolic space. Subsequently, the identities of the dogs associated with the seven unknown urine samples were predicted using DA with Bayesian-determined thresholds (as described above). The predicted individuals for the seven unknown samples are summarised in Table 3. For five out of seven of the unknowns the model predicted that each urine sample was associated with a specific dog. For one of the unknowns (sample 40) it predicted that it could arise from either of two dogs, although one of these predictions was only just above the threshold. We therefore assigned the urine sample to the more likely animal that was significantly above the threshold (LR1). For the one remaining unknown (sample 42), the model predicted that it did not arise from any of the seven dogs. However, one of these predictions was only just below the threshold and so we assigned that urine sample to the most likely animal (MS3). These final results were then communicated to the WALTHAM Centre for Pet Nutrition who confirmed that seven out of seven of these predictions were correct (Table 3). Considering the results from the entire study, this confirms that NMR fingerprinting of urine followed by supervised multivariate analysis is capable of identifying both breed and individual dogs. This therefore suggests that a single urine sample (rather than a temporal series of samples) may be considered representative of an individual. Furthermore, in the future this approach may allow us to identify breed-specific standard ranges for specific metabolites in urine and to study the physiological implications of these metabolite levels in urine.

PLS scores plot from analysis of the pre-processed pJRES NMR spectra of 37 known urine samples (1 outlier removed), where individual dogs were used for the class descriptors. Dotted lines have been drawn around the clusters for each Labrador retriever (LR1 (△), LR2 (□), LR3 (○)) and solid lines around each miniature Schnauzer (MS1 (✱), MS2 (▴), MS3 (■), MS4 (•)). The seven unknown urine samples are labelled 38 to 44 and were not included in the training of the PLS model
4 Concluding Remarks
We believe that this study represents one of the first NMR-based metabolomics investigations of canine urine samples. Whilst it has not employed large numbers of dogs, but instead is a proof of principle, there are a number of comments and conclusions that can be made. Our results suggest that the non-invasive collection (using “free catch”) of urine and its NMR metabolic fingerprint is a potentially valid approach to undertake biologically relevant studies in dogs. Furthermore, we have determined that breed may be a major determinant of urine metabolic fingerprints in healthy dogs. Our results also suggest that a spot sample may be representative of an individual, and so there appears to be no need for whole day collections and repeated samplings, the latter assuming that an appropriate multivariate model with suitable replication has already been built. It is important to realise, however, that the dogs in this study are pure-bred dogs living in the same environment, with the most consistent and best care possible. As such these dogs are clearly representative of healthy individuals and environmental noise will be relatively low compared to privately owned dogs. In this regard it is not necessarily surprising to find that dog breeds are able to be distinguished from each other, but the fact that in this dataset we also see separation between individual animals indicates that the urinary profile may be particularly information-rich. To develop a better understanding of what factors need to be controlled when designing and interpreting experiments using a metabolomics approach we require further work to investigate the importance of other factors such as diet, age and gender, as well as extending the number and diversity of breeds screened. With this information a more accurate development of specific nutritional care for an individual or breed will become possible.
References
Aranibar, N., Ott, K. H., Roongta, V., & Mueller, L. (2006). Metabolomic analysis using optimized NMR and statistical methods. Analytical Biochemistry, 355, 62–70.
Bijlsma, S., Bobeldijk, L., Verheij, E. R., Ramaker, R., Kochhar, S., Macdonald, I. A., van Ommen, B., & Smilde, A. K. (2006). Large-scale human metabolomics studies: A strategy for data (pre-) processing and validation. Analytical Chemistry, 78, 567–574.
Brindle, J. T., Antti, H., Holmes, E., Tranter, G., Nicholson, J. K., Bethell, H. W. L., Clarke, S., Schofield, P. M., McKilligin, E., Mosedale, D. E., & Grainger, D. J. (2002). Rapid and non-invasive diagnosis of the presence and severity of coronary heart disease using H-1 NMR-based metabonomics. Natural Medicine, 8, 1439–1444.
Clutton-Brock, J. (1999). A natural history of domesticated mammals (2nd ed.). UK: Cambridge University Press.
Coen, M., O’Sullivan, M., Bubb, W. A., Kuchel, P. W., & Sorrell, T. (2005). Proton nuclear magnetic resonance-based metabonomics for rapid diagnosis of meningitis and ventriculitis. Clinical Infectious Diseases, 41, 1582–1590.
Dumas, M. E., Maibaum, E. C., Teague, C., Ueshima, H., Zhou, B., Lindon, J. C., Nicholson, J. K., Stamler, J., Elliott, P., Chan, Q., & Holmes, E. (2006). Assessment of analytical reproducibility of (1)H NMR spectroscopy based metabonomics for large-scale epidemiological research: The INTERMAP study. Analytical Chemistry, 78, 2199–2208.
