
Abstract
A β-agarase gene hz2 with 2,868 bp was cloned from the marine agarolytic bacterium Agarivorans sp. HZ105. It encoded a mature agarase HZ2 of 102,393 Da (920 amino acids). Based on the amino acid sequence similarity, agarase HZ2 was assigned to the glycoside hydrolase family 50. The β-agarase shared a gene sequence identity of 98.6% with the reported but much less characterized β-agarase agaB from Vibrio sp. JT0107. Its recombinant agarase rHZ2 was produced in E. coli cells and purified to homogeneity. The agarase rHZ2 degraded agarose and neoagarooligosaccharides with degrees of polymerization above four, to yield neoagarotetraose as the dominant product, which was different from β-agarase agaB of Vibrio sp. JT0107. The agarose hydrolysis pattern suggested that rHZ2 was an endo-type β-agarase. Beta-mercaptoethanol (90 mM) and dithiothreitol (9 mM) increased the agarase activity of rHZ2 by 72.9% and 17.3% respectively, while SDS (9 mM) inhibited the activity completely. The agarase activity was independent of Na+, K+, Mg2+ and Ca2+. The maximal enzyme activity was observed at 40°C and pH 7. The kinetic parameters K m, V max, K cat, and K cat/K m values toward agarose of agarase rHZ2 were 5.9 mg ml−1, 235 U mg−1, 401 s−1 and 6.8 × 105 M−1 s−1, respectively. Agarase rHZ2 could have a potential application in the production of bioactive neoagarotetraose.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Agar, which consists of agarose and agaropectin, is a component present in the cell walls of some red algae. Agarase is a kind of enzyme that can degrade agarose and mainly comes from agar-degrading bacteria. Agarases are classified into two groups according to their mode of action on agarose, namely α-agarase (EC 3.2.1.158) and β-agarase (EC3.2.1.81). α-agarases cleave α-1,3 linkages in agarose to produce agarooligosaccharides, while β-agarases hydrolyze β-1,4 linkages in agarose to yield neoagarooligosaccharides (NAOS) with various degrees of polymerization (DP).
NAOS have been reported to have various bioactive effects such as skin-moisturizing and whitening effects (Kobayashi et al. 1997; Lee et al. 2008; Jang et al. 2009), antioxidative effects (Wang et al. 2004) and prebiotic effects (Hu et al. 2006).
According to the CAZy database (http://www.cazy.org), β-agarases have been mainly classified into three glycoside hydrolase (GH) families, GH16, GH50 and GH86 based on the amino acid sequence similarity. To date, only a few β-agarases belong to the GH50 and GH86 families while most β-agarases belong to the GH16 family which has most abundant members such as agarase, carrageenase, glucanase, galactosidase and laminarinase (Fu and Kim 2010). In the GH50 family, only partial β-agarases have been characterized (Sugano et al. 1993, 1994; Ohta et al. 2005; Lee et al. 2006; Kim et al. 2010; Nikapitiya et al. 2010). Most of these GH50 agarases produced neoagarobiose as the main final product of agarose degradation.
The agarolytic marine bacterium Agarivorans sp. HZ105 which could produce multi-agarases as described by Hu et al. (2009) was isolated from marine sediments in the seacoast area of Xiamen, China. Based on the mass spectrum (MS) results of the purified agarases, all of the three extracellular agarases from strain Agarivorans sp. HZ105 matched the GH50 β-agarases. In this study we reported the gene cloning, expression and characterization of a neoagarotetraose-producing β-agarase from Agarivorans sp. HZ105.
Materials and methods
Bacterial strain, plasmid, medium, culture conditions
The bacterium Agarivorans sp. HZ105 was cultured on agar medium at 25°C. E. coli DH5α and plasmid pET-32a(+) (Novagen) were used for preparation of recombinant plasmids. E. coli BL21 (DE3) was used for enzyme expression. E. coli cells were cultured in Luria–Bertani (LB) broth with 100 μg ampicillin/ml when necessary. Agarose (Agarose LE) was purchased from MDbio, Inc.
Cloning of the agarase gene
The MS results of the agarases produced by strain Agarivorans sp. HZ105 (Hu et al. 2009) indicated that strain Agarivorans sp. HZ105 may have similar agarases to Vibrio sp. JT0107. Based on the gene sequence of agarase agaB of Vibrio sp. JT0107 (GenBank accession number = D21202), a pair of PCR primers was designed to amplify the β-agarase gene hz2 of Agarivorans sp. HZ105. The forward primer was fhz2f (5′-AACGGATCGTCCAGTACTG-3′). The reverse primer was fhz2r (5′-TTCAGCATTGTTTTCAGAGC-3′). DNA sequencing was performed at the Beijing Genomics Institute. The reading frames, theoretical isoelectric point and molecular weight (MW) were predicted using the DNAstar program. Database searching was performed with BLAST. Prediction of signal peptide sequence was performed on the SignalP 3.0 server (http://www.cbs.dtu.dk/services/SignalP/).
