
Abstract
The α-glucosidase (AGL) from Aspergillus niger has been applied to produce isomaltooligosaccharides. In the present study, various factors which affect the yield of recombinant AGL, produced by engineered Pichia pastoris, were investigated. The expression level reached 5.5 U ml−1 in bioreactor after optimization of parameters of initial induction cell density, induction temperature and methanol concentration. In addition, it was found that coexpression of protein disulfide isomerase (PDI) inhibited the growth of the engineered P. pastoris strains and had an adverse effect on the production of AGL, while codon optimization of native A. niger α-glucosidase encoding gene (aglu) resulted in a significant enhancement of enzyme production, which reached 10.1 U ml−1. We believe that yield of AGL is increased by codon optimization as a result of enhanced translation efficiency as well as more stable mRNA secondary structure. In contrast, PDI coexpression under the control of alcohol oxidase promoter (PAOX1) seems to be less efficient in helping disulfide bond formation in AGL while probably induce unfolded protein response, which further leads to cell apoptosis and increased protein degradation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
α-Glucosidases (EC3.2.1.20, AGL) are a group of typical exo-type carbohydrases, which catalyze the liberation of α-glucose from non-reducing terminals of substrates (Kimura et al. 1992). Aspergillus niger AGL can not only hydrolyze glycosides but also transfer a glucosyl residue to the 6-OH of the accepting unit and yield isomaltose, panose, isomaltotriose and tetrasaccharides from maltose (Duan et al. 1994; McCleary et al. 1989). In the industry of functional oligosaccharides, the transglycosylation activity of A. niger AGL has been applied to produce isomaltooligosaccharides (IMOs). However, the low yield of AGL from wild-type A. niger makes it uneconomical for large-scale industrial application. Genetic engineering approach has been utilized to heterogeneously express this enzyme. A. niger AGL was expressed in A. nidulans (Nakamura et al. 1997) and E. nidulans (Ogawa et al. 2006), however, both of the expression levels were reported to be less than 1 U ml−1.
The yeast Pichia pastoris expression system is widely used for the expression of various proteins due to its high production and secretion efficiency (Romanos et al. 1992). It is commonly known that overexpression of heterologous proteins exerts stress on the host cells thus largely influencing the production of an expression system (Mattanovich et al. 2004). In various cases, physiological limitations that reduce the final yield were identified. Potential limiting factors are the poor codon usage bias of the expressed gene (Hu et al. 2006; Li et al. 2008), the gene copy number (Clare et al. 1991), inappropriate promoters (Hohenblum et al. 2004), translocation determined by the secretion signal peptide (Koganesawa et al. 2001), inefficiency of protein folding and assembly in the endoplasmic reticulum (ER) (Xu et al. 2005), as well as protein turnover by proteolysis (Jahic et al. 2003). Many strategies, including fermentation optimization, high heterologous gene copy number, codon optimization, coexpressing chaperones, using strong promoters, etc., have been developed to improve heterologous expression of proteins in P. pastoris. However, so far, there is no conclusion for that which kinds of strategies benefits for selected target protein since that expression ability of each gene is unique in heterologous host.
Overexpression of heterologous proteins can be seen as an intracellular stress inducer leading to overloading of the secretory pathway, typically in the ER, and, generally, the rate-limiting step is protein transportation from ER to Golgi apparatus (Chapman et al. 1998). It has previously been demonstrated that overexpression of protein disulfide isomerase (PDI), which is an essential eukaryotic protein from the thioredoxin super family that catalyzes the oxidation, reduction, and isomerization of disulfide bonds, resulted in increased secretion of various heterologous proteins in P. pastoris including malaria vaccine candidate, bovine follicle-stimulating hormone and single chain antibody fragments (Damasceno et al. 2007; Huo et al. 2007; Tsai et al. 2006).
In addition, it has now been indicated that the difference in codon usage between the wild-type cell and the expression host can affect the expression level of recombinant proteins (Teng et al. 2007). The excessive presence of rare codons which are correlated with low levels of their cognate tRNA species in host cells can result in ribosome stalling, slow translation, and translation errors (Roche and Sauer 1999). Therefore, the codon optimization technique has been widely used to increase the expression of foreign proteins such as xylanase, glucose oxidase and anti-ErbB2 single-chain antibody (Gao et al. 2012; Hu et al. 2006; Jia et al. 2012) and usually results in several-fold increase in target protein production.