Eriksson, L., Johansson, E., Kettaneh-Wold, N., & Wold, S. (2001). Multi- and megavariate data analysis—principles and applications In L. Eriksson, E. Johansson, N. Kettaneh-Wold, & S. Wold (Eds.), Multi- and megavariate data analysis—principles and applications, Sweden: Umetrics, Umea.
Gunther, U. L., Ludwig, C., & Ruterjans, H. (2000). NMRLAB—advanced NMR data processing in MATLAB. Journal of Magnetic Resonance, 145, 201–208.
Hines, A., Oladiran, G. S., Bignell, J. P., Stentiford, G. D., & Viant, M. R. (2007). Direct sampling of organisms from the field and knowledge of their phenotype: Key recommendations for environmental metabolomics. Environmental Science & Technology 41, 3375–3381.
Hwang, T. L., & Shaka, A. J. (1995). Water suppression that works—excitation sculpting using arbitrary wave-forms and pulsed-field gradients. Journal of Magnetic Resonance Series A, 112, 275–279.
Keun, H. C., Beckonert, O., Griffin, J. L., Richter, C., Moskau, D., Lindon, J. C., & Nicholson, J. K. (2002). Cryogenic probe C-13 NMR spectroscopy of urine for metabonomic studies. Analytical Chemistry, 74, 4588–4593.
Lauridsen, M., Hansen, S. H., Jaroszewski, J. W., & Cornett, C. (2007). Human urine as test material in H-1 NMR-based metabonomics: Recommendations for sample preparation and storage. Analytical Chemistry, 79, 1181–1186.
Lee, G. C., & Woodruff, D. L. (2004). Beam search for peak alignment of NMR signals. Analytica Chimica Acta, 513, 413–416.
Lin, C. Y., Viant, M. R., & Tjeerdema, R. S. (2006). Metabolomics: Methodologies and applications in the environmental sciences. Journal of Pesticide Science, 31, 245–251.
Markley, J. L., Anderson, M. E., Cui, Q., Eghbalnia, H. R., Lewis, I. A., Hegeman, A. D., Li, J., Schulte, C. F., Sussman, M. R., Westler, W. M., Ulrich, E. L., & Zolnai, Z. (2007). New bioinformatics resources for metabolomics. Pacific Symposium on Biocomputing, 12, 157–168.
Matsumoto, M., Zhang, C. H., Kosugi, C., & Matsumoto, I. (1995). Gas-chromatography mass-spectrometric studies of canine urinary metabolism. Journal of Veterinary Medical Science, 57, 205–211.
Nicholson, J. K., Connelly, J., Lindon, J. C., & Holmes, E. (2002). Metabonomics: A platform for studying drug toxicity and gene function. Nature Review Drug Discovery, 1, 153–161.
Parsons, H. M., Ludwig, C., Günther U. L., & Viant, M. R. (2007). Improved classification accuracy in 1- and 2-dimensional NMR metabolomics data using the variance stabilising generalized logarithm transformation. BMC Bioinformatics, 8, 234.
Patterson, D. F. (2000). Companion animal medicine in the age of medical genetics. Journal of Veterinary Internal Medicine, 14, 1–9.
Pelczer, I. (2005). High-resolution NMR for metabomics. Current Opinion in Drug Discovery & Development, 8, 127–133.
Plumb, R. S., Granger, J. H., Stumpf, C. L., Johnson, K. A., Smith, B. W., Gaulitz, S., Wilson, I. D., & Castro-Perez, J. (2005). A rapid screening approach to metabonomics using UPLC and oa-TOF mass spectrometry: Application to age, gender and diurnal variation in normal/Zucker obese rats and black, white and nude mice. Analyst, 130, 844–849.
Purohit, P. V., Rocke, D. M., Viant, M. R., & Woodruff, D. L. (2004). Discrimination models using variance stabilizing transformation of metabolomic NMR data. OMICS, 8, 118–130.
Rosenblum, E. S., Tjeerdema, R. S., & Viant, M. R. (2006). Effects of temperature on host-pathogen-drug interactions in red abalone, haliotis rufescens, determined by 1H NMR metabolomics. Environmental Science & Technology, 40, 7077–7084.
Savolainen, P., Zhang, Y. P., Luo, J., Lundeberg, J., & Leitner, T. (2002). Genetic evidence for an East Asian origin of domestic dogs. Science, 298, 1610–1613.
Shaka, A. J., Lee, C. J., & Pines, A. (1988). Iterative schemes for bilinear operators—application to spin decoupling. Journal of Magnetic Resonance, 77, 274–293.
