
Abstract
Realised gains in stand volume of Eucalyptus globulus Labill. families at 7–9 years were compared with gains predicted at 3.5 years. Gain predictions were based on height, diameter at breast height (DBH), sectional area, and stem volume for 153 full-sib families and 18 commercial checks in five-tree line plots on three West Australian sites. Single-site narrow-sense heritability estimates were 0.12–0.24 for height, 0.08–0.12 for DBH, 0.09–0.13 for sectional area, and 0.14–0.19 for stem volume. Genetic dominance effects were significant (p < 0.05) in most cases, and the estimated dominance/additive genetic variance ratio was heterogeneous for height and DBH. Stand volume was measured for 93 of the same families and checks in 40-tree block plots on four sites. Heritability of stand volume was 0.25–0.76, with an across-sites estimate of 0.41. Dominance effects were statistically absent at two sites. Estimated region-wide additive genetic correlations between selection traits (in line plots) and stand volume (in block plots) ranged from 0.86 to 0.90. Estimated stand volume gain was 23 % of the mean for the best 12 % of families and 14 % of the mean for the best 24 % of families. Realised gain was under-estimated by predictions based on height, DBH, and sectional area, which had smaller coefficients of additive variation than did stem volume. It is concluded that although BLUP analysis of early-age height and DBH can provide for indirect selection on E. globulus stand volume, analysis of stem volume is required to predict genetic gain at an appropriate scale.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
A common goal of forest tree improvement is to increase harvest stand volume, usually by indirect selection on stem size early in the rotation. Trials with small plots such as lines or single trees are recommended for this purpose because they allow evaluation of many genetic entries while providing good replication and control of environmental variation (Magnussen 1993). The optimum selection age for pulpwood eucalypts has been estimated at between 3 and 4 years (Greaves et al. 2003). However, such early data from small-plot trials might not provide for an accurate prediction of genetic gains in harvest stand volume. The scale of genetic effects may change through time and with trial design (Foster 1989; Carson et al. 1999), and competition between entries in small plots can either inflate heritability and/or gain estimates (Foster 1989; Vergara et al. 2004; Stanger et al. 2011) or cause size-related mortality leading to artificially depressed heritability (Stackpole et al. 2010). Realised gains in stand volume must therefore be determined using trials with large plots (‘block plots’) which represent the growing environment of a commercial stand (Foster 1992; Foster and Knowe 1995).
Predicted genetic gains in harvest stand volume are commonly used for projecting yields and wood supply, implementing index selection, and estimating the financial returns from breeding. For softwood species with rotations of 30 years or more, early-rotation genetic gains have been translated into harvest volume increments by incorporating genetic values into empirical growth models (e.g. Hamilton and Rehfeldt 1994; Carson et al. 1999; Adams et al. 2006; Gould et al. 2008). A simpler method is to calculate genetic gain as a percentage of the population mean in the selection trait. This approach been applied to Eucalyptus globulus Labill. on a 10-year rotation (Sanhueza et al. 2002) and to other species for comparing the predicted gain with realised gain at mid-rotation (Vergara et al. 2004; Weng et al. 2008; Verryn et al. 2009; Ye et al. 2010).
Early-rotation measurements can be used to predict stand volume gain if genetic correlations are large enough and the traits are expressed at an equivalent genetic scale. There are few published estimates of genetic correlations between predicted and realised gain in forest trees. Ye et al. (2010) studied 15-year-old Pseudotsuga menziesii var. menziesii (Mirb.) Franco gains trials and reported a correlation of 0.77 between predicted and realised gain amongst 20 families. This estimate was based on best-linear unbiased predictions (BLUPs) representing predicted breeding values for stem volume and least-square means (LSMs) for stand volume in block plots. Jansson et al. (1998) used linear mixed models to jointly model Pinus sylvestris L. family data from single-tree plots and block plots and estimate the correlations between them. They reported additive genetic correlations between stem volume in single-tree plots and stand volume in block plots ranging from 0.63 to 0.99, with an average of about 0.87.
Predicted and realised gains have been compared using LSM volume of seed orchard entries in block plots and predictions based on the BLUPs of parents represented in the deployment. In these cases, the seed orchard entries are comprised of seed from a whole orchard which may have been rouged or they represent different classes of genetic gain collected from an orchard. Following this approach, Verryn et al. (2009) reported an average of 14 % realised gain in stem volume of Eucalyptus grandis Hill ex Maiden. per generation over three generations compared with a predicted 13 % gain. Ye et al. (2010) reported a realised gain in stand volume of P. menziesii var. menziesii of 17 % compared with a predicted 16 % from analysis of single-tree plots. Poorer correspondence between predicted and realised gain has also been reported. Weng et al. (2008) found that realised gain in stand volume of Pinus banksianna Lamb. following seed orchard rouging was nearly one-third lower than predicted (realised gain of 13 % versus predicted gain of 18 %). Similarly, average genetic gains in mean annual increment of Pinus elliottii Engelm, var elliottii across 38 block-plot trials in southern USA were found to be almost one fifth lower than predicted for two breeding populations (Vergara et al. 2004).
E. globulus is a highly valued pulpwood plantation species and is the subject of advanced genetic improvement programs. Growth improvement of E. globulus is commonly achieved by selection on diameter at breast height (DBH) and height (e.g. Li et al. 2007; Costa e Silva et al. 2009; Callister et al. 2011). We measured stand volume in a series of four block-plot progeny trials of full-sib families in Western Australia after 7–9 years of a 10-year pulpwood rotation. The same families were represented in line-plot trials in the same region, which were measured for DBH and height at 3.5 years. The objectives of the study were to (1) quantify the genetic control of stand volume at late rotation in block-plot trials, (2) estimate the genetic correlations between early-rotation stem size in line plots and late-rotation stand volume in block plots, and (3) compare predicted and realised gain estimates using a range of individual-tree traits for the prediction.
Materials and measurements
Breeding population and test sites
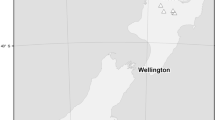
This study was based on a second-generation breeding population which was previously described as ‘EGP1’ by Callister et al. (2011). The population consisted of 153 full-sib E. globulus families representing 86 parents from seven Australian provenances (SE Tasmania, W Tasmania, NE Tasmania, Furneaux Group, King Island, Gippsland, and Otways) and a Portuguese and a Californian land race. The parents were first-generation selections from 84 open-pollinated (OP) families collected from native trees and land-race plus trees. Two parents were selected from the same OP family in two instances. The parents were mated using an incomplete factorial design which included crosses within and between provenances/land races. There was no reciprocal crossing. Five commercial bulk collections and 13 unselected open-pollinated native families were included as ‘checks’. The trials were established at seven locations in the south of Western Australia in 1999; three line-plot (or row-plot) trials (L1-3) and four block-plot trials (B1-4; Fig. 1; Table 1). The trial sites were within 200 km of each other and had previously been used for agriculture. All full-sib families and checks were represented in the line-plot series and a subset of 86 control-pollinated families and seven checks were established in the block-plot series. From 22 to 86 full-sib families were measured at each block plots site (Table 1), and a total of 647 block-plots contributed data to this analysis. The representation of genetic entries in block-plot trials was highly unbalanced. For example, four families were each represented by 35 to 64 plots across the four trials, while 30 families were represented by only one plot at one of the trials.