Previously, the cDNA of α-glucosidase was cloned from the total RNA of A. niger by RT-PCR and expressed in P. pastoris in our laboratory, the activity of the recombinant enzyme in culture supernatant of a 3 l bioreactor reached to 3.51 U ml−1 (Chen et al. 2010). In the present study, to investigate the rate-limiting factors in the overexpression of AGL in P. pastoris and further enhance the expression level, we optimized the fermentation parameters of engineered P. pastoris, coexpressed ER resident chaperone PDI, and optimized the native gene based on the preferred codon usage of P. pastoris. Our results showed that the yield of recombinant protein reached 10.1 U ml−1, which is the highest expression level of recombinant AGL reported so far.
Materials and methods
Strains, vectors, reagents and media
Pichia pastoris strain KM71/pPIC9K-aglu and recombinant plasmid pPICZ A-PDI was previously constructed in our lab (Zhang et al. 2011). The EZ-10Spin Column Plasmid Mini-Preps kit, agarose gel DNA purification kit, restriction enzymes, and T4 DNA ligase were obtained from TakaRa (Dalian, China). p-Nitrophenyl-α-d-glucopyranoside (pNPG) was obtained from Seebio Biotech. Inc (Shanghai, China). Other chemicals were obtained from Sinopharm Chemical Reagent Co. Ltd. (Shanghai, China). DNA primers were synthesized by Shanghai Sangon Biological Engineering Technology & Services Co. Ltd. (Shanghai, China). DNA sequencing was performed by Shanghai Sangon Biological Engineering Technology & Services Co. Ltd. (Shanghai, China). YPD, MD, BMGY, BMMY, BSM were prepared according to the EasySelect™ Pichia Expression Kit for the cultivation of yeast.
Fermentation optimization of P. pastoris strain KM71/pPIC9K-aglu
The strategy of 2-phase cultivation was utilized and further optimized for the production of AGL in P. pastoris KM71/pPIC9K-aglu in a 3 l bioreactor (BIOFLO 110, America). The fermentation began at batch growth phase at 30 °C and pH 5, and the pH was maintained with ammonium hydroxide. The level of dissolved oxygen was maintained about 20 % by a cascaded control of agitation rate and aeration rate. With a rapid increase of dissolved oxygen, the exponential glycerol feeding was carried out to achieve high cell density. Different initial cell concentrations were obtained (38.4, 76.8, 115.2, 153.6 g l−1) during the first glycerol feeding phase. Then 2 % volume of methanol solution was added to the bioreactor at the beginning of the second methanol induction phase. Effect of different induction temperature (22, 26, 30 °C) and different levels of methanol concentration (0.1, 1, 2 %) controlled by the methanol online control station (FC2002, East China University of Science and Technology) on the expression of AGL were studied. The optimization processes were carried out step by step.
Coexpression of PDI in P. pastoris KM71/pPIC9K-aglu
In our laboratory, the PDI gene cloned from the genome of P. pastoris was previously integrated into pPICZ A under the control of the strong alcohol oxidase promoter (PAOX1) (Zhang et al. 2011). P. pastoris KM71/pPIC9K-aglu was transformed with SacI-linearized pPICZ A-PDI and selected in MD/G418 plates, further screening on the YPD plate containing 100 μg ml−1 Zeocin (Invitrogen). The presence of the PDI gene in the transformants was confirmed by bacterial colony PCR using GACTGGTTCCAATTGACAAGC as forward primer and GCAAATGGCATTCTGACATCC as reverse primer. For expression in shake flasks, single colonies were grown in 10 ml of YPD for 24 h then inoculated in 50 ml BMGY medium. Cells were harvested, resuspended in 25 ml BMMY with an initial methanol addition of 4 % (v/v). Cultures were grown in a shaker at 30 °C and 200 rpm. Methanol was added to a final concentration of 0.5 % (v/v) every 24 h to maintain induction. Samples were centrifuged at 12,000g for 10 min, the supernatant and the cell pellet were kept at −80 °C for further analysis. Then the cells were washed and resuspended in 50 mmol l−1 phosphate buffer (pH 7.0), disrupted by high pressure homogenization for cell-free extracts. The intracellular PDI expression level was detected by SDS-PAGE. All induction experiments were performed in triplicates. Fermentation experiments in 3 l bioreactor were carried out using the optimized conditions.