Shi, H. L., Vigneau-Callahan, K. E., Shestopalov, A. I., Milbury, P. E., Matson, W. R., & Kristal, B. S. (2002a). Characterization of diet-dependent metabolic serotypes: Primary validation of male and female serotypes in independent cohorts of rats. Journal of Nutrition, 132, 1039–1046.
Shi, H. L., Vigneau-Callahan, K. E., Shestopalov, A. I., Millbury, P. E., Matson, W. R., & Kristal, B. S. (2002b). Characterization of diet-dependent metabolic serotypes: Proof of principle in female and male rats. Journal of Nutrition, 132, 1031–1038.
Stanley, E. G., Bailey, N. J. C., Bollard, M. E., Haselden, J. N., Waterfield, C. J., Holmes, E., & Nicholson, J. K. (2005). Sexual dimorphism in urinary metabolite profiles of Han Wistar rats revealed by nuclear-magnetic-resonance-based metabonomics. Analytical Biochemistry, 343, 195–202.
Strasinger, S. K., & Di Lorenzo, M. S. (2001). Urinalysis and body fluids (4th ed.). Philadelphia: F.A. Davis Company.
Viant, M. R. (2003). Improved methods for the acquisition and interpretation of NMR metabolomic data. Biochemical and Biophysical Research Communications, 310, 943–948.
Viant, M. R., Rosenblum, E. S., & Tjeerdema, R. S. (2003). NMR-based metabolomics: A powerful approach for characterizing the effects of environmental stressors on organism health. Environmental Science & Technology, 37, 4982–4989.
Vigneau-Callahan, K. E., Shestopalov, A. I., Milbury, P. E., Matson, W. R., & Kristal, B. S. (2001). Characterization of diet-dependent metabolic serotypes: Analytical and biological variability issues in rats. Journal of Nutrition, 131, 924S-932S.
Walsh, M. C., Brennan, L., Malthouse, J. P. G., Roche, H. M., & Gibney, M. J. (2006). Effect of acute dietary standardization on the urinary, plasma, and salivary metabolomic profiles of healthy humans. American Journal of Clinical Nutrition, 84, 531–539.
Wang, Y. L., Lawler, D., Larson, B., Ramadan, Z., Kochhar, S., Holmes, E., & Nicholson, J. K. (2007). Metabonomic investigations of aging and caloric restriction in a life-long dog study. Journal of Proteome Research, 6, 1846–1854.
Whitfield, P. D., Noble, P.-J. M., Major, H., Beynon, R. J., Burrow, R., Freeman, A. I., & German, A. J. (2005). Metabolomics as a diagnostic tool for hepatology: validation in a naturally occurring canine model. Metabolomics, 1, 215–225.
Wise, B. M., Gallagher, N. B., Bro, R., Shaver, J. M., Windig, W., & Koch, R. S. (2004). PLS_Toolbox version 3.5 manual. In B. M. Wise, N. B. Gallagher, R. Bro, J. M. Shaver, W. Windig, & R. S. Koch (Eds.), PLS_Toolbox version 3.5 manual, Manson, US: Eigenvector Research.
Wishart, D. S., Tzur, D., Knox, C., Eisner, R., Guo, A. C., Young, N., Cheng, D., Jewell, K., Arndt, D., Sawhney, S., Fung, C., Nikolai, L., Lewis, M., Coutouly, M. A., Forsythe, I., Tang, P., Shrivastava, S., Jeroncic, K., Stothard, P., Amegbey, G., Block, D., Hau, D. D., Wagner, J., Miniaci, J., Clements, M., Gebremedhin, M., Guo, N., Zhang, Y., Duggan, G. E., MacInnis, G. D., Weljie, A. M., Dowlatabadi, R., Bamforth, F., Clive, D., Greiner, R., Li, L., Marrie, T., Sykes, B. D., Vogel, H. J., & Querengesser, L. (2007). HMDB: The human metabolome database. Nucleic Acids Research, 35, D521–D526.
Acknowledgments
The authors are indebted to Chenomx Inc. for use of their NMR metabolomics software, and to all pet carers and other staff who helped in the collection of urine at the WALTHAM Centre for Pet Nutrition. Financial support: This work was performed at the Henry Wellcome Building for Biomolecular NMR Spectroscopy (HWB•NMR) and was supported by the Wellcome Trust.
Author information
Authors and Affiliations
Corresponding author
Additional information
An erratum to this article is available at http://dx.doi.org/10.1007/s11306-009-0172-4.
Rights and permissions
About this article
Cite this article
Viant, M.R., Ludwig, C., Rhodes, S. et al. Validation of a urine metabolome fingerprint in dog for phenotypic classification. Metabolomics 3, 453–463 (2007). https://doi.org/10.1007/s11306-007-0092-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11306-007-0092-0