
Abstract
The distribution of genetic diversity among natural populations is significantly shaped by geographical and environmental heterogeneity. The key objectives of this study were to outline the population genetic structure and to investigate the effects of historical and current factors in shaping the population structure of an endemic tropical tree, Hagenia abyssinica. We used 11 polymorphic microsatellites to estimate genetic variability and evaluate gene flow among natural populations of H. abyssinica. Further, we employed ecological niche modeling approaches, to analyze the demographic history and map potential distributions of H. abyssinica during the Last Glacial Maximum and the present. Significant levels of genetic diversity (H O = 0.477, H E = 0.439) were observed among the sampled locations. High coefficient of genetic differentiation (F ST = 0.32) and considerable genetic variation within the sampled locations (68.01%) were detected. Our results indicated the existence of three genetic groups with limited gene exchange and revealed positive correlations (r = 0.425, P < 0.05) between genetic diversity and geo-graphic distance. The ecological niche modeling (ENM) results support the existence of three distribution zones during the Last Glacial Maximum (LGM), with high probability of occurrence (0.8–1.0), and indicated slight distribution disturbances during and after the LGM. The fundamental patterns of genetic diversity and population structuring of H. abyssinica result from a combination of both environmental and geographical factors, including long-term isolation by distance and characteristic life history of this species. Our ENM results identified three zones that could have served as glacial refugia for this species and lay a foundation for further studies, outlining demographic histories and population structures of Afromontane species.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The outstanding geographic features and the diversified climate of the tropical eastern Africa have resulted in a biodiversity hotspot. The pattern of species distributions currently observed in this region results from a combination of historical factors, and the high level (80%) of species endemism observed on the tropical mountains of eastern Africa indicates that these plants have been in isolation for a long period (Hedberg 1969; Assefa et al. 2007; Linder 2014). Besides geographical isolation, environmental diversity in these regions had considerably influenced the distribution of both plant and animal species. Surprisingly, there is a significant similarity in biodiversity on different mountain massifs (Hall 1973; Kadu et al. 2013), suggesting the existence of historical links that connected some of the populations, probably during the Pleistocene era when climatic changes favored species dispersal across existing patches of flora, and the subsequent extinction of these patches as they sought refuge in the mountains (Hewitt 1999; White 1981). Presently, most of the species inhabiting these mountains such as Hagenia abyssinica (Bruce) J. F. Gmel, (Rosaceae), display local endemism and exist in isolated population patches.
Several studies have been conducted on widespread and rare endemic species of montane forests of Ethiopian highlands and the tropical mountains of eastern Africa, e.g., Arabis alpina (Koch et al. 2006; Ehrich et al. 2007; Assefa et al. 2007), Lobelia giberroa (Kabede et al. 2007), Prunus africana (Kadu et al. 2011, 2013; Mihretie et al. 2015), and Erica spp. (Gizaw et al. 2013). Among the remarkable findings of these studies are the effects of past climate and environmental heterogeneity on species genetic diversity, phylogeographic and recolonization histories, and implication on genetic conservation. During the Pleistocene epoch, climate fluctuations caused expansion and contraction of the montane forests, eventually resulting into more fragmented montane forests (Kabede et al. 2007). Recently, the incorporation of ecological niche models (ENM) in phylogeographic and genetic structure studies to map the past and current and to predict the future geographic distribution of species has led to better understanding of evolutionary history and the role played by combined historical processes in shaping genetic structure in present populations. ENM is based on interactions between biotic and abiotic factors within specified known habitats of a given species (Guisan and Zimmermann 2000; Peterson 2003a).
In Africa, only a few studies have applied ENM to investigate the influence of climate fluctuations on species distribution, e.g., in Vitellaria paradoxa (Allal et al. 2011) and in Chasmanthera dependens Hochst (Iloh and Ogundipe 2016), highlighting probable refugial areas and potential future distribution patterns for species in West Africa. However, despite eastern Africa being a diversity hotspot and a potential Quaternary glacial refugia, ecological niche models are yet to be conducted on species inhabiting the “sky islands.” Besides climate changes, anthropogenic activities (e.g., expansion of agricultural lands) caused by tremendous increase in human population over the last century have also had an impact on the patterns of vegetation distribution. Habitat loss led to rapid decline in population sizes, prompting Hedberg (1969) to observe that human impact caused disjunction of east African montane forests on Mt. Elgon, Cherang’ani hills, Aberdare Ranges, and Mt. Kenya.
H. abyssinica, a long-lived tree species in a monotypic genus, is found in most Afromontane forests, from the northern parts of Ethiopia and Sudan to the southern regions of Malawi and Zambia, displaying a disjunct distribution (Hedberg 1969). This species is dioecious, anemogamous, and anemochorous (Negash 1995). Its medicinal benefits, mainly anthelmintic, have been relied on for decades by most African communities for treating gastrointestinal parasites in both man and livestock (Woldemariam et al. 1992; Scantlebury et al. 2013; Feyssa et al. 2015). Other uses include source of fuel and timber for construction (Feyissa et al. 2007). H. abyssinica is ecologically linked to a rich biodiversity with a record of stable coexistence with other Afromontane flora (Seburanga et al. 2014). However, overexploitation and habitat destruction have led to reduction in its population sizes and is now classified as an endangered species in Ethiopia and Rwanda (Negash 1995; Feyissa et al. 2007; Seburanga et al. 2014). It is endemic to Afromontane forests, some of which are separated by large plains and grasslands, with several being on the opposite sides of the Great Rift Valley. Therefore, it is an interesting species to study the patterns of gene flow and population history.