Codon optimization and synthesis of the modified gene
The codon usage of aglu (NCBI 4991096) from A. niger was analyzed using Graphical Codon Usage Analyser (http://gcua.schoedl.de/), and optimized by replacing the rare codons in P. pastoris with preferred ones by DNAworks software (http://mcl1.ncifcrf.gov/dnaworks/) while preserving the protein sequence. The mRNA structure and free energy of the folding mRNA was analyzed by the RNA Structure 5.2 Program. DNAMAN was used to analyze the restriction enzyme sites of the resulting DNA sequence. The optimized gene (aglu OP) was synthesized by Shanghai Sangon Biological Engineering Technology & Services Co. Ltd. (Shanghai, China).
Expression of aglu OP in P. pastoris
The synthetic DNAs were amplified by PCR using appropriate sets of forward primer (ATTAATGCGGCCGCGTCCACCACTGCCCCTTCG) and reverse primer (AGCACTAGCGGCCGCCCATTCCAATACCCAGTTTTCC), NotI restriction site (underlined) was designed into the primers. The purified PCR product was ligated into pPIC9K yielding the recombinant plasmid pPIC9K-aglu OP, which was identified by restriction analysis and sequencing. The recombinant plasmid was linearized with BglII and then electroporated into P. pastoris KM71. The single colonies of recombinant P. pastoris clone were isolated from 1.0 mg ml−1 YPD/G418 plate. Expression in shake flasks and 3 l bioreactor were performed as described above.
Assay of AGL activity
The hydrolytic activity of AGL was determined as the amount of p-nitrophenol (pNP) released from pNPG. The reaction mixture contained 1 ml of 100 mmol l−1 sodium acetate buffer (pH 5.5), 0.05 ml of 10 mmol l−1 pNPG, and 0.05 ml of enzyme appropriately diluted by sodium acetate buffer (pH 5.5). The reaction was incubated at 50 °C for 15 min and terminated by 1 ml of 1 mol l−1 sodium carbonate solution. One unit (U) of enzyme activity was defined as the amount of 1 μmol l−1 pNP produced per min under the above conditions (Chen et al. 2010).
Results
Fermentation optimization of P. pastoris strain KM71/pPIC9K-aglu
The yield of AGL in our preliminary scale-up from shake flask to 3 l bioreactor cultures was 3.51 U ml−1 (Chen et al. 2010). To increase the expression level, fermentation parameters in a 3 l bioreactor were investigated. The results indicated that initial induction cell density, induction temperature and methanol concentration had different effects on AGL expression and the initial induction cell density of the culture was shown to be the most important factor (Fig. 1). As shown in Fig. 1a, to some degree, the AGL activity was in proportion to the initial induction cell density. When the initial cell density increased from 76.8 to 115.2 g l−1, the enzyme activity showed a corresponding increase from 3.51 to 4.73 U ml−1. However, the enzyme activity declined sharply to 3.22 U ml−1 when further increasing the initial induction cell density to 153.6 g l−1. The optimized condition was obtained as following: when the initial cell concentration reached 115.2 g l−1, the induction phase began during which temperature and methanol concentration were maintained at 26 °C and 1 %, respectively.

Time courses of AGL activity and protein content in the induction phase by P. pastoris KM71/pPIC9K-aglu in a 3 l bioreactor. a Effect of initial induction cell density on the activity of AGL. 38.4 g l−1 (filled square), 76.8 g l−1 (filled circle), 115.2 g l−1 (filled triangle), 153.6 g l−1 (open square). b Effect of induction temperature on the activity of AGL. 22 °C (filled square), 26 °C (filled circle), 30 °C (filled triangle). c Effect of methanol concentration on the activity of AGL. 0.1 % (filled square), 1 % (filled circle), 2 % (filled triangle). d Time courses of AGL activity (filled square) and protein content (filled circle) in the induction phase by P. pastoris KM71/pPIC9K-aglu in a 3 l bioreactor after fermentation optimization
Coexpression of PDI in P. pastoris KM71/pPIC9K-aglu
PDI is an essential eukaryotic protein of the ER which helps in rearrangement of incorrect disulfide pairings by isomerase activity. According to the prediction of the disulphide bonds by the DiANNA webserver (http://clavius.bc.edu/~clotelab/DiANNA/), A. niger α-glucosidase contains three pair of disulphide bonds, which was confirmed based on the analysis of chemical modification using fluorescent SH reagents by a previous report (Kimura 1998). Therefore, in the present study, in order to improve the production of recombinant A. niger α-glucosidase, PDI was selected to coexpress with the enzyme.