Map of the line-plot trial locations (triangles) and block-plot trial locations (circles) relative to population centres in southern Western Australia (stars)
The line-plot trials consisted of five replicates of ten incomplete blocks. Each block contained 18 to 21 five-tree plots to which control-pollinated families and checks were randomly assigned. The block-plot trials were established with incomplete blocks of 16 to 20 block plots. Each block plot was five rows wide and eight planting spaces long (40 planting spaces per plot). Tree spacing throughout was 5 × 2.5 m (800 stems ha−1) except at site B1 where it was 5 × 2.0 m (1,000 stems ha−1). Soils were typical for the region, classified as duplex or gravelly duplex and greater than 3 m deep (Table 1). Anaerobic subsoils occurred within 3 m depth at sites B2 and B4.
Climate information for each trial site was obtained by spatial interpolation of available information from nearby weather stations using ‘Data Drill’ (Jeffrey et al. 2001). The climate at all sites could be described as ‘Mediterranean’ with hot, dry summers and cool, wet winters. However, rainfall across the region in the years following trial establishment was less than the 30-year average and ranged from an average of 461 mm year−1 at site L2 to 694 mm year−1 at site B3 (Table 1). Pan evaporation was 2.3 to 3.3 times greater than rainfall (Table 1).
Data measurement and augmentation
Every tree in the line-plot trials was assessed for total height and diameter at 1.3 m (DBH) at 3.5 years. On trees with two stems (incidence less than 4 %), the DBH of both stems was measured and entered as the square root of the sum of the squares. Sectional area at breast height was calculated from DBH assuming each stem was circular. Block plots were measured at 9 years at sites B1, B3, and B4 and at 7 years at site B2. DBH of all trees was measured. At site B2, the height of all plot trees in the 18 central planting positions was measured. At B1, B3, and B4, heights of a subsample of nine trees per plot were measured, comprising the six trees in the plot centre and the three largest remaining trees (total n = 4,650). The following fixed-effects model was fitted to these data using Genstat Version 13 (VSN International, UK):
where HT ijk and DBH ijk are height and DBH of the kth tree from the ith family in the jth incomplete block, Fam i and block/site j are direct effects of the ith family and the jth incomplete block (nested within site) on height, Fam i ·DBH ijk and block/site j ·DBH ijk are effects of the ith family and the jth incomplete block (nested within site) on the slope of relationship between HT and DBH, and e ijk is random error. Model (1) explained 75 % of observed variation in height, and all fitted effects were significant (p < 0.001). Residual plots confirmed that that the DBH-HT relationship was linear and residuals were homoscedastic and normally distributed. The unmeasured heights of 14,746 trees at sites B1, B3, and B4 were then predicted using their DBH measurements and the coefficients estimated in fitting Model (1).
Total over-bark stem volume of each measured tree in each trial was calculated using the following volume function which had been parameterised with data from West Australian E. globulus trees ranging from 30 to 400 dm3 in volume:
where V over-bark is expressed in cubic decimetres, DBHover-bark is measured in centimetres, and tree height (H) is measured in metres.
Trees in the central 18 planting positions of each block plot were identified at sites B2, B3, and B4 and formed the basis for all subsequent analyses at those sites. Trees on the periphery of block plots were excluded to remove potential competition from adjacent plots. The within-plot positions of trees were poorly identified during the measurement at site B1, so all trees at site B1 contributed to subsequent analyses. Stand volume (in cubic metres per hectare) was calculated for each block plot by summing tree volumes and dividing by the plot area.
Analyses and results
The general statistical framework of genetic analyses
Genetic analyses were conducted using ASReml version 3.0 (Gilmour et al. 2009) within the framework of the general linear mixed model:
where y is the vector of observed values for the response variable, sorted by site for multi-site analyses; b and u are vectors of fixed and random effects, respectively; Z and X are incidence matrices relating observations to model effects; and e is the vector of residuals. The distribution of random effects was assumed to be multivariate normal with means and (co)variances defined by:
where G and R are the (co)variance matrices relating to u and e, respectively, and 0 is a null matrix. Each of the j effects in u were assumed to be mutually independent with (co)variance G i , so that \( G = ⊕ {_i}{_{{ = {1}}}^{\mathrm{j}}}{G_i} \), where ⊕ denotes the direct sum operation. Standard errors of functions of variance components were calculated in ASReml using an approximation of a first-order Taylor series expansion (Gilmour et al. 2009).
Single-site analyses of stem size in line-plot trials
Methods
Height, DBH, sectional area, and stem volume data from line-plot trials were first analysed using univariate, single-site mixed models to understand the genetic architecture for selection (line-plot) traits. We expected the variance functions for DBH and sectional area to be almost identical, but that sectional area might be expressed on a larger scale. In each model, the terms in b comprised fixed effects for the mean, replicates, and checks. The random terms in u represented provenance effects of parents with the design matrices for the maternal and paternal provenances overlaid upon each other, general combing ability (GCA) given by overlaying the design matrices for the maternal and paternal parents, specific combining ability (SCA) given by family-specific effects, plot effects, trial design effects of factors blocks, rows, and columns, and a term for additional within-plot variance of checks. Residuals were generally decomposed into spatially dependent (ξ) and spatially independent (η) components according to: \( R = {\sigma_{\xi }}^{2}\left[ {{\mathrm{AR}1}\left( {{\rho_{\mathrm{col}}}} \right) \otimes {\mathrm{AR}1}\left( {{\rho_{\mathrm{row}}}} \right)} \right] + {\sigma_{\eta }}^{2}{I_n} \), where \( \sigma_{\xi}^2 \) and \( \sigma_{\eta}^2 \) are spatially dependent and spatially independent residual variances, respectively, ⊗ is the Kronecker product and AR1(ρ) represents a first-order autoregressive correlation matrix (Cullis and Gleeson 1991; Dutkowski et al. 2006). No variance was explained by the spatial autocorrelation process for DBH and sectional area at site L1. The significance of SCA effects was determined with one-tailed likelihood ratio tests (LRTs) with \( \chi_{{{0}{.5}}}^2 \) (Stram and Lee 1994). Additive genetic variance (\( \sigma_{\mathrm{A}}^2 \)) was estimated as \( 4\widehat{\sigma}_{\mathrm{GCA}}^2 \), where \( \widehat{\sigma}_{\mathrm{GCA}}^2 \) is the estimated GCA variance, dominance genetic variance (\( \sigma_{\mathrm{D}}^2 \)) was estimated as\( 4\widehat{\sigma}_{\mathrm{SCA}}^2 \), where \( \widehat{\sigma}_{\mathrm{SCA}}^{2} \)is the estimated SCA variance, and phenotypic variance (\( \sigma_{\mathrm{P}}^2 \)) was estimated as \( 2\widehat{\sigma}_{\mathrm{GCA}}^2 + \widehat{\sigma}_{\mathrm{SCA}}^2 + \widehat{\sigma}_{\mathrm{plot}}^2 + {\sigma}_{\mathrm{block}}^2 + \widehat{\sigma}_{\mathrm{row}}^2 + \widehat{\sigma}_{\mathrm{col}}^2 + \widehat{\sigma}_{\eta}^2 \), where \( {\sigma}_{\mathrm{plot}}^2 \)is the variance due to plots, \( {\sigma}_{\mathrm{block}}^2 \) is the variance due to incomplete blocks, \( {\sigma}_{\mathrm{row}}^2 \) is the variance due to trial rows, and \( {\sigma}_{\mathrm{col}}^2 \) is the variance due to trial columns. Coefficients of additive genetic variation (CVA) were estimated for each trait and site by \( {\mathrm{C}}{\widehat{\mathrm{V}}_{\mathrm{A}}} = {{{\widehat{\sigma}_{\mathrm{A}}}} \left/ {{\overline X }} \right.} \), where \( \overline X \) is the mean. Narrow-sense heritabilities (h 2) and dominance proportions (d 2) were estimated for each trait at each site by \( {\widehat{h}^2} = {{{\widehat{\sigma}_{\mathrm{A}}^2}} \left/ {{\widehat{\sigma}_{\mathrm{P}}^2}} \right.} \) and \( {\widehat{h}^2} = {{{\widehat{\sigma}_{\mathrm{D}}^2}} \left/ {{\widehat{\sigma}_{\mathrm{P}}^2}} \right.} \).
Results
\( {\mathrm{C}}{\widehat{\mathrm{V}}_{\mathrm{A}}} \) was smallest for DBH (mean, 0.057), followed by height (mean, 0.073), sectional area (mean, 0.108), and stem volume (mean, 0.141; Tables 2 and 3). Single-site \( {\widehat{h}^2} \) ranged from 0.12 to 0.24 for height, 0.08 to 0.12 for DBH, 0.09 to 0.13 for sectional area, and from 0.14 to 0.19 for stem volume (Tables 2 and 3). Estimated dominance genetic effects were considerably smaller at site L2, where it was only significant for height (p < 0.05; Table 2). Although \( {\widehat{h}^2} \) and \( {\widehat{d}^2} \) were nearly equivalent for DBH and sectional area, the quadratic relationship between the traits resulted in \( {\mathrm{C}}{\widehat{\mathrm{V}}_{\mathrm{A}}} \) for sectional area that were nearly double those for DBH. Similarly, \( {\widehat{h}^2} \) and \( {\widehat{d}^2} \) for stem volume were generally intermediate between those for height and DBH, while \( {\mathrm{C}}{\widehat{\mathrm{V}}_{\mathrm{A}}} \) for stem volume were more than double the average \( {\mathrm{C}}{\widehat{\mathrm{V}}_{\mathrm{A}}} \) for height and DBH.
The mean \( {{{\widehat{\sigma}_{\mathrm{D}}^2}} \left/ {{\widehat{\sigma}_{\mathrm{A}}^2}} \right.} \) was 1.31 for height, 0.72 for DBH, 0.57 for sectional area, and 0.64 for stem volume (Tables 2 and 3). SCA effects did not suggest the presence of between-provenance heterosis. Provenance variance \( \left( {\widehat{\sigma}_{\mathrm{PROV}}^2} \right) \) was relatively small for all traits, as indicated by the mean ratio \( {{{\widehat{\sigma}_{\mathrm{PROV}}^2}} \left/ {{\widehat{\sigma}_{\mathrm{GCA}}^2}} \right.} \) of 0.15 (Tables 2 and 3). No provenance variance was found for height and stem volume at site L2, and in most cases the standard error was larger than the provenance variance estimate (Tables 2 and 3).
Multi-site analyses of stem size in line-plot trials
Methods
Multi-site models were fitted to selection trait data to determine inter-site correlations and heterogeneity in genetic variance ratios. These results were used to determine the best model for data standardisation and to assist the interpretation of results from region-wide analyses. Data for multi-site analyses were adjusted for spatial trends by subtracting from y the row and column effects and spatial residuals. Five models (Lms-1 to Lms-5) were then fitted to full-sib family data for each trait (Table 4). They contained effects of site means and replicates in b; parent provenances (overlaid design matrices), GCA (overlaid design matrices), SCA, blocks, and plots in u. Genetic correlations were constrained to be uniform between pairs of sites (as in Li et al. 2007; Costa e Silva et al. 2009). Further constraints on genetic (co)variances were varied between models to test the hypotheses that inter-site SCA or GCA correlations were 1 using LRTs with \( \chi_{{{0}{.5}}}^2 \) and to test heterogeneity in the ratios \( {{{\widehat{\sigma}_{\mathrm{PROV}}^2}} \left/ {{\widehat{\sigma}_{\mathrm{GCA}}^2}} \right.} \) and \( {{{\widehat{\sigma}_{\mathrm{SCA}}^2}} \left/ {{\widehat{\sigma}_{\mathrm{GCA}}^2}} \right.} \) using LRTs with \( \chi_{2}^2 \) (Table 4). There was insufficient provenance variance at site L2 to fit this term with inter-site correlation.
Results
Uniform amongst-sites correlation estimates in GCA ranged from 0.71 for sectional area to 0.87 for height, and they were significantly different from 1 in each case (Table 5). On the other hand, uniform correlations in SCA were only significantly different to 1 for height (r SCA, 0.63; Table 5). Ratios of \( {{{\widehat{\sigma}_{\mathrm{PROV}}^2}} \left/ {{\widehat{\sigma}_{\mathrm{GCA}}^2}} \right.} \) were not significantly heterogeneous amongst sites for any trait (p > 0.05) and uniform estimates ranged from 0.04 for height to 0.15 for sectional area (Table 5). Ratios of \( {{{\widehat{\sigma}_{\mathrm{SCA}}^2}} \left/ {{\widehat{\sigma}_{\mathrm{GCA}}^2}} \right.} \) were significantly heterogeneous for height, DBH, and sectional area due to smaller SCA variance at site L2 (Tables 2, 3, and 5). Nevertheless, \( {{{\widehat{\sigma}_{\mathrm{SCA}}^2}} \left/ {{\widehat{\sigma}_{\mathrm{GCA}}^2}} \right.} \) was not significantly heterogeneous for stem volume (p > 0.05), for which the uniform estimate was 0.55 (Table 5).
Region-wide analyses of stem size in line-plot trials
Methods
The region-wide analysis of each selection trait provided the breeding values for use in selection and genetic-gain estimation. Site additive standard deviations from Model Lms-5 (Table 4) were used to standardise the spatially adjusted data for scale effects. The variance constraints in Lms-5 made it as close as possible to the region-wide model, in which the standardised data were used. Re-scaled data (x adj) were also centred to the respective trial mean \( (x{{p}_{{\text{adj}}}})/{{x}_{{\text{adj}}}}=\left( {x-{{{\bar{x}}}_{i}}} \right)/{{\hat{\sigma }}_{{{{\text{A}}_{i}}}}} \). This standardisation procedure is convenient because back-transformed breeding values (as a percentage of the population mean) are obtained by simply multiplying BLUPs by \( {\mathrm{C}}{\widehat{\mathrm{V}}_{\mathrm{A}}} \).
The region-wide models of height, DBH, sectional area, and stem volume each contained effects of replicates within sites, and checks in b. Effects of parent provenances, provenance × site, GCA, GCA × site, SCA, SCA × site, blocks-in-sites, plots-in-sites, and additional within-plot effects for checks were included in u (using overlaid design matrices for provenance, GCA, and their site interaction effects). Residual variances were heterogeneous. Average narrow-sense heritabilities (\( h_{\mathrm{ave}}^2 \)) and dominance proportions (\( d_{\mathrm{ave}}^2 \)) were estimated for each trait by:
where genetic variances are estimated across sites, \( \widehat{\sigma}_{{{\mathrm{bloc}}{{\mathrm{k}}_i}}}^{2} \) is block variance of the ith site, \( \widehat{\sigma}_{{{\mathrm{plo}}{{\mathrm{t}}_i}}}^{2} \) is plot variance of the ith site, and \( \widehat{\sigma}_{{{\mathrm{erro}}{{\mathrm{r}}_i}}}^2 \) is the residual variance of the ith site.
Results
Region-wide average heritability estimates (\( \widehat{h}_{\mathrm{ave}}^2 \)) were 0.18 for height, 0.07 for DBH and sectional area, and 0.14 for stem volume (Table 6). Region-wide \( {{{\widehat{\sigma}_{\mathrm{D}}^2}} \left/ {{\widehat{\sigma}_{\mathrm{A}}^2}} \right.} \) ranged from 0.38 for stem volume to 0.81 for DBH. Provenance effects were small at the region-wide scale, with \( {{{\widehat{\sigma}_{\mathrm{PROV}}^2}} \left/ {{\widehat{\sigma}_{\mathrm{GCA}}^2}} \right.} \) ranging from 0.10 for height to 0.22 for DBH (Table 6).
Survival in block plots
Methods and results
Variable survival in block plots was examined for genetic effects and to provide a basis for removing stand volume data that were significantly impacted by poor survival. The proportion of surviving trees within each block plot ranged from 23 to 100 % with a mean of 87 %. It was assumed to follow a binomial distribution and modelled, site by site, with a generalised linear mixed model using a logit link function:
where η ijk is the link function, π is the proportion of surviving trees, μ is the conditional mean, B k is the fixed effect of the kth incomplete block, GCA i and GCA j are the random effects of the ith maternal parent and jth paternal parent with overlaid design matrices, SCA ij is the random effect of specific combining ability between parents i and j, and e ijkl is the random residual with N (0, σ 2e I). Data were excluded from plots in two drainage lines which cut across site B4.
There was no detectable variance associated with parental or family genetic effects at any of the block-plot sites. We therefore considered variable survival to be a nuisance in the analysis of stand volume, and we sought to remove its effect by imposing site-specific survival thresholds for the inclusion of stand volume data. A fixed-effects model was fitted to data from each site separately using Genstat version 13 to determine the significance of survival to plot stand volume:
where V nkl is the stand volume of the nth plot, μ is the mean, S n is the survival of the nth plot, F k is the random effect of the kth family, Bl is the random effect of the lth block, and e nkl is random error.
Survival was a significant determinant of stand volume at each site when all measured plots were considered (p < 0.05). Data from plots with the poorest survival were then excluded and the remaining data were re-analysed with Model (6). If survival remained a significant effect in the remaining data, plots with the next-poorest survival were excluded and the remaining data were re-analysed. This process was repeated until survival was not a significant effect in the analysis of remaining data (p < 0.05). The poorest plot survival in the remaining data at the completion of this process was considered to be the survival threshold for the site.
Survival thresholds were found to be 63 % at B1 and 78 % at B2, B3, and B4. Stand volume data from 5 to 11 % of measured plots at each site were excluded from further analyses because their survival was poorer than these thresholds.
Single-site analyses of stand volume in block-plot trials
Methods
The analysis of stand volume commenced at the single-site level to understand the genetic architecture of this trait. Trial means and checks were included as fixed effects in b, while random effects of the parent provenance, GCA, SCA, and incomplete block effects were generally included in u. Design matrices were overlaid for provenance and GCA effects. SCA was not fitted at site B2, where only 22 families were measured. An additional random term was fitted to within-plot variance of checks at sites B1 and B3, the only trials in which checks were represented sufficiently to estimate this term. Residuals at sites B1, B3, and B4 were decomposed into spatially dependent (ξ) and spatially independent (η) components using the same first-order autoregressive structure as for the line-plot trials (AR1 × AR1), except that in this case the experimental units were plots rather than trees. Spatial analysis was not possible at site B2, where measured plots were widely scattered amongst unmeasured plots.
Heritability of stand volume in block plots was estimated by \( \widehat{h}_{\mathrm{STAND}}^{2} = {{{4\widehat{\sigma}_{\mathrm{GCA}}^2}} \left/ {{\left( {2\widehat{\sigma}_{{GCA}}^2 + \widehat{\sigma}_{{SCA}}^2 + \widehat{\sigma}_{{block}}^2 + \widehat{\sigma}_{\xi}^2 + \widehat{\sigma}_{\eta}^2} \right)}} \right.} \), where terms are defined as for line-plot analyses above. Dominance proportion of stand volume in block plots was estimated by: \( \hat{d}_{{\text{STAND}}}^{2}=4\hat{\sigma }_{{\text{SCA}}}^{2}/\left( {2\hat{\sigma }_{{GCA}}^{2}+\hat{\sigma }_{{SCA}}^{2}+\hat{\sigma }_{{block}}^{2}+\hat{\sigma }_{\xi }^{2}+\hat{\sigma }_{\eta }^{2}} \right) \) . \( \widehat{h}_{\mathrm{STAND}}^2 \) and \( \widehat{d}_{\mathrm{STAND}}^{2} \)differ from narrow-sense heritability and dominance proportion estimates because amongst-trees size variation was averaged within plots and did not contribute to phenotypic variance. Although spatial residual variance is not customarily included in the phenotypic variance component (e.g. Ye and Jayawickrama 2008), it was included here to produce estimates of variance components at the whole-of-site scale, which we considered to be appropriate for deployed gain in stand volume.
Results
Mean stand volume was 138, 79, 97, and 92 m3 ha−1 at sites B1 to B4, respectively, corresponding to mean annual increment of 15.3, 11.3, 10.8, and 10.2 m3 ha−1 y−1, respectively. At sites B2, B3, and B4 genetic variation in stand volume was overwhelmingly attributed to additive effects (GCA), with nil estimated variance for SCA at sites B3 and B4, and nil or negligible variance for provenance effects (Table 7). However, at site B1 genetic control of stand volume followed a different pattern, whereby estimated provenance variance was nearly two thirds of \( \widehat{\sigma}_{\mathrm{GCA}}^2 \)and \( \widehat{\sigma}_{\mathrm{SCA}}^2 \)was large but not significant (Table 7). \( \widehat{h}_{\mathrm{STAND}}^{2} \) ranged from 0.25 to 0.76 (Table 7), indicating that a substantial proportion of variance in late-rotation stand volume was attributed to additive genetic variance. \( {\mathrm{C}}{\widehat{\mathrm{V}}_{\mathrm{A}}} \) for stand volume ranged from 0.138 to 0.217 (mean, 0.178).
Multi-site analyses of stand volume in block-plot trials
Methods
Multi-site models were fitted to stand volume to determine inter-site correlations and explore the best way to accommodate the larger provenance and SCA variances at site B1. These results were used to determine the best model for data standardisation and which terms to include in the region-wide and unified analyses. Data for multi-site analyses of stand volume were adjusted for spatial trends at sites B1, B3, and B4 by subtracting spatial residuals from y. Six models (Bms-1 to Bms-6) were then fitted to full-sib family data (Table 8). The models each included site mean effects in b; heterogenous GCA effects (with overlaid design matrices) and block effects in u. The inconsistent partitioning of genetic variance at site B1 was examined by comparing the Akaike information criterion (AIC) of models that included provenance and SCA only at B1 with models that included provenance and SCA variance at each site as a uniform ratio of GCA variance. The model sequence also allowed testing whether a uniform between-site additive genetic correlation was significantly different from 1.
Results
The best multi-site model by AIC included provenance and SCA effects only at site B1 (Model Bms-1; Table 8). The uniform inter-site GCA correlation from this model was 0.44, which was significantly different from 1 (p < 0.001, LRT Bms-2 versus Bms-1 using \( \chi_{{{0}{.5}}}^2 \)). The omission of the provenance term at site B1 resulted in only a minor increase of 0.5 in AIC (Model Bms-3; Table 8), so Bms-3 was adopted as the most parsimonious multi-site model to underpin further analyses. Model Bms-4, which included provenance and SCA effects as uniform proportions of GCA variance, had an AIC 4.3 greater than model Bms-1, which included provenance and SCA only at site B1 (Table 8). Comparison of models Bms-3 and Bms-6 showed that SCA variance at site B1 was significant (p = 0.02, LRT using \( \chi_{{{0}{.5}}}^2 \)) when included with correlated GCA effects in analysis of multi-site data.
Region-wide analysis of stand volume in block-plot trials
Methods
Region-wide analysis of stand volume was conducted to estimate the proportion of phenotypic variance represented by additive genetic variance at a regional scale and as a precursor to the unified GCA analyses. Full-sib family data were spatially adjusted and standardised for scale effects using additive standard deviations from Model Bms-3. The region-wide model included site means in b and a single variance for GCA, SCA at site B1, GCA × site interaction, and site-specific block effects in u. Site residuals were heterogeneous. Across-sites average heritability in stand volume (\( h_{{{\mathrm{STAN}}{{\mathrm{D}}_{\mathrm{ave}}}}}^2 \)) was estimated as: \( \widehat{h}_{{{\text{STAN}}{{\text{D}}_{{{\text{ave}}}}}}}^{2} = {{{4\widehat{\sigma }_{{GCA}}^{2}}} \left/ {{\left( {2\widehat{\sigma }_{{GCA}}^{2} + \widehat{\sigma }_{{GCA \times site}}^{2} + \left[ {\widehat{\sigma }_{{SCA\,(site B1)}}^{2} + \sum\nolimits_{{i = 1}}^{4} {\left( {\widehat{\sigma }_{{bloc{k_{i}}}}^{2} + \widehat{\sigma }_{{{\text{erro}}{{\text{r}}_{{\text{i}}}}}}^{{\text{2}}}} \right)} } \right]/4} \right)}} \right.} \).
Results
Additive genetic variance in stand volume was an appreciable proportion of total variance at the regional scale, with \( \widehat{h}_{{{\mathrm{STAN}}{{\mathrm{D}}_{\mathrm{ave}}}}}^2 \) of 0.41 (Table 9). GCA × site variance was more than 50 % greater than GCA variance, which is expected from the uniform across-sites GCA correlation estimate of 0.44 reported above.
Unified GCA analyses of line- and block-plot data
Methods
Joint analyses of GCA using data from both plot types (‘unified GCA’ analyses) were undertaken to estimate genetic correlations between individual-tree selection traits at age 3.5 years and stand volume at age 7–9 years. Four models were fitted with correlations at the site scale, each including one selection trait and stand volume. Line- and block-plot data representing full-sib families were adjusted only for spatial trend, as previously described. The models included all trial means and replicates within line-plot trials in b, while u contained provenance effects for each line-plot trial, GCA for each trial, SCA for each line-plot trial and at site B1, and block and plot effects at sites where they were present. Provenance variance at line-plot trials was fitted as heterogeneous without inter-site correlation and SCA correlations were heterogeneous between line-plot trials. Design matrices were overlaid for provenance and GCA effects. GCA was modelled with: (1) uniform correlation in the selection trait between sites, (2) uniform correlation in stand volume between sites, and (3) uniform correlation between sites, between traits.
Four unified GCA models were fitted to estimate genetic correlations at the regional scale. Each model included one selection trait and stand volume. The full-sib family data were adjusted for spatial trend and for scale effects by additive standard deviation, as previously described. All trial means and replicates within line plots were included in b, while u contained one variance for provenance effects across line-plot trials, a variance for GCA across each trial type, SCA at each individual line-plot trial and at site B1, and block and plot effects at sites where they were present. SCA correlations were heterogeneous between line-plot trials. Design matrices were overlaid for provenance and GCA effects. GCA effects were modelled with one inter-trait correlation which was compared with 1 using LRT with \( \chi_{{{0}{.5}}}^2 \).
Results
The uniform additive genetic correlations between 3.5-year selection traits and late-rotation stand volume at the site scale were 0.65 for height, 0.59 for DBH, 0.58 for sectional area, and 0.65 for stem volume (Table 10). Between-site genetic correlations within traits were similar to those reported above. The additive genetic correlations between 3.5-year selection traits and late-rotation stand volume at the regional scale ranged from 0.86 for height to 0.90 for stem volume (Table 10). Each correlation was significantly different to 1 (p < 0.05).
Comparison of predicted and realised genetic gains
Methods
Although the above results demonstrate good genetic correlation between early-age selection traits and late-rotation stand volume, differences in the scale of genetic effects could result in very different estimates of genetic gain. Our final analysis was therefore intended to compare the genetic gain of the best families in selection traits (i.e. predicted gain) with realised gain of the same families in late-rotation stand volume.
BLUPs from region-wide models of line-plot data were used to estimate values for provenance, GCA and SCA in units of additive standard deviation. The predicted genetic value of each family was calculated for height, DBH, sectional area, and stem volume by summing the relevant provenance, GCA, and SCA values and multiplying the result by the mean \( {\mathrm{C}}{\widehat{\mathrm{V}}_{\mathrm{A}}} \) of the three line-plot trials (0.073 for height, 0.057 for DBH, 0.108 for sectional area, and 0.141 for stem volume).
Genetic entries in block-plot trials were considered (perhaps counterintuitively) to be fixed effects represented by LSMs for stand volume. This distinction from our preceding analyses of random genetic effects is important because LSMs are less conservative than BLUPs and they will produce larger estimates of genetic gain when sample sizes are relatively small (as is the case here). The statistical literature does not provide a consistent view on how to judge an effect to be considered fixed or random, or even how the terms are defined (Gelman 2005). McCulloch et al. (2011; Sect 1.6b) provide the following guidance:
‘In endeavoring to decide whether a set of effects is fixed or random, the context of the data, the manner in which they were gathered and the environment from which they came are the determining factors… are the levels of the factor going to be considered a random sample from a population of values which have a distribution?’
In our case, the families that were entered into block-plot trials could be considered as random representations of a distribution representing all possible full-sib E. globulus families, and this formed the basis of the preceding analyses. However, for estimating genetic gain, we considered the ‘context of the data’ differently. We considered a hypothetical scenario in which the full range of families was tested in line plots, measured at 3.5 years and represented by random effects, or BLUPs. In this scenario, the best-ranked families were then deployed commercially (mimicked by the block plots in our experiment), prompting the question ‘how well did we predict the realised genetic gain in our commercial stands?’ In this case, once those best families were selected for deployment they were no longer considered to be drawn randomly from a distribution but rather they represented a particular treatment with a fixed effect. It should be also noted that the experimental errors in the block-plot analyses were independent of the errors adjusted for when making BLUP estimates in line plots. Treating families as fixed effects is comparable to an assessment of inventory plots in which ‘selected’ and ‘average’ families were established.
Stand volume data were adjusted for spatial trend (as described above) and then divided by the respective site mean. This simple transformation largely overcame site productivity differences and the intuitive representation of stand volume as a percent of the site mean is useful for making comparisons with predicted levels of genetic gain. Representation of better- and poorer-performing families across sites was sufficiently balanced for bias to be negligible. The statistical model contained fixed effects for genetic entries and random effects for incomplete blocks. Families and checks with only one or two block plots were excluded from realised gain estimation as their LSMs were considered to be too imprecisely estimated. The LSMs of the remaining 42 full-sib families and four checks were compared with their predicted gain in height, DBH, sectional area, and stem volume. The predicted and realised genetic gains of the best 12, 24, and 50 % of the 42 full-sib families were calculated as:
where x i is the genetic value (either BLUP or LSM) of the ith family in an elite group of n families on the basis of the selection variable (height, DBH, sectional area, or stem volume), and x j is the genetic value (either BLUP or LSM) of the jth family in the entire group \( \left( {i \subseteq j} \right) \).
Results
Realised genetic gain was around 23 % for the best five families, 14 % for the best ten families, and 7 % for the best half of the group of 42 full-sib families (Table 11). Predicted genetic gains varied with \( {\mathrm{C}}{\widehat{\mathrm{V}}_{\mathrm{A}}} \) of the selection trait from 4.7 % DBH gain to 14.6 % stem volume gain in the best five families (Table 11). This scale effect resulted in under-predictions of genetic gain based on height, DBH, or sectional area (Table 11). Genetic gain predictions were closest to realised gain estimates for stem volume; realised gain was 8 percentage points greater than predicted for the best five families, 1.2 percentage points greater than predicted for the best ten families, and 0.3 percentage points smaller than predicted for the best 21 families. The correlation between realised gain and predicted gain in stem volume of individual full-sib families and checks was 0.82 (Fig. 2). Agreement between predicted and realised gain was better for families and checks with ten or more block plots (Fig. 2), suggesting that some of the imprecision was due to small sample size of block plots.

Relationship between realised genetic gain in stand volume of families in block plots (as a percentage of the site mean) and predicted genetic gain in stem volume in line plots at 3.5 years (as a percentage of the population mean on a site where CVA = 0.141) for full-sib families (circles) and checks (triangles) represented by more than ten block plots (solid symbols) and three to ten block plots (open symbols). The line represents parity
Three checks were represented by more than ten block plots and realised gain for these entries ranged from −5.4 to −1.5 % of the population mean (Fig. 2). Two of these entries were bulk collections from open-pollinated seed orchards and one was a collection from a native stand which had performed well in previous trials.
Although we stand by our use of LSM estimates for realised gain, some readers will no doubt be interested in the gains estimated if we had treated families as random effects. As expected, we found that treating families as random resulted in smaller estimates of gain, and that this reduction was dependent on the family sample size. Gain estimates for families represented by more than ten plots were reduced by less than 1.5 % when treating families as random. Gain estimates for families represented by three to eight plots were reduced by up to 13.3 %. The BLUP-based realised gain was 17.4 % for the best 5 families, 10.2 % for the best 10 families, and 5.6 % for the best 21 families ranked by 3.5-year stem volume.
Discussion
Single-site \( {\widehat{h}^2} \)and \( {\widehat{d}^2} \) for DBH were comparable to previous estimates for full-sib E. globulus in Australia (Volker 2002; Li et al. 2007) and DBH \( {\widehat{h}^2} \) was similar to the average value of 0.12 from 40 Portuguese sites reported by Araujo et al. (2012). Single-site \( {\widehat{h}^2} \) and \( {\widehat{d}^2} \) for height were within the range reported by Li et al. (2007). Previous reports of genetic parameters for sectional area and stem volume are lacking for full-sib E. globulus families. Inter-site r GCA estimates in selection traits between 0.71 and 0.87 suggest that additive genetic × site interaction is a not a major concern for the population in this region, even though it may be statistically significant. Previous estimates of additive genetic correlation in full-sib E. globulus DBH have been reported between 0.60 and 0.83 (Volker 2002; Li et al. 2007; Costa e Silva et al. 2009; Araujo et al. 2012).
The genetic architecture of our population in Western Australia appears broadly similar to that of another full-sib E. globulus population studied by Li et al. (2007) across sites ranging from Tasmania and Victoria to Western Australia. Our estimate of 1.31 for a uniform \( {{{\widehat{\sigma}_{\mathrm{D}}^2}} \left/ {{\widehat{\sigma}_{\mathrm{A}}^2}} \right.} \) for height was similar to the value of 1.20 reported by Li et al. (2007) although our estimate of 0.64 for DBH was smaller than their reported value of 1.00. Like Li et al. (2007), we found that heterogeneity in \( {{{\widehat{\sigma}_{\mathrm{D}}^2}} \left/ {{\widehat{\sigma}_{\mathrm{A}}^2}} \right.} \) was more significant for height than for DBH and we also found that SCA effects were more site-dependent for height than for DBH. The \( {{{\widehat{\sigma}_{\mathrm{PROV}}^2}} \left/ {{\widehat{\sigma}_{\mathrm{GCA}}^2}} \right.} \) estimates that we obtained were far smaller than similar statistics reported by Li et al. (2007), who used the subrace classification of Dutkowski and Potts (1999). This suggests that some of the variance attributed to additive effects in our analyses may have been distributed to subrace effects if we had used the Dutkowski and Potts (1999) classification rather than coarser geographical provenances and land races.
We found that stand volume in block plots was under substantial additive genetic control, with region-wide additive variance representing 41 % of the phenotypic variance (given by \( \widehat{h}_{{{\mathrm{STAN}}{{\mathrm{D}}_{\mathrm{ave}}}}}^2 \)). This result is crucial to commercial E. globulus improvement programs which aim to increase harvest yield. \( {\mathrm{C}}{\widehat{\mathrm{V}}_{\mathrm{A}}} \) for stand volume in our study (mean, 0.18) corresponded with those of Jansson (2007), who reported \( {\mathrm{C}}{\widehat{\mathrm{V}}_{\mathrm{A}}} \) between 0.14 and 0.24 (mean, 0.17) for 15 block-plot family trials of P. sylvestris. \( \widehat{h}_{\mathrm{STAND}}^2 \) of stand volume at sites B2 and B4 were larger than expected for \( {\widehat{h}^2} \) of a growth trait, due probably to the lack of between-trees variance in the denominator. Unusually large heritability estimates for stand volume were also calculated by Jansson et al. (1998) who reported a mean of 0.59 from full-sib families of P. sylvestris at five block-plot trials.
Although the genetic correlations between early-age selection traits and stand volume are of critical importance for effective yield improvement, they have rarely been quantified due to the cost of implementing block-plot family trials. The uniform between-site r GCA estimates of 0.59 to 0.65 that we found between selection traits and stand volume (Table 10) are at the low end of the range reported for the same traits in P. sylvestris (0.53 to 0.99) (Jansson et al. 1998). The larger r GCA estimates of 0.86 to 0.90 at the regional scale (Table 10) are more meaningful measures of the inter-trait correlations because they relate to the breeding value estimates of stem size and of stand volume at the scale of deployment.
Our inter-trait r GCA results suggest that height or DBH of line-plot entries were nearly as good as stem volume for indirect selection on harvest stand volume. On the other hand, predicted gains based on height were about half the realised gain and those based on DBH were about a third of the realised gain. CVA of selection traits determines the scale of predicted genetic gain from parent selection. For example, sectional area had a larger \( {\mathrm{C}}{\widehat{\mathrm{V}}_{\mathrm{A}}} \) than did DBH and it produced a less biased estimate of stand volume gain. Nevertheless, we found that only stem volume was expressed on a scale suitable for predicting stand volume gain. E. globulus breeders have published variances and heritability estimates for DBH (e.g. Costa e Silva et al. 2009; Stackpole et al. 2010; Araujo et al. 2012) and sometimes for DBH and height (e.g. Lopez et al. 2002; Li et al. 2007), but rarely for stem volume (cf. Sanhueza et al. 2002). Our findings prompt a call for publication of genetic parameters (including CVA) for individual stem volume, as it appears to be expressed on a scale more similar to that of stand volume.
Most of the published studies comparing predicted and realised gain have confounded the issues of genetic correlation and scale by using a handful of seed orchard entries to represent different levels of realised gain. In two cases volume gains have met or surpassed predicted values (Verryn et al. 2009; Ye et al. 2010), and the correlation of 0.77 between predicted and realised gains for families reported by Ye et al. (2010) is similar to our correlation of 0.82 (see Fig. 2). In other cases, predicted gains have not been achieved in comparable gains trials (e.g. Vergara et al. 2004; Weng et al. 2008). One reason that realised gains might not meet predicted gains is that between-tree competition in intimate genetic mixtures is not representative of block plantings where individuals compete with genetically similar material. This complication may be particularly evident amongst clones (e.g. Sharma et al. 2008; Stanger et al. 2011) and its effects were perhaps not apparent in our study due to the early assessment of line-plot trials just before canopy closure and the onset of stronger competition. Another potential problem with early-age predictions of gain is that percentage gain can decrease over time when absolute gain is unchanging (e.g. see the gain predictions of Carson et al. 1999). The trial sites in our study may not have expressed this phenomenon because they were strongly water-limited and would not have grown much in the last half of the rotation.
E. globulus can be feasibly reproduced in large full-sib families (Collins and Callister 2010), presenting the prospect of commercial deployment of individual families. This strategy would be predicated on an assumption that gain predictions from line-plot progeny trials were unbiased and precise. Although we found that region-wide stem volume BLUPs formed unbiased estimates of genetic gain in stand volume, the precision of individual family gain predictions may be insufficient for individual-family deployment. For example, there were cases in which realised gain of a well-represented family was more than 10 % below predicted gain (see Fig. 2). Seed-based deployment of E. globulus should therefore continue as family mixtures or as individual families only if a reasonable number of families are represented in each commercial stand. These guidelines will help to counter the risk that the genetic value of particular families is over-predicted.
We found that variable survival in block plots was not under measurable genetic control and we removed its effect by setting thresholds for data exclusion. Another option was to apply a model combining genetic effects and stocking (e.g. Jansson et al. 1998), but we found that too few plots were significantly affected by mortality to adequately parameterise such a model. Stand volume of E. globulus in the trial region has previously been found to be insensitive to stocking rates above about 600 stems ha−1 (White et al. 2009). This is consistent with the survival thresholds that we applied, which corresponded to around 630 stems ha−1.
A number of outstanding issues could be addressed in further studies with improved designs. We had too few block-plot sites to adequately understand the GCA × site interaction in stand volume, and our study was limited to sites that were relatively poor for first-rotation E. globulus. Nevertheless, such sites were representative of a large portion of the West Australian estate in the first rotation and they are likely to be more typical of second-rotation sites due to water and nutrient depletion in the first rotation (Mendham et al. 2011). An improved study design may feature adjacent line- and block-plot family trials along productivity gradients and across regions, to better quantify the effects of site and plot type on genetic correlations and the scale of genetic variation. Results from a large experiment such as this may be suitable for developing a gain-prediction tool based on integrated selection-age measurements and growth models. On the other hand, our results do not suggest that a growth model is needed for genetic gain prediction within a more limited environment. We were satisfied that the gain predictions of the best 25 and 50 % of families based on stem volume were unbiased, and the gain under-prediction for the best 12 % of families could be due to random variation at the family level considering that only five families were represented in that stratum. Our study was too small to form any reliable conclusions about the degree of non-additive genetic control of stand volume. An experiment that aimed to reliably define SCA effects on stand volume would need to be extremely large, considering that the estimation of the dominance variance requires about 20 times as much data as for additive variance, for equivalent accuracy (Misztal 1997).
Conclusions
We found that late-rotation stand volume was under substantial additive genetic control, with \( \widehat{h}_{{{\mathrm{STAN}}{{\mathrm{D}}_{\mathrm{ave}}}}}^2 \) of 0.41 at the regional scale. Our results suggest that BLUP analysis of early-rotation height, DBH, or stem volume from line-plot progeny data provides adequately for indirect selection on stand volume in E. globulus and that predicted gains in stem volume are reasonable measures of stand volume gain. Predicted gains in height or DBH were found to under-estimate stand volume gains due to scale differences. Therefore, we recommend expressing E. globulus progeny trial data as stem volume if unbiased genetic gain predictions are required, and we encourage the publication of genetic parameters for stem volume in addition to DBH and height.
References
Adams JP, Matney TG, Land SB, Belli KL, Duzan HW (2006) Incorporating genetic parameters into a loblolly pine growth-and-yield model. Can J For Res 36(8):1959–1967
Araujo JA, Borralho NMG, Dehon G (2012) The importance and type of non-additive genetic effects for growth in Eucalyptus globulus. Tree Genet Genom 8(2):327–337
Callister AN, England N, Collins S (2011) Genetic analysis of Eucalyptus globulus diameter, straightness, branch size, and forking in Western Australia. Can J For Res 41:1333–1343
Carson SD, Garcia O, Hayes JD (1999) Realized gain and prediction of yield with genetically improved Pinus radiata in New Zealand. For Sci 45(2):186–200
Collins SL, Callister AN (2010) Genetic and environmental influences on capsule retention following hand pollination of Eucalyptus globulus flowers. Aust For 73(3):198–203
Costa e Silva J, Borralho NMG, Araujo JA, Vaillancourt RE, Potts BM (2009) Genetic parameters for growth, wood density and pulp yield in Eucalyptus globulus. Tree Genet Genom 5:291–305
Cullis BR, Gleeson AC (1991) Spatial analysis of field experiments—an extension to two dimensions. Biometrics 47:1449–1460
Dutkowski GW, Potts BM (1999) Geographic patterns of genetic variation in Eucalyptus globulus ssp. globulus and a revised racial classification. Aust J Bot 47:237–263
Dutkowski GW, Silva JCE, Gilmour AR, Wellendorf H, Aguiar A (2006) Spatial analysis enhances modelling of a wide variety of traits in forest genetic trials. Can J For Res 36(7):1851–1870. doi:10.1139/x06-059
Foster GS (1989) Inter-genotypic competition in forest trees and its impact on realized gain from family selection. Paper presented at the 20th Biennial Southern Tree Breeding Conference, Charleston, SC, 26–30 June 1989
Foster GS (1992) Estimating yield: beyond breeding values. In: Finns L, Friedman ST, Brotschol JV (eds) Handbook of quantitative forest genetics. Kluwer Academic Publishers, Dordrecht, pp 229–269
Foster GS, Knowe SA (1995) Deployment and genetic gains. Paper presented at the Eucalyptus Plantations: Improving Fibre Yield and Quality, Hobart, 19–24 Feb
Gelman A (2005) Analysis of variance—why it is more important than ever. Ann Stat 33(1):1–31. doi:10.1214/009053604000001048
Gilmour AR, Gogel BJ, Cullis BR, Thompson R (2009) ASReml User Guide Release 3.0. VSN International Ltd, Hemel Hempstead, HP1 1ES, UK,
Gould P, Johnson R, Marshall D, Johnson G (2008) Estimation of genetic-gain multipliers for modeling Douglas-fir height and diameter growth. For Sci 54(6):588–596
Greaves BL, Borralho NMG, Raymond CA (2003) Early selection in eucalypt breeding in Australia—optimum selection age to minimise the total cost of kraft pulp production. New For 25(3):201–210
Hamilton DA, Rehfeldt GE (1994) Using individual tree growth projection models to estimate stand-level gains attributable to genetically improved stock. For Ecol Man 68:189–207
Jansson G (2007) Gains from selecting Pinus sylvestris in southern Sweden for volume per hectare. Scand J For Res 22:185–192
Jansson G, Danell Ö, Stener L-G (1998) Correspondence between single-tree and multiple-tree plot genetic tests for production traits in Pinus sylvestris. Can J For Res 28:450–458
Jeffrey SJ, Carter JO, Moodie KM, Beswick AR (2001) Using spatial interpolation to construct a comprehensive archive of Australian climate data. Environ Model Softw 16(4):309–330
Li Y, Dutkowski GW, Apiolaza LA, Pilbeam DJ, Costa e Silva J, Potts BM (2007) The genetic architecture of a Eucalyptus globulus full-sib breeding population in Australia. For Genet 12:167–179
Lopez GA, Potts BM, Dutkowski GW, Apiolaza LA, Gelid PE (2002) Genetic variation and inter-trait correlations in Eucalyptus globulus base population trials in Argentina. For Genet 9(3):217–231
Magnussen S (1993) Design and analysis of tree genetic trials. Can J For Res 23:1144–1149
McCulloch E, Searle SR, Neuhaus JM (2011) Generalized, linear, and mixed models, 2nd edn. Wiley, New York
Mendham DS, White DA, Battaglia M, McGrath JF, Short TM, Ogden GM, Kinal J (2011) Soil water depletion and replenishment during first- and early second-rotation Eucalyptus globulus plantations with deep soil profiles. Agric For Meteorol 151:1568–1579
Misztal I (1997) Estimation of variance components with large-scale dominance models. J Dairy Sci 80:965–974
Sanhueza RP, White TL, Huber DA, Griffin AR (2002) Genetic parameters estimates, selection indices and predicted genetic gains from selection of Eucalyputs globulus in Chile. For Genet 9(1):19–29
Sharma RK, Mason EG, Sorensson CT (2008) Productivity of radiata pine (Pinus radiata D. Don.) clones in monoclonal and clonal mixture plots at age 12years. For Ecol Man 255(1):140–148. doi:10.1016/j.foreco.2007.08.033
Stackpole DJ, Vaillancourt RE, de Aguiar M, Potts BM (2010) Age trends in genetic parameters for growth and wood density in Eucalyptus globulus. Tree Genet Genom 6(2):179–193
Stanger TK, Galloway GM, Retief EC (2011) Final results from a trial to test the effect of plot size on Eucalyptus hybrid clonal ranking in Coastal Zululand, South Africa. South For 73(3–4):131–135
Stram DO, Lee JW (1994) Variance components testing in the longitudinal mixed effects setting. Biometrics 57:1138–1147
Vergara R, White TL, Huber DA, Shiver BD, Rockwood DL (2004) Estimated realized gains for first-generation slash pine (Pinus elliottii var. elliottii) tree improvement in the southeastern United States. Can J For Res 34:2587–2600
Verryn SD, Snedden CL, Eatwell KA (2009) A comparison of deterministically predicted genetic gains with those realised in a South African Eucalyptus grandis breeding program. South For 71(2):141–146
Volker PW (2002) Quantitative genetics of Eucalyptus globulus, E. nitens and their F1 hybrid. Ph.D. thesis, Department of Botany, The University of Tasmania, Hobart, Tasmania
Weng YH, Tosh K, Adam G, Fullarton MS, Norfolk C, Park YS (2008) Realized genetic gains observed in a first generation seedling seed orchard for jack pine in New Brunswick, Canada. New For 36(3):285–298. doi:10.1007/s11056-008-9100-0
White DA, Battaglia M, Bruce J, Benyon R, Beadle C, McGrath J, Kinal J, Crombie S, Doody T (2009) Water-use efficient plantations—separating the wood from the leaves. Forest and Wood Products Australia Limited, Melbourne, Victoria
Ye TZ, Jayawickrama KJS (2008) Efficiency of using spatial analysis in first-generation coastal Douglas-fir progeny tests in the US Pacific Northwest. Tree Genet Genom 4(4):677–692. doi:10.1007/s11295-008-0142-4
Ye TZ, Jayawickrama KJS, St Clair JB (2010) Realized gains from block-plot coastal Douglas-fir trials in the northern Oregon Cascades. Silvae Genet 59(1):29–39
Acknowledgements
The authors acknowledge and thank the field staff who have assisted with establishing, managing, and measuring the 1999 Elders Forestry E. globulus trials; Wayne Davis, Keith Smith, Shanu Nunn, Kristy Ratcliffe, and Jacinta Baird. We are also grateful to two anonymous reviewers who made important contributions to the improvement of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by R. Burdon
Rights and permissions
About this article
Cite this article
Callister, A.N., England, N. & Collins, S. Predicted genetic gain and realised gain in stand volume of Eucalyptus globulus . Tree Genetics & Genomes 9, 361–375 (2013). https://doi.org/10.1007/s11295-012-0558-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11295-012-0558-8