Previously, H. abyssinica has been studied in effort to outline its genetic diversity, using randomly amplified polymorphic DNA (RAPD) (Kumilign 2005), inter-simple sequence repeats (ISSRs) (Feyissa et al. 2007), chloroplast microsatellites (cpSSR) (Ayele et al. 2009), and amplified fragment length polymorphism (AFLP) (Ayele et al. 2011). However, these studies focused only on the Ethiopian highlands. The current study sampled 11 natural populations of H. abyssinica distributed in Afromontane forests in Ethiopian highlands and tropical mountains of east Africa. The principal objectives were to quantify levels of gene flow and current genetic diversity and to outline the population structure of natural populations of H. abyssinica across the region using microsatellite markers. We estimated historical and current factors that shape population structure, e.g., correlation between genetic and geographic distances, and used ENM to map the potential distribution zones of Hagenia during the LGM and present. Information on population genetic patterns of this species lays a foundation for further studies on endemic species and deepens our understanding on the roles of historical dynamics on species distribution and dispersal processes.
Materials and methods
Sampling
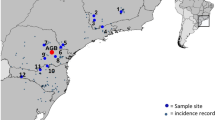
H. abyssinica has a disjunct distribution in the scattered high mountains of East Africa (Hedberg 1969). Our focus was on Ethiopian highlands and eastern Africa (Kenya, Tanzania, and Uganda), where anthropogenic activities (farming and logging) have negatively impacted on the populations. We collected samples from fragmented natural populations on major mountains where this species is known to grow at a narrow distribution range. Leaf samples were obtained from 168 individuals in 11 natural populations (Fig. 1, Table 1). Individuals in each population were sampled at a distance of between 100 and 250 m depending on the size of the population. Due to inaccessibility of some individuals in certain populations, our sample sizes varied among populations. A voucher specimen was collected from each sampling site and deposited at the Herbarium of Wuhan Botanical Garden, CAS (HIB). Young leaves were put in manila envelopes, dried in silica gel, and stored at −20 °C until DNA was extracted.

A map showing 11 sampling sites of Hagenia abyssinica. The sites are represented by circles while the colors represent the genetic groups to which individuals were assigned to based on the STRUCTURE results (K = 3)
DNA extraction and SSR genotyping
Total genomic DNA was extracted using the MagicMag Genomic DNA Micro Kit (Sangon Biotech Co., Shanghai, China) following the manufacturer’s instructions. Eleven species-specific SSR markers (Gichira et al. 2016) were used to genotype the 168 samples. The forward primers were labeled with 6-FAM fluorescent dye on the 5′ end. The 25 μL PCR mixture contained 2 μL 50 ng genomic DNA, 2.5 μL 10× Taq buffer (plus Mg2+), 0.25 mM of each dNTP, 0.25 μM of each primer, 0.2 U Taq polymerase (TaKaRa Bio, Dalian, China), and PCR water. PCR amplification was conducted using T100™ Thermal Cycler (Bio-Rad, CA, USA) with the following cycling conditions: an initial step of 4 min at 95 °C, followed by 30 cycles of 30 s at 95 °C, 30 s at 53–58 °C, 30 s at 72 °C, and a final extension step for 10 min at 72 °C. The quality of PCR products was checked on 2.0% agarose and then separated using an ABI 3730 XL automated sequencer (TsingKe Biotech, Beijing, China). Alleles were called using GeneMarker 2.2.0 system (Soft Genetics).
Data analyses
Genetic diversity
Micro-Checker 2.2.3 (Van Oosterhout et al. 2004) was used to detect null alleles. The polymorphic information content (PIC) and deviations from the Hardy-Weinberg equilibrium (HWE) with the Bonferroni correction for each locus were checked in Cervus 3.0 (Kalinowski et al. 2007). GenePop 4.0 (Rousset 2008) was used to check for deviations from the HWE at population level. Genetic diversity details including observed heterozygosity (H O), expected heterozygosity (H E), the number of private alleles(A P), observed alleles (N a), effective number of alleles (N e), Shannon’s diversity index (I), and inbreeding coefficient(F IS) were done in GeneAlex 6.5 (Peakall and Smouse 2012).
Population structure
Global genetic differentiation coefficient (F ST) and the analysis of molecular variance (AMOVA) were conducted in Arlequin 3.5 (Excoffier and Lischer 2010). The principal coordinate analysis (PCoA) and pairwise genetic matrix were done using GeneAlex 6.5 (Peakall and Smouse 2012). A Mantel’s test for isolation by distance was conducted using the IBDWS 3.23 (Jensen et al. 2005), a web-based program using Nei’s genetic distance (Nei 1972) and geographic distance (km) with 999 permutations.
Population structure and individual-based differentiation analyses were carried out using the Bayesian clustering method implemented by STRUCTURE 2.3.3 (Pritchard et al. 2000). STRUCTURE was run using the admixture model, assuming uncorrelated allele frequencies (independent) and without prior information about the populations. Preliminary simulations for K ranged from 1 to 10, each with 10 iterations. One hundred thousand replicates were used for the Markov chain Monte Carlo with a burn-in period of 50,000. Population clusters (K) were calculated using Structure Harvester (Earl 2012), based on ∆K statistics (Evanno et al. 2005).