To obtain recombinant P. pastoris with coexpression of PDI, the plasmid of pPICZ A-PDI, which containing the encoding gene of PDI from P. pastoris, was transformed into P. pastoris KM71/pPIC9K-aglu. Three resulting colonies screened on YPD/zeocin plate were chosen for further cultivation in shake flasks after confirmation of the insertion of PDI by bacterial colony PCR. SDS-PAGE analysis showed that there was an increased expression level of a 57 kDa protein (Fig. 2), which was in agreement with the calculated molecular weight of PDI and also that from a previous report (Gilbert 1998). However, the coexpression of PDI seemed to have an adverse effect on AGL production, especially in the third strain (Fig. 3a). For further confirmation, the first recombinant P. pastoris strain KM71/pPIC9K-aglu/pPICZ A-PDI which showed the highest enzyme activity in flasks was cultivated in a 3 l bioreactor using the above optimized fermentation conditions. As expected, the AGL expression level in the new strain showed no significant difference from P. pastoris KM71/pPIC9K-aglu, however, the cell growth was obviously inhibited. The final cell density decreased from 203.5 to 169.0 g l−1 (Fig. 3b).

SDS-PAGE analysis of expressed intracellular protein of PDI after shake-flask cultivation for 96 h. Lane 1 protein weight marker; lane 2 intracellular proteins of P. pastoris KM71/pPIC9K-aglu; lanes 3–5 intracellular proteins of P. pastoris KM71/pPIC9K-aglu/pPICZ A-PDI

Effect of PDI coexpression on AGL production. a AGL secretion in shake flasks by PDI coexpression. Control: P. pastoris KM71/pPIC9K-aglu; 1–3: P. pastoris KM71/pPIC9K-aglu/pPICZ A-PDI. The values given are the average of separate experiments performed in triplicate. The error bars indicate standard deviations. b Comparison of cell growth and AGL production in the induction phase in a 3 l bioreactor between P. pastoris KM71/pPIC9K-aglu and KM71/pPIC9K-aglu/pPICZ A-PDI. Symbols biomass concentration (filled square) and AGL activity (filled circle) of P. pastoris KM71/pPIC9K-aglu; biomass concentration (open square) and AGL activity (open circle) of KM71/pPIC9K-aglu/pPICZ A-PDI. The values given are the average of separate experiments performed in triplicate. The error bars indicate standard deviations
Synthesis of codon-optimized gene and expression in P. pastoris
Analysis by Graphical Codon Usage Analyser revealed that the native aglu is harboring almost 30 % of rare codons such as TCG (Ser), CUC (Leu), UGG (Ser) and GCG (Ala), which shared less than 10 % of usage percentage in P. pastoris. The rare codons in aglu were replaced by preferred ones (Table 1), and the G+C content was correspondingly adjusted from 50.0 to 44.2 %. In addition, the mRNA secondary structure around the start codon was avoided. The free energy of folding mRNA was increased from −992.1 to −810.2 kcal/mol compared with the native gene according to RNA Structure 5.2 Program, which indicated more stable mRNA secondary structure after codon optimization.
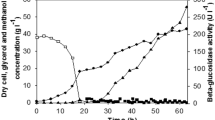
The modified gene was cloning into P. pastoris KM71. The recombinant P. pastoris KM71/pPIC9K-aglu OP, showing higher AGL activity in shake flask culture, was chosen to scale-up fermentation in a 3 l bioreactor. The AGL activity in the supernatant reached 10.1 U ml−1 after 110 h induction (Fig. 4). In contrast to the expression of aglu in P. pastoris (5.5 U ml−1), the expression level of codon optimized gene was increased by 1.8-fold. SDS-PAGE revealed a 140 kDa band in the supernatant of the KM71/pPIC9K-aglu OP, which was consistent in size with the expected molecular mass of AGL (Fig. 5).

Time courses of AGL activity (filled square) and protein content (filled circle) in the induction phase by P. pastoris KM71/pPIC9K-aglu OP in a 3 l bioreactor. The values given are the average of separate experiments performed in triplicate. The error bars indicate standard deviations

SDS-PAGE analysis of culture supernatant of P. pastoris KM71/pPIC9K-aglu OP in a 3 l bioreactor in the induction phase. Lanes 1–6 culture supernatant of recombinant P. pastoris after methanol induction for 20, 40, 60, 80, 100, 110 h, respectively; lane 7 protein weight marker
Discussion
AGL has been widely applied to produce IMOs for its transglycosylation activity in the industry of functional oligosaccharides (Duan et al. 1994). In the present study, the effect of fermentation conditions, coexpression of PDI, as well as codon usage on heterogeneous expression of AGL in P. pastoris were investigated.
Usually, the optimization of fermentation conditions is a basic principle to enhance the enzyme production. In the present study, the effect of the fermentation parameters, including initial induction cell density, induction temperature, as well as methanol concentration, on the production of AGL by P. pastoris KM71/pPIC9K-aglu in a 3L bioreactor were first studied. It was found that, among the three parameters, the initial induction cell density was the major factor. In the optimum condition, AGL activity in the culture supernatant was increased to 5.5 U ml−1, which was 1.57-fold to that of 3.5 U ml−1 attained before fermentation optimization. This is a very successful strategy to increase expression of recombinant proteins in P. pastoris. Nevertheless, the ER environment involving disulfide bond formation has to be considered as an important limiting step to protein secretion (Damasceno et al. 2007). Therefore, we wanted to investigate the effects of PDI coexpression to further improve secretion of the AGL.
The results of the effect of PDI coexpression on the growth of P. pastoris cell showed that the PDI coexpression strains displayed different growth characteristics and their growth seemed to be significantly repressed. This phenomenon was unexpected and had not been previously observed even though there were certain amounts of studies about coexpression of PDI with foreign proteins in P. pastoris (Huo et al. 2007; Tsai et al. 2006). Previously, it was reported that overexpression of PDI under the control of the strong alcohol oxidase promoter (PAOX1) seemed to induce stress and the cell may respond by further increasing levels of chaperone and foldase in the ER through unfolded protein response (UPR) (Damasceno et al. 2007; Schröder and Kaufman 2005). It was found that, in metazoan cells, under prolonged ER stress, the UPR can ultimately trigger apoptotic cell death (Bravo et al. 2012). According to the observation of depressed cell growth in the present study, it is assumed that there might be a connection between UPR and apoptosis in PDI coexpression system of P. pastoris. Further studies should be performed to study the mechanisms.
In addition, it was found that PDI coexpression decreased AGL expression when compared with that of control (P. pastoris KM71/pPIC9K-aglu). This result was unexpected because AGL contains three disulfide bonds and it was expected that PDI involvement would increase the rate of disulfide bond formation which benefited recombinant protein folding. The possible reason for this phenomenon might be that: firstly, coexpression of PDI leads to depression of cell growth in the engineered P. pastoris as mentioned above, and secondly, an induced increase in chaperone level in the ER lumen through UPR can also lead to increased protein degradation through ER associated protein degradation (ERAD) (Travers et al. 2000), even though the degradation of misfolded protein by itself will not reduce the yield, retrotranslocation of protein back to the cytosol can be considered as a competition for protein translocation, which would limit the product entering the secretion pathway (Mattanovich et al. 2004).
In contrast with the negative results of AGL production after coexpression of PDI, the AGL expression was significantly enhanced (10.1 U ml−1) after codon optimization. According to the translational efficiency hypothesis related to translation initiation and elongation rates for explaining the codon usage bias in the organisms (Xia 1998), it was speculated that the increased AGL expression by codon optimization would be mainly due to the enhanced translation efficiency. Furthermore, considering the promotion of expression level of AGL by codon optimization corresponding to increased demand of foldase, we also coexpressed PDI in KM71/pPIC9K-aglu OP and it was found that there is no further improvement of AGL production (data not shown). These results suggested, compared with protein folding, the inefficient translation is the most important factor limiting the yield of AGL production. The overexpression of the foldase PDI may not be the only solution for poor secretion of recombinant proteins in yeast, as it fails to promote the secretion of a rich cysteine-containing protein such as AGL.
It has been reported that the AGL was subjected to a limited proteolysis after secretion from the A. niger and the mature wild-type enzyme was actually a heterosubunit protein, in which the two heterosubunits were composed of residues 26–252 and 267–985 of aglu, respectively (Nakamura et al. 1997). In the engineered P. pastoris expression system, this kind of protease was also suggested to exist (Chen et al. 2010). If this is the truth, considering the enhanced expression level of AGL obtained in the present study, it may be considered that the required amount of protease, which hydrolyzes the AGL to form its mature form, is sufficient in P. pastoris, thus not being the limiting factor with the increased expression capacity of AGL.