Expression of the agarase gene
The primers of 2chf and 2chr were used to amplify the agarase gene hz2 without signal peptide sequence (5′–3′, 2chf, GCTCATATGGAAGGCGCTGTTGAAGAT; 2chr, CGCCTCGAGTTTTTTGTAACGAAGCATG). The termination codon was removed in the reverse primer in order to add His-tag to the C-terminal of the recombinant agarase. After gel purification, the PCR products were digested with NdeI and XhoI, and ligated into vector pET-32a(+), yielding the recombinant plasmids, which were then transferred into E. coli BL21(DE3). Transformed E. coli BL21(DE3) were grown at 37°C to mid-growth phase in LB medium containing 100 μg ml−1 ampicillin. The culture temperature was shifted to 25°C and the expression of recombinant agarases was induced with 1 mM IPTG for 12 h.
Purification of recombinant agarase
The induced E. coli cells were harvested by centrifugation and suspended in buffer A (50 mM NaH2PO4, 0.3 M NaCl, pH 8) containing 20 mM imidazole and disrupted on ice by a sonicator. The supernatant of the cell lysate was collected by centrifugation. The His6-tagged agarase rHZ2 was purified with nickel-nitrilotriacetic acid (Ni–NTA) agarose column chromatography (Ni–NTA His Bind Resin 70666, Novagen) under native conditions according to the recommendations of the manufacturer. The purified His6-tagged protein was concentrated and desalted against buffer B (20 mM Na2HPO4/NaH2PO4, pH 7) with centrifugal filter devices. The purity of the recombinant agarase was checked by SDS-PAGE.
Determination of agarase activity
The agarase activity was measured by the release of the reducing sugar equivalent using the 3,5-dinitrosalicylic acid (DNS) method (Miller 1959). A 100 μl sample was added to 900 μl of substrate solution (in 20 mM Tris/HCl, pH 8 buffer) and incubated at 40°C for 20 min. The enzyme reaction was stopped by boiling for 10 min. Then the 1 ml reaction solution was mixed with 2 ml DNS reagent. After being heated at 100°C for 15 min and then cooled, the mixture was diluted to 10 ml with deionized water. Optical density was read at 540 nm, and values for reducing sugars were expressed as d-galactose equivalents. One unit of agarase activity (1 U) was defined as the amount of enzyme that released 1 μmol of reducing sugar (measured as d-galactose) from agarose per minute under the above conditions.
Enzymatic properties of the recombinant agarase
The temperature dependence test for the activity of the recombinant agarase rHZ2 was assayed with 0.25% (w/v) agarose at pH 8 at different temperatures for 15 min. The optimum pH for the activity of rHZ2 was assayed with 0.25% (w/v) agarose at 40°C and various pH for 15 min. The effects of various reagents on rHZ2 activity were examined by determining the activity with 0.25% (w/v) agarose in 20 mM Tris/HCl buffer (pH 8) at 40°C for 20 min in the presence of 9 or 90 mM of various reagents (listed in Table 2). All reactions were performed in three independent experiments.
To determine the kinetic parameters, the enzyme assay was performed at agarose concentrations ranging from 1 to 15 mg ml−1. Reaction mixtures were incubated at 40°C for 10 min, and aliquots (0.5 ml) were taken every 1 min. K m and V max values were calculated using GraphPad Prism version 4.00 for Windows, GraphPad Software, San Diego California USA, http://www.graphpad.com.
Agarose and neoagarooligosaccharides degradation
Thin-layer chromatography (TLC) was used to identify products. Enzymatic hydrolysis of agarose and NAOS was carried out at 40°C in 20 mM Tris/HCl, pH 8 buffer. For the degradation of agarose, 200 μl solution of purified agarase (7.4 U ml−1) was added into the equivalent volume of agarose substrate (1%, w/v, prepared in 20 mM Tris/HCl, pH 8 buffer). For the degradation of NAOS, 50 μl solution of purified agarase (7.4 U ml−1) was added into the equivalent volume of NAOS substrate (prepared in 20 mM Tris/HCl, pH 8 buffer). The reaction mixtures were applied to silica gel 60 TLC plates (Shenghai Inc, Qingdao, China) which were developed using a solvent system composed of 1-butanol/acetic acid/water (2:1:1, by vol.). Spots of oligosaccharides resulting from the hydrolysis of the substrate were visualized by spraying with 10% (v/v) H2SO4 and heating at 85°C.