Gene flow estimation
We used the Bayesian inference strategy with thermodynamic integration implemented in MIGRATE-n program, which uses coalescent and Markov chain Monte Carlo (MCMC) (Beerli 2006) to estimate the rates of gene flow between the three clusters outlined by STRUCTURE. In this analysis, we applied the Brownian approximation model assuming a constant mutation rate across all loci and turned on MCMC static heating in four chains, with no swapping. The different temperatures were set at 1, 1.5, 3, and 104 while the initial parameters for θ and M were generated by the program’s inbuilt F ST calculations (Beerli and Felsenstein 1999). The analysis was set to run in a single long chain with 1,000,000 burn-in, 500,000 sampled steps, and 5000 recorded steps. MIGRATE-n calculates the mutation-scaled effective population size Θ (4N e μ; N e is the effective population size and μ is the mutation rate per locus in a generation), and the mutation-scaled migration rate M (m/μ; m is the immigration rate among populations in a generation) (Beerli and Palczewski 2010). To get the actual effective number of migrants (Nm) per generation, the mutation-scaled migration rate (M) is multiplied with the mutation-scaled effective population size (Θ) and then divided by four (Nm j→i = [Θ i × M j→i]/4).
Environmental data
Nineteen bioclimatic variables (BIOCLIM) were obtained from the WorldClim database (http://www.worldclim.org), derived from annual and monthly mean temperatures and precipitation for the period between 1950 and 2000 (Hijmans et al. 2005). The 19 environmental variables were subjected to a jackknife analysis in MAXTENT, in order to account for correlation among data and to select variables that showed considerable influence on the distribution of the species. Eventually, the following five environmental variables: temperature seasonality (Bio4), temperature annual range (Bio7), isothermality (Bio3), mean temperature of the wettest quarter (Bio8), and annual precipitation (Bio12) were selected for the present and Last Glacial Maximum (LGM, −21 ka, MIROC 3.2) analysis at 2.5 arcmin resolutions. The occurrence data of H. abyssinica was obtained during our field survey, from Ayele et al. (2009) and from the potential natural vegetation (PNV) map (www.vegetationmap4africa.org; van Breugel et al. 2015). In total, 109 presence points (Online Resource 1) were recorded from the northern parts of Ethiopia to the southern parts of Africa in Zambia and Malawi.
Ecological niche modeling
ENM approaches require continuous layers of environmental variables and geographic positioning of species occurrence data (Stockwell 2006). Based on these bioclimatic variables and the obtained presence data, ENM was conducted in MaxEnt program 3.3.3 (Phillips et al. 2006), a species distribution modeling tool which has been credited on its simplicity in use and ability to analyze data from relatively small sample sizes (Wisz et al. 2008), resulting in high model accuracy (Merrow et al. 2013). The parameters were set at default with 500 iterations without duplicates. We selected the random seed option and used 70% of the data in training and 30% in model testing.
MaxEnt output is in the form of a cumulative prediction map that is interpreted based on receiver-operator curve (AUC). AUC is an independent measure used to assess model suitability and it ranges from 0.5, interpreted as random accuracy, to 1.0, perfect discrimination (Elith et al. 2006). ENM models with AUC values >0.7 are considered to be strong and ideal for the data. The predictions were mapped using DIVA-GIS 7.5 (http://www.diva-gis.org).
Results
Genetic diversity
Null alleles were detected at loci Hgssr1, Hgssr5, Hgssr6, and Hgssr10 in some populations. The average PIC was 0.601. Except for Hgssr 3 and Hgssr 11, significant deviations from the HWE were observed at all the other loci (P < 0.001) (Table 2). However, at the population level, only two populations significantly deviated from the HWE. In total, 71 alleles were recorded at all the 11 microsatellite loci. The number of alleles observed per locus (N a) varied from 2 in Hgssr5 and Hgssr10 to 13 in Hgssr6 and Hgssr8 (average = 6.5); values of gene diversity per locus were recorded in Table 2. The mean observed heterozygosity (H O) and mean expected heterozygosity (H E) were estimated at 0.477 and 0.439, respectively. Details of genetic diversity including the number of alleles observed (N a), effective number of alleles (N e), Shannon’s diversity index (I), and inbreeding coefficient (F IS) per population are shown in Table 1. Nine private alleles were discovered in five populations at varying frequencies. The Simiens population had the highest number of private alleles (4) - three at locus Hgssr8, with frequencies of 0.038, 0.154, and 0.077, and one allele at locus Hgssr6, with a frequency of 0.077. Two private alleles were found in the Elgon population at loci Hgssr2 and Hgssr8.
Population structure and gene flow
Hierarchical AMOVA revealed strong population structure (F ST = 0.32) and there was limited inbreeding among populations (Online Resource file 2 Table S1). PCoA shows clear differentiation among geographic regions along the first three axes accounting for 69.89% of the variation (the scatter plot of coordinates 1 and 2 is shown in Fig. 2). The pairwise F ST values between pairs of the 11 analyzed populations ranged from 0.024 (BLI and BLII) to 0.379 (MK1 and RW). Generally, Ethiopian populations appeared to be less differentiated, with the least F ST, while Rwenzori (RW) was more differentiated against most of the other populations (Table 3). Bayesian clustering, based on ∆K statistics, defined three genetic clusters (K = 3) of H. abyssinica, with few individuals displaying mixed ancestry. This clustering placed RW-BLI-BLII-SM sample sites in one cluster (P1), KM-MR in a second cluster (P2), and CH-EL-AB-MKI-MKII in a third cluster (P3) (Fig. 3, Online Resource 2; S2, Fig. S2.1).

A scatter plot of the first two PCoA axes showing the relationship of the 168 individuals from 11 populations of Hagenia abyssinica. The first and the second coordinates account for 30.41 and 22.69% of the variation, respectively

Population genetic structure of Hagenia abyssinica, showing the proportion of membership of the 168 individuals in each of the three inferred clusters based on Bayesian probabilities. PI (red) had populations BLI-BLII-SM-RW, PII (green) KM-MR, and PIII had populations AB-MK I-MKII-CH-EL
Mantel’s test revealed significant correlations between Nei’s genetic distance and geographic distances (km) r = 0.425, P < 0.05 (Fig. 4). We estimated the mutation-scaled migration rate (M) as well as the mutation-scaled effective population size (Θ) for the three genetic clusters, based on an unconstrained migration model which allowed genes to flow towards any direction. The values of M and Θ were used to calculate the effective number of migrants (Nm) per generation. Θ values for genetic clusters I, II, and III were estimated at 0.0985 (97.5%, confidence interval 0.0958–0.1000), 0.0965 (0.0836–0.1000), and 0.0152 (0.0075–0.0195), respectively. Accordingly, the migration rates between the genetic clusters in each generation were recorded as follows: P2–1 = 0.5, P3–1 = 0.9, P1–2 = 0.4, P3–2 = 0.6, P1–3 = 0.4, and P2–3 = 0.1 (Online Resource 2; Table S3, Fig. S2.2).

Scatterplot showing positive correlation between the geographic distance (km) and Nei’s genetic distance (Mantel test P < 0.05)
Past and present potential distribution of H. abyssinica
Jackknife test results indicated that “temperature seasonality” (Bio4) had the highest gain and therefore contributed most of the information to the ENM. The environmental variable “annual precipitation” (Bio12) had the most unique information and, therefore, eliminating it from the analysis resulted in a reduction in the model’s gain. The model’s evaluation test displayed high efficiency and robustness based on the algorithm’s values (AUC 0.992).
MAXTENT proved the suitability of the used occurrence zones for H. abyssinica, whose predicted geographical distribution is consistent with the currently known habitation zones. Based on the past climate (LGM) model, high species density was observed in the eastern African region (Fig. 5a). This region is dominated by some of the highest and largest mountains in Africa and represents our sampling sites for population genetic study. There were slight but noteworthy changes on species distribution in some areas, specifically the southern regions of eastern Africa, which were unfit for Hagenia during the LGM but became suitable under the current climate models (Fig. 5b).

The potential geographical distribution zones of Hagenia abyssinica in the past (a) and present (b) climates, based on ecological niche modeling using MaxEnt. Red and orange colors indicate zones with high probability of occurrence
Discussion
Gene diversity (H E) varied among collection sites with the highest observed in Meru (0.536) and the lowest in Rwenzori (0.316), revealing variation in levels of diversity in populations on different geographic regions. Despite the recent reduction and the highly fragmented populations, H. abyssinica has managed to maintain a significant level of genetic diversity (H E = 0.439 and H O = 0.477) in our area of study. This discovery is contrary to what was expected; while populations of this species have had a history of isolation and its populations are greatly fragmented, it is probable that the effects of genetic drift and excessive inbreeding could have negatively affected the genetic diversity of this species. Comparably, high levels of genetic diversity have been recorded in other species inhabiting the Afromontane forests in tropical Africa, e.g., in P. africana H E = 0.66 (Kadu et al. 2013), and generally in isolated Afromontane forest patches. This observation is in line with the Aguilar et al. (2008) report that negative effects of recent population reduction and habitat fragmentation on genetic diversity may not be felt on the current mature individuals but will be reflected in their future generations.
Although endemic species are ecologically restricted, they may tend to exhibit almost the same levels of genetic diversity as widespread species (Hamrick and Godt 1989). The low and negative inbreeding coefficient (F IS) values observed in most of the populations support the fact that H. abyssinica, being a dioecious and a long-lived species, is primarily an outcrossing species. Species exhibiting these life forms tend to display a higher mean of genetic diversity (Hamrick and Godt 1996). In general, most of the populations displayed heterozygote excess or deficit although only populations MR and RW deviated significantly (P < 0.05) under heterozygote deficit. Homozygosity excess could be a result of biased sampling or existence of null alleles which were notable in four of the 11 loci used.
High levels of within-population diversity (68.01%), compared to the variation expressed among the populations (31.99%), may be attributed to the life history of this species. It is a dioecious and predominantly outcrossing, long-lived, anemophillic, and anemochory tree species, all of which are characteristics that favor high genetic variation within populations (Aguinagalde et al. 2005). AMOVA results reflected a strong variation structure and were supported by a high mean value of genetic differentiation (F ST = 0.32) recorded among all collection sites. The genetic relationship among the 168 individuals was shown by the scatter PCoA. The first three axes accumulatively showed a total of 69.89%, with axis 1 accounting for 30.41% while the second axis accounted for 22.69%, supporting the existence of a strong population structure.
Genetic isolation by distance is a dynamic process taking place in space and time, altering the species’ genetic makeup (Epperson and Li 1996). The Mantel test revealed the existence of positive correlations between genetic and geographic distances among the sampled populations (r = 0.425), indicating that isolation by distance (IBD) has an effect on the genetic differentiation of H. abyssinica. Generally, genetic variation is influenced by natural processes such as natural selection, drift, hybridization, and gene flow. The larger the geographic distance between two populations, the higher the genetic differentiation due to restricted gene exchange. This is further accelerated by the presence of obstacles to gene dispersal, e.g., mountains, rivers, and lakes. IBD is widely accepted as one of the causes of population divergence.
STRUCTURE analysis revealed the existence of a significant genetic structure among the populations by clustering the 11 populations into three distinct groups. The mean admixture α was less than 1, indicating that an individual belonged to a specific population as defined by Falush et al. (2003). However, a few individuals displayed mixed ancestry. This mode of clustering, where populations from distant mountain massifs group together, is common in Afro-alpine species in this region (Wondimu et al. 2014; Gizaw et al. 2013). The life history of H. abyssinica and the existence of geographical barriers, e.g., long distances and high mountains, might explain the strong genetic differentiation and structure displayed by this species.
We further conducted analyses to determine the amount of gene exchange across the genetic clusters defined by STRUCTURE. Although each of the genetic group had a few admixed individuals, the values of gene flow (N m ) estimated by MIGRATE-n were not significantly different from 0 and the highest flow of migrants (0.9) was recorded from cluster III to cluster I followed by 0.6 from cluster III to cluster II. Cross-mountain dispersals in eastern African mountains are mainly facilitated by air (wind) and birds through seed ingestion or which may also be embedded on their feathers and in the mud on their feet (Hedberg 1970). Long distance dispersal is feasible over many kilometers and most plant species have seeds that are highly specialized for wind dispersal. H. abyssinica is a wind-dispersed (seeds and pollen) dioecious tree species and although there is reduced gene flow recorded, a fundamental pattern was observed in that dispersal appears to have occurred in the north-south direction and rarely in the opposite direction. Gene exchange has pronounced implications on genetic differentiation among populations. Greater levels of gene flow (N m > 1) reduce genetic diversity among populations and prevent genetic drift (Slatkin 1985). However, and as revealed by the relatively low gene flow level in this study, dispersal of both pollen and seeds by wind is ineffective among populations separated by large areas of land (Loveless and Hamrick 1984). Therefore, the displayed genetic structure of this species might be due to the existence of remnant populations that took refuge in these mountains during the Pleistocene era.
Using the jackknifing procedure, we realized that predictions of the distribution of H. abyssinica are strongly influenced by both temperature and precipitation variables. ENM revealed slight but notable changes in distribution of this species during the LGM and the present. For instance, during the LGM, the model predicted strong (0.8–1.0) occurrence of the species in the southwestern region although this appear to have presently shifted towards the southeastern zone. In addition, the species has experienced expansions towards the southern part of our study site where areas that were unsuitable during the LGM displayed a 0.2–0.4 probability of occurrence. The LGM was characterized by low temperatures and reduced precipitation (Wu et al. 2007). Therefore, it is possible that the increased dryness barred this species from spreading all over the lowlands and instead it inhabited only some areas (may be along riverbanks, swamps, and/or lakes); this could explain the sparse patches.
The ENM distribution results appear to be consistent with recorded palynological data which show that H. abyssinica existed in the southwestern parts (Burundi) of our study area from as early as 34,000 years before present (BP) (Bonnefille et al. 1995); in the southeastern parts (Mt. Kenya), it existed 33,350 years BP (Olago et al. 1999) while in the north (Ethiopian highlands), records suggest a recent occurrence (Lamb et al. 2004). These pollen records were, in most places, found at altitudes lower than where this species currently inhabits. Consequently, the patterns of population clustering observed in East Africa (MKI-MKII-AB-CH-EL) could be attributed to gradual migrations along the bottom of the GRV, probably during the ice ages when most high-altitude species survived at the low altitude (McCarty 2001; Gottelli et al. 2004), and before the anthropogenic effects when the Afromontane forests were more widespread (Hedberg 1969). Dispersal bridges could have been more effective for gradual dispersal of species between populations on close mountain massifs such as Mt. Kenya and the Aberdares. However, gene exchange between populations at long distances apart, such as Kilimanjaro-Meru (south of our study region) and Rwenzori (southwest), could have been a result of random dispersal events, e.g., by cyclones (Hedberg 1969).
Our findings strongly suggest that past climate oscillations during the interglacial and the LGM did not break down genetic structuring of H. abyssinica in Ethiopian highlands and eastern Africa. There appear to be three major distribution zones with a high probability of occurrence (0.8–1.0) of Hagenia during the LGM (Fig. 5a), that correspond to areas with major mountains in eastern Africa (Young and Hastenrath 1987). ENM results show that climate variations since the LGM have had little impact on the present distribution of this species on these mountains (Fig. 5); therefore, we hypothesize that these “sky islands” served as glacial refugial area for Hagenia. To further support this hypothesis, the results from our genetic differentiation analyses appeared to group H. abyssinica’s populations into three genetic clusters based on geographical regions which lie within the distribution zones that existed since the LGM. Furthermore, the results revealed high genetic connectivity among populations from the same distribution zone and strong genetic (high F ST) differentiation among distanced populations, as revealed by the matrix of pairwise F ST, which is a product of long-term isolation and restricted gene flow. The presence of private alleles in some of the sampled sites and the Mantel test suggested that differentiation and population structuring observed in H. abyssinica are results of long-term isolation facilitated by long distances and a complex topography.
Conclusion
Climate fluctuations, geographical complexity, and physical distances are the main drivers of genetic diversity in tree species (Gugger et al. 2013; e Souza et al. 2016). By integrating genetic diversity analysis with ecological niche modeling, we observed that the population structure of Hagenia results from a combination of geographical factors, isolation by distance, and characteristic life-forms of this species. Reduced/absence of gene flow limits the chances of gene exchange among populations and may also hinder the colonization of new suitable habitats in the event of further climate change. Currently, although H. abyssinica is yet to be assessed for the IUCN Red List (www.iucnredlist.org 29/09/2016), it is endangered in some regions and has been mentioned under the Red List of Endemic Trees (Vivero et al. 2005). Anthropogenic impingements, e.g., deforestation to create land for cultivation and illegal charcoal burning, are on the rise in eastern Africa and, therefore, it is expected that further loss of rare alleles will lead to reduction in genetic diversity. Consequently, appropriate conservation measures are needed to effectively conserve this species and preserve its natural habitats.
The precision and exactness of genetic diversity (heterozygosity) are mainly affected by polymorphism of individual loci (Hale et al. 2012). In addition, F ST values were observed to be the approximate determinants of genetic distances; when F ST values are >0.05, sampling 20 or less individuals is enough to estimate population genetic distances (Kalinowski 2005). Therefore, although the sample size used in this study was less in some populations and may not have adequately outlined the allele diversity of the populations, it was efficient to capture the population structure and differentiation in H. abyssinica. We also focused on a smaller region compared to the entire distribution zone of this species and thus, in order to outline a conclusive genetic structure of this endangered endemic tree, we propose further future studies with more samples [25–30 individuals per population are ideal to efficiently reveal the genetic diversity of a species (Hale et al. 2012)] from all regions where H. abyssinica is known to grow.
References
Aguilar R, Quesada M, Ashworth L, Herrerias‐Diego Y, Lobo J (2008) Genetic consequences of habitat fragmentation in plant populations: susceptible signals in plant traits and methodological approaches. Mol Ecol 17:5177–5188
Aguinagalde I, Hampe A, Mohanty A, MartínJP DJ, Petit RJ (2005) Effects of life‐history traits and species distribution on genetic structure at maternally inherited markers in European trees and shrubs. J Biogeogr 32:329–339
Allal F, Sanou H, Millet L, Vaillant A, Camus-Kulandaivelu L, Logossa Z, Lefevre F, Bouvet J (2011) Past climate changes explain the phylogeography of Vitellaria paradoxa over Africa. Heredity 107:174–186
Assefa A, Ehrich D, Taberlet P, Nemomissa S, Brochmann C (2007) Pleistocene colonization of afro-alpine ‘sky islands’ by the arctic-alpine Arabis alpina. Heredity 99:133–142
Ayele TB, Gailing O, Umer M, Finkeldey R (2009) Chloroplast DNA haplotype diversity and postglacial recolonization of Hagenia abyssinica (Bruce) J. F. Gmel. in Ethiopia. Plant Syst Evol 280:175–185
Ayele TB, Gailing O, Finkeldey R (2011) Assessment and integration of genetic, morphological and demographic variation in Hagenia abyssinica (Bruce) J. F. Gmel to guide its conservation. J Nat Conserv 19:8–17
Beerli P (2006) Comparison of Bayesian and maximum-likelihood inference of population genetic parameters. Bioinformatics 22:341–345
Beerli P, Felsenstein J (1999) Maximum-likelihood estimation of migration rates and effective population numbers in two populations using a coalescent approach. Genetics 152:763–773
Beerli P, Palczewski M (2010) Unified framework to evaluate panmixia and migration direction among multiple sampling locations. Genetics 185:313–326
Bonnefille R, Riollet G, Buchet G, Icole M, Lafont R, Arnold M, Jolly D (1995) Glacial anterglacial record from intertropical Africa, high resolution pollen and carbon data at Rusaka, Burundi. Quat Sci Rev 14:917–936
e Souza HAV, Collevatti RG, Lima-Ribeiro MS, De Lemos-Filho JP, Lovato MB (2016) A large historical refugium explains spatial patterns of genetic diversity in a Neotropical savanna tree species. Ann Bot, mcw096
Earl DA (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361
Ehrich D, Gaudeul M, Assefa A, Koch MA, Mummenhoff K, Nemomissa S, Consortium I, Brochmann C (2007) Genetic consequences of Pleistocene range shifts: contrast between the Arctic, the Alps and the East African mountains. Mol Ecol 16:2542–2559
Elith J, Graham CH, Anderson RP, Dudı’k M, Ferrier S, Guisan A, Hijmans RJ, Huettmann F, Leathwick JR et al (2006) Novel methods improve prediction of species’ distributions from occurrence data. Ecography 29:129–151
Epperson BK, Li TQ (1996) Measurement of genetic structure within populations using Moran’s spatial autocorrelation statistics. Proc Natl Acad Sci USA 93:10528–10532
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Excoffier L, Lischer HE (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567
Falush D, Stephens M, Pritchard JK (2003) Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164:1567–1587
Feyissa T, Nybom H, Bartish IV, Welander M (2007) Analysis of genetic diversity in the endangered tropical tree species Hagenia abyssinica using ISSR markers. Genet Resour Crop Evol 54:947–958
Feyssa DH, Abdeta C, Berhan T, Sharma M (2015) Medicinal plants use and conservation practices in Jimma Zone, South West Ethiopia. Int J Biodivers Conserv 7:202–210
Gichira AW, Long ZC, Hu GW, Gituru RW, Wang QF, Chen JM (2016) Development of microsatellite markers in Hagenia abyssinica (Bruce) J. F. Gmel, an endangered tropical tree of eastern Africa, using next-generation sequencing. Genetics and molecular research: GMR, 15
Gizaw A, Kebede M, Nemomissa S, Ehrich D, Bekele B, Mirré V, Popp M, Brochmann C (2013) Phylogeography of the heathers Erica arborea and E. trimera in the afro-alpine ‘sky islands’ inferred from AFLPs and plastid DNA sequences. Flora-Morphology, Distribution, Functional Ecology of Plants 208:453–463
Gottelli D, Marino J, Sillero‐Zubiri C, Funk SM (2004) The effect of the last glacial age on speciation and population genetic structure of the endangered Ethiopian wolf (Canis simensis). Mol Ecol 13:2275–2286
Gugger PF, Ikegami M, Sork VL (2013) Influence of late Quaternary climate change on present patterns of genetic variation in valley oak, Quercus lobata Née. Mol Ecol 22:3598–3612
Guisan A, Zimmermann NE (2000) Predictive habitat distribution models in ecology. Ecol Model 135:147–186
Hale ML, Burg TM, Steeves TE (2012) Sampling for microsatellite-based population genetic studies: 25–30 individuals per population is enough to accurately estimate allele frequencies. PLoS One 9:e45170
Hall JB (1973) Vegetational zones on the southern slopes of Mount Cameroon. Vegetation 27:49–69
Hamrick JL, Godt MJW (1989) Allozyme diversity in plant species. In: Brown AHD, Clegg MT, Kahler AL, Weir BS (eds) Plant population genetics, breeding and genetic resources. Sinauer Associates, Sunderland, MA, pp 43–63
Hamrick JL, Godt M (1996) Effects of life history traits on genetic diversity in plant species. Philos Trans R Soc Lond B 351:1291-1298
Hedberg O (1969) Evolution and speciation in a tropical high mountain flora. Biol J Linn Soc 1:135–148
Hedberg O (1970) Evolution of the afroalpine flora. Biotropica:16–23
Hewitt GM (1999) Post‐glacial re‐colonization of European biota. Biol J Linn Soc 68:87–112
Hijmans RJ, Cameron SE, Parra JL, Jones PG, Jarvis A (2005) Very high resolution interpolated climate surfaces for global land areas. Int J Climatol 25:1965–1978
Iloh AC, Ogundipe OT (2016) Using ecological niche modelsto plan conservation in a changing environment: a case for the plant Chasmanthera dependens Hochst (Menispermaceae) in West Africa. J Ecol Nat Environ 8:1–8
Jensen JL, Bohonak AJ, Kelley ST (2005) Isolation by distance, web service. BMC Genet 6:1
Kabede M, Ehrich D, Taberlet P, Nemomissa S, Brochmann C (2007) Phylogeography and conservation genetics of a giant lobelia (Lobelia giberroa) in Ethiopian and tropical East African mountains. Mol Ecol 16:1233–1243
Kadu C, Schueler S, Konrad H, Muluvi G, Eyog‐Matig O, Muchugi A, Williams V, Ramamonjisoa L, Kapinga C, Foahom B (2011) Phylogeography of the Afromontane Prunus africana reveals a former migration corridor between East and West African highlands. Mol Ecol 20:165–178
Kadu CA, Konrad H, Schueler S, Muluvi GM, Eyog-Matig O, Muchugi A, Williams VL, Ramamonjisoa L, Kapinga C, Foahom B (2013) Divergent pattern of nuclear genetic diversity across the range of the Afromontane Prunus africana mirrors variable climate of African highlands. Ann Bot 111:47–60
Kalinowski ST (2005) HP-RARE 1.0: a computer program for performing rarefaction on measures of allelic richness. Mol Ecol Notes 5:187–189
Kalinowski ST, Taper ML, Marshall TC (2007) Revising how the computer program CERVUS accommodates genotyping error increases success in paternity assignment. Mol Ecol 16:1099–1106
Koch MA, Kiefer C, Ehrich D, Vogel J, Brochmann C, Mummenhoff K (2006) Three times out of Asia Minor: the phylogeography of Arabis alpina L.(Brassicaceae). Mol Ecol 15:825–839
Kumilign A (2005) Estimation of sex-related genetic diversity of Hagenia abyssinica (Bruce) J.F. Gmel using random amplified polymorphic DNA (RAPD) markers. aau
Lamb AL, Leng MJ, Mohammed MU, Lamb HF (2004) Holocene climate and vegetation change in the Main Ethiopian Rift Valley, inferred from the composition (C/N and δ 13 C) of lacustrine organic matter. Quat Sci Rev 23:881–891
Linder HP (2014) The evolution of African plant diversity. Front Ecol Evol 2:1–14
Loveless MD, Hamrick JL (1984) Ecological determinants of genetic structure in plant populations. Annu Rev Ecol Syst 15:65–95
Mccarty JP (2001) Consecuencias Biológicas de Cambios Climáticos Recientes. Conserv Biol 15:320–331
Merrow C, Smith MJ, Silander JA (2013) A practical guide to MaxEnt for modeling species’ distributions: what it does, and why inputs and settings matter. Ecography 36:1058–1069
Mihretie Z, Schueler S, Konrad H, Bekele E, Geburek T (2015) Patterns of genetic diversity of Prunus africana in Ethiopia: hot spot but not point of origin for range-wide diversity. Tree Genet Genomes 11:1–13
Negash L. (1995) Indigenous trees of Ethiopia: biology, uses and propagation techniques. Addis Ababa: Department of Biology, Addis Ababa University 285p
Nei M (1972) Genetic distance between populations. Am Nat:283–292
Olago D, Street-Perrott F, Perrott R, Ivanovich M, Harkness D (1999) Late Quaternary glacial-interglacial cycle of climatic and environmental change on Mount Kenya, Kenya. J Afr Earth Sci 29:593–618
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research—an update. Bioinformatics 28:2537–2539
Peterson AT (2003a) Predicting the geography of species’ invasions via ecological niche modeling. Q Rev Biol 78:419–433
Phillips SJ, Anderson RP, Schapire RE (2006) Maximum entropy modeling of species geographic distributions. Ecol Model 190:231–259
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Rousset F (2008) genepop’007: a complete re‐implementation of the genepop software for Windows and Linux. Mol Ecol Resour 8:103–106
Scantlebury CE, Peachey L, Hodgkinson J, Matthews JB, Trawford A, Mulugeta G, Tefera G, Pinchbeck GL (2013) Participatory study of medicinal plants used in the control of gastrointestinal parasites in donkeys in eastern Shewa and Arsi zones of Oromia region, Ethiopia. BMC Vet Res 9:1
Seburanga J, Nsanzurwimo A, Folega F (2014) Status of Hagenia forest in the Parc National des Volcans, Rwanda: a review of historical records. J Ecol Nat Environ 6:170–182
Slatkin M (1985) Gene flow in natural populations. Annu Rev Ecol Syst:393–430
Stockwell D (2006) Niche modeling: predictions from statistical distributions, CRC Press
van Breugel P, Kindt R, Lillesø JPB, Bingham M, Demissew S, Dudley C, Friis I, Gachathi F, Kalema J, Mbago F, Moshi HN, Mulumba J, Namaganda M, Ndangalasi HJ, Ruffo CK, Védaste M, Jamnadass R, Graudal L (2015) Potential natural vegetation map of eastern Africa (Burundi, Ethiopia, Kenya, Malawi, Rwanda, Tanzania, Uganda and Zambia). Version 2.0. Forest & Landscape Denmark and World Agroforestry Centre (ICRAF). URL: http://vegetationmap4africa.org
Van Oosterhout C, Hutchinson WF, Wills DP, Shipley P (2004) MICRO‐CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538
Vivero JL, Demissew S, Kelbessa E (2005) The red list of endemic trees et shrubs of Ethiopia and Eritrea. Fauna et Flora International
White F (1981) The history of the Afromontane archipelago and the scientific need for its conservation. Afr J Ecol 19:33–54
Wisz MS, Hijmans R, Li J, Peterson AT, Graham C, Guisan A (2008) Effects of sample size on the performance of species distribution models. Divers Distrib 14:763–773
Woldemariam TZ, Fell AF, Linley PA, Bibby MC, Phillips RM (1992) Evaluation of the anti-tumour action and acute toxicity of kosins from Hagenia abyssinica. J Pharm Biomed Anal 10:555–560
Wondimu T, Gizaw A, Tusiime FM, Masao CA, Abdi AA, Gussarova G, Popp M, Nemomissa S, Brochmann C (2014) Crossing barriers in an extremely fragmented system: two case studies in the afro-alpine sky island flora. Plant Syst Evol 300:415–430
Wu H, Guiot J, Brewer S, Guo Z (2007) Climatic changes in Eurasia and Africa at the Last Glacial Maximum and mid-Holocene: reconstruction from pollen data using inverse vegetation modelling. Clim Dyn 29:211–229
Young JAT, Hastenrath S (1987) Glaciers of the Middle East and Africa - Glaciers of Africa, In: Williams RG. Ferrigno JG (ed), Satellite image atlas of glaciers of the world, U.S Government Printing Office 1386-G-3; U.S. Geological Survey, G49-G70
Acknowledgments
The authors acknowledge Geoffrey Mwachala and Daniel Miyawa of the National Museums of Kenya for their support in fieldwork, Zhicheng Long for his assistance in data analysis, Cornelius Mulili for proofreading, and Paul Gugger for his valuable comments and suggestions that improved an earlier version of this manuscript. This research was funded by the Sino-Africa Joint Research Center (Nos. Y323771W07 and SAJC201322).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Communicated by V. Decroocq
Data archiving declaration: The dataset of microsatellite loci used in this study is given in the Online Resource 1
Electronic supplementary material
Additional Supporting Information may be found in the online version of this article:
Online Resource 1
Occurrence points of Hagenia abyssinica and microsatellite dataset used in this study. (XLSX 26.4 kb)
Online Resource 2
Supporting details of AMOVA, STRUCTURE and MIGRATE-n analyses. (DOCX 54.1 kb)
Rights and permissions
About this article

Cite this article
Gichira, A.W., Li, ZZ., Saina, J.K. et al. Demographic history and population genetic structure of Hagenia abyssinica (Rosaceae), a tropical tree endemic to the Ethiopian highlands and eastern African mountains. Tree Genetics & Genomes 13, 72 (2017). https://doi.org/10.1007/s11295-017-1156-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11295-017-1156-6