In conclusion, after fermentation optimization and further codon optimization, the expression level of AGL reached 10.1 U ml−1, which is the highest yield reported so far. Our studies may provide the foundation for the application of AGL in the industry of IMOs production as well as a model to enhance expression level of other heterologous proteins in P. pastoris.
References
Bravo R, Gutierrez T, Paredes F, Gatica D, Rodriguez AE, Pedrozo Z, Chiong M, Parra V, Quest AFG, Rothermel BA, Lavandero S (2012) Endoplasmic reticulum: ER stress regulates mitochondrial bioenergetics. Int J Biochem Cell B 44(1):16–20. doi:10.1016/j.biocel.2011.10.012
Chapman R, Sidrauski C, Walter P (1998) Intracellular signaling from the endoplasmic reticulum to the nucleus. Annu Rev Cell Dev Biol 14(1):459–485
Chen DL, Tong X, Chen SW, Chen S, Wu D, Fang SG, Wu J, Chen J (2010) Heterologous expression and biochemical characterization of α-glucosidase from Aspergillus niger by Pichia pastroris. J Agric Food Chem 58(8):4819–4824
Clare JJ, Romanes MA, Rayment FB, Rowedder JE, Smith MA, Payne MM, Sreekrishna K, Henwood CA (1991) Production of mouse epidermal growth factor in yeast: high-level secretion using Pichia pastoris strains containing multiple gene copies. Gene 105(2):205–212
Damasceno LM, Anderson KA, Ritter G, Cregg JM, Old LJ, Batt CA (2007) Cooverexpression of chaperones for enhanced secretion of a single-chain antibody fragment in Pichia pastoris. Appl Microbiol Biotechnol 74(2):381–389. doi:10.1007/s00253-006-0652-7
Duan KJ, Sheu DC, Lin MT, Hsueh HC (1994) Reaction mechanism of isomaltooligosaccharides synthesis by α-glucosidase from Aspergillus carbonarious. Biotechnol Lett 16(11):1151–1156
Gao Z, Li Z, Zhang Y, Huang H, Li M, Zhou L, Tang Y, Yao B, Zhang W (2012) High-level expression of the Penicillium notatum glucose oxidase gene in Pichia pastoris using codon optimization. Biotechnol Lett 34(3):507–514
Gilbert HF (1998) Protein disulfide isomerase. Methods Enzymol 290:26–50
Hohenblum H, Gasser B, Maurer M, Borth N, Mattanovich D (2004) Effects of gene dosage, promoters, and substrates on unfolded protein stress of recombinant Pichia pastoris. Biotechnol Bioeng 85(4):367–375
Hu S, Li L, Qiao J, Guo Y, Cheng L, Liu J (2006) Codon optimization, expression, and characterization of an internalizing anti-ErbB2 single-chain antibody in Pichia pastoris. Protein Expr Purif 47(1):249–257. doi:10.1016/j.pep.2005.11.014
Huo X, Liu Y, Wang X, Ouyang P, Niu Z, Shi Y, Qiu B (2007) Co-expression of human protein disulfide isomerase (hPDI) enhances secretion of bovine follicle-stimulating hormone (bFSH) in Pichia pastoris. Protein Expr Purif 54(2):234–239
Jahic M, Gustavsson M, Jansen AK, Martinelle M, Enfors SO (2003) Analysis and control of proteolysis of a fusion protein in Pichia pastoris fed-batch processes. J Biotechnol 102(1):45–53
Jia H, Fan G, Yan Q, Liu Y, Yan Y, Jiang Z (2012) High-level expression of a hyperthermostable Thermotoga maritima xylanase in Pichia pastoris by codon optimization. J Mol Catal B Enzym 78:72–77
Kimura A (1998) Structure and catalytic mechanism of crystalline α-glucosidase from Aspergillus niger. J Appl Glycosci 45(1):71–79
Kimura A, Takata M, Sakai O, Matsui H, Takai N, Takayanagi T, Nishimura I, Uozumi T, Chiba S (1992) Complete amino acid sequence of crystalline alpha-glucosidase from Aspergillus niger. Biosci Biotechnol Biochem 56(8):1368–1370
Koganesawa N, Aizawa T, Masaki K, Matsuura A, Nimori T, Bando H, Kawano K, Nitta K (2001) Construction of an expression system of insect lysozyme lacking thermal stability: the effect of selection of signal sequence on level of expression in the Pichia pastoris expression system. Protein Eng 14(9):705–710