Because of the lack of commercial products, NAOS mixtures with different DP were prepared by enzymatic hydrolysis of agarose. The 1% (w/v) agarose was hydrolyzed by other β-agarases studied in our lab. The hydrolysis products were recovered from the TLC plates. TLC was performed again to confirm the success of recovery. LC–MS/MS (AB4700 Proteomic Analyzer) was used to determine the molecular weights of self-made oligosaccharides.
Accession number of agarase gene
The nucleotide sequence of agarase gene hz2 was deposited into the GenBank with an accession number of HQ625021.
Results and discussion
Sequence analysis of the agarase gene
A DNA fragment of 3,320 bp was obtained with the primers fhz2f and fhz2r. In this 3,320 bp fragment an ORF of 2,868 bp was found and designated gene hz2 (Fig. 1). The deduced product of gene hz2 is a protein of 955 amino acids with an estimated molecular mass of 106 kDa and an isoelectric point of 4.87. According to the BLAST search results, the encoded protein designated HZ2 showed a high identity with the β-agarases in the Genbank database (Table 1): 99.8% to agaD02 from Agarivorans sp. QM38 (GenBank accession no. ABM90422), 99.4% to agaB from Vibrio sp. JT0107 (GenBank accession no. BAA04744), 99% to agaE from Vibrio sp. PO-303 (GenBank accession no. BAG71428), then only 50% to AgaA from Vibrio sp. LA1 (GenBank accession no. AEB71854). Up to date there are few reports about the matched agarases. Du et al. (2011) reported the gene cloning of agarase agaD02 from Agarivorans sp. QM38, but there was no study on the characterization of the enzyme. Sugano et al. (1994) reported the expression of the partial gene of agarase agaB from Vibrio sp. JT0107 and there was little description about the enzymatic properties. Furthermore, no reports about the agarase agaE from Vibrio sp. PO-303 could be found.

Nucleotide sequence of agarase HZ2 from Agarivorans sp.HZ105 and its deduced amino acid sequence. Nucleotide sequence is shown in uppercase letters. The italic letters of ATG and TAA represent the start codon and stop codon, respectively. The deduced amino acids are shown above the nucleotide sequence. The signal peptide is underlined. The middle part of the sequence is not displayed and replaced by dashed line to save space
The SignalP 3.0 server predicted a signal peptide at the N-terminal of HZ2 and the most likely cleavage site was between the amino acids Ala35 and Glu36 (Fig. 1). When the predicted signal peptide was retained in the recombinant vector, the recombinant protein showed low agarase activity (data not shown). On the other hand, when the signal peptide was removed, a recombinant protein rHZ2 with higher agarase activity was obtained.
Purification of agarase rHZ2
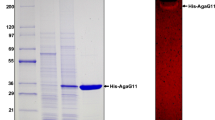
The recombinant agarase rHZ2 was purified to homogeneity from the supernatant of the cell lysate of E. coli cells (Fig. 2). The molecular mass of the rHZ2 was estimated to be 107 kDa by SDS-PAGE analysis, which was close to that estimated from the gene sequence.

SDS-PAGE of purified agarase rHZ2. Lane M, protein markers. Lane 1, purified recombinant agarase rHZ2. SDS-PAGE was performed with 12% (w/v) polyacrylamide gel. Gels were stained with coomassie brilliant blue R-250
Enzymatic properties of the recombinant agarase
The optimal pH for rHZ2 was 7 (Fig. 3). Similar to some reported agarases (Ohta et al. 2005; Lee et al. 2006; Fu et al. 2008), agarase rHZ2 exhibited the maximum agarase activity at 40°C which is above the gelling temperature of agar (around 38°C). Agarase is usually more efficient in solution than in gel state. Therefore, although thermostable agarases would be useful to avoid such gel state, an agarase with a maximum activity at 40°C is already an enzyme which would add some cost to a reactor running at that temperature. Nevertheless, now that the sequence is known (Fig. 1), genetic engineering could be used to reduce the enzyme’s optimum parameters to just above the gel state temperature to avoid such a potential drawback. Alternatively, running the reaction at a lower temperature but with the enzyme still displaying a significant activity could be a viable possibility.