Li ZX, Hong GQ, Wu ZH, Hu B, Xu J, Li L (2008) Optimization of the expression of hepatitis B virus e gene in Pichia pastoris and immunological characterization of the product. J Biotechnol 138(1–2):1–8. doi:10.1016/j.jbiotec.2008.07.1989
Mattanovich D, Gasser B, Hohenblum H, Sauer M (2004) Stress in recombinant protein producing yeasts. J Biotechnol 113(1–3):121–135. doi:10.1016/j.jbiotec.2004.04.035
McCleary BV, Gibson TS, Sheehan H, Casey A, Horgan L, O’Flaherty J (1989) Purification, properties, and industrial significance of transglucosidase from Aspergillus niger. Carbohydr Res 185(1):147–162
Nakamura A, Nishimura I, Yokoyama A, Lee DG, Hidaka M, Masaki H, Kimura A, Chiba S, Uozumi T (1997) Cloning and sequencing of an alpha-glucosidase gene from Aspergillus niger and its expression in A. nidulans. J Biotechnol 53(1):75–84
Ogawa M, Nishio T, Minoura K, Uozumi T, Wada M, Hashimoto N, Kawachi R, Oku T (2006) Recombinant alpha-glucosidase from Aspergillus niger. Overexpression by Emericella nidulans, purification and Characterization. J Appl Glycosci 53(1):13
Roche ED, Sauer RT (1999) SsrA-mediated peptide tagging caused by rare codons and tRNA scarcity. EMBO J 18(16):4579–4589
Romanos MA, Scorer CA, Clare JJ (1992) Foreign gene expression in yeast: a review. Yeast 8(6):423–488. doi:10.1002/yea.320080602
Schröder M, Kaufman RJ (2005) ER stress and the unfolded protein response. Mutat Res 569(1):29–63
Teng D, Fan Y, Yang YL, Tian ZG, Luo J, Wang JH (2007) Codon optimization of Bacillus licheniformis beta-1,3-1,4-glucanase gene and its expression in Pichia pastoris. Appl Microbiol Biotechnol 74(5):1074–1083. doi:10.1007/s00253-006-0765-z
Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P (2000) Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. CELL-CAMBRIDGE MA- 101(3):249–258
Tsai CW, Duggan PF, Shimp RL, Miller LH, Narum DL (2006) Overproduction of Pichia pastoris or Plasmodium falciparum protein disulfide isomerase affects expression, folding and O-linked glycosylation of a malaria vaccine candidate expressed in P. pastoris. J Biotechnol 121(4):458–470. doi:10.1016/j.jbiotec.2005.08.025
Xia X (1998) How optimized is the translational machinery in Escherichia coli, Salmonella typhimurium and Saccharomyces cerevisiae? Genetics 149(1):37–44
Xu P, Raden D, Doyle FJ III, Robinson AS (2005) Analysis of unfolded protein response during single-chain antibody expression in Saccaromyces cerevisiae reveals different roles for BiP and PDI in folding. Metab Eng 7(4):269–279
Zhang J, Wu D, Chen J, Wu J (2011) Enhancing functional expression of β-glucosidase in Pichia pastoris by co-expressing protein disulfide isomerase. Biotechnol Bioprocess Eng 16(6):1196–1200
Acknowledgments
This work was financially supported by the Fundamental Research Funds for the Central Universities (JUSRP211A05), the National Natural Science Foundation of China (30970057 and 31100048), the open program for key laboratory of industrial biotechnology ministry of education (KLIB-KF200904) and the 111 Project (No. 111-2-06).
Author information
Authors and Affiliations
Corresponding author
Additional information
Xu Liu and Dan Wu contributed equally to this study and share first authorship.
Rights and permissions
About this article
Cite this article
Liu, X., Wu, D., Wu, J. et al. Optimization of the production of Aspergillus niger α-glucosidase expressed in Pichia pastoris . World J Microbiol Biotechnol 29, 533–540 (2013). https://doi.org/10.1007/s11274-012-1207-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11274-012-1207-y