Effects of temperature and pH on agarase activity. The agarase solution having an activity of 2.2 U/ml was used. a Temperature profile of the agarase activity. The reaction was carried out with 0.25% (w/v) agarose at pH 8 in 20 mM Tris/HCl buffers for 15 min. The values on the ordinate are shown as percentages of the agarase activity (100%) observed at 40°C. b pH profile of the agarase activity. The reaction was carried out with 0.25% (w/v) agarose at 40°C for 15 min. The values on the ordinate are shown as percentages of the agarase activity (100%) observed at pH 7. The buffers used were citric acid/Na2HPO4 buffers (0.1 M, pH 4–6), Tris/HCl buffers (20 mM, pH 7–9) and glycine/NaOH buffers (40 mM, pH 10). All reactions were performed in three independent experiments
Table 2 shows that cations of Na+, K+, Mg2+ and Ca2+ displayed no increase but inhibition of the agarase activity suggesting that the enzyme activity of rHZ2 did not depend on them. The activities of some reported agarases required cations. Potin et al. 1993 reported an α-agarase from Alteromonas agarlyticus strain GJ1B whose enzyme activity was dependent upon the presence of calcium ions. Furthermore, the glycoside hydrolase family 86 β-agarase AgrA showed low activity in buffers without CaCl2 (Ohta et al. 2004). It has been known that GH16 agarases have a Ca2+ ion-binding site and Ca2+ ion binding is necessary for the formation of the active form of the enzyme (Allouch et al. 2003). Nevertheless, the activity of GH16 agarase AgaB34 from Agarivorans albus YKW-34 was not affected by metal ions commonly existing in seawater (Fu et al. 2009). 0.1 M NaCl and 10 mM CaCl2 could stimulate the activity of the GH50 agarase agaA11 from the strain Agarivorans sp. JAMB-A11 by 20% and 30% respectively, while the other metal ions abundant in seawater, such as KCl and MgCl2, did not affect the enzyme activity. The GH50 agarase AgaA34, from Agarivorans albus YKW-34, did not require metal ions for its activity (Fu et al. 2008). Therefore, it would seem that the metal ions abundant in seawater have less effect on the activity of GH50 agarases. In this study, the GH50 agarase rHZ2 is inhibited distinctly by 90 mM of Mg2+ and Ca2+ (Table 2) which make it different from other reported agarases.
SDS (9 mM) inhibited the agarase activity of rHZ2 completely, while beta-mercaptoethanol (90 mM) and dithiothreitol (9 mM) increased the activity by 72.9% and 17.3% respectively (Table 2). This indicated the possible existence of thiol in the catalytic site, as the reducing reagent could protect thiol from being oxidized to a disulfide bond.
To understand the mechanism of catalysis of agarases especially the agarases belonging to family 50, determination of the three-dimensional structures of agarase is required.
The kinetic parameters K m, V max, K cat, and K cat/K m values of agarase rHZ2 were 5.9 mg ml−1, 235 U mg−1, 401 s−1 and 6.8 × 105 M−1 s−1, respectively (Table 3). Compared with other agarases listed in Table 3 except agarase AgaB34, rHZ2 had a higher K cat/K m value. Based on the low K m value and high V max, K cat, and K cat/K m values of agarase rHZ2, agarose may be the optimal substrate for rHZ2.
Agarose and NAOS degradation
NAOS with various DP are confirmed by comparing the observed molecular weights with their calculated values. The mass spectra of self-made NAOS were displayed in the supplementary materials (Supplementary Figure 1–7).
As shown in Fig. 4a, in the initial stage of agarose hydrolysis agarase rHZ2 decomposed agarose to yield oligosaccharides with various DP. And after 5 min the dominant end product was neoagarotetraose (NA4) with only a low amount of neoagarobiose (NA2), as well as other oligosaccharides. After 15 min, the dominant end product was neoagarotetraose with low amounts of neoagarobiose. This hydrolysis pattern suggested that rHZ2 was an endo-type β-agarase. Among the agarases that had high homology with rHZ2, only the agarose degradation of agaB from Vibrio sp. JT0107 had been reported. But rHZ2 was different in agarose degradation from agaB which hydrolyzed agarose to yield predominantly NA4 and neoagarohexaose (NA6) (Sugano et al. 1994). And there was no report about the NAOS degradation of agaB.

Agarose and NAOS degradation of rHZ2. a Time course of agarose degradation of rHZ2. Reactions were carried out at 40°C. Two hundred μl solution of purified agarase (7.4 U/ml) was added into the equivalent volume of agarose substrate (1%, w/v, prepared in 20 mM pH 8 Tris/HCl buffer). ST, self-made standards of NAOS confirmed by mass spectrometry. NA2, neoagarobiose; NA4, neoagarotetraose; NA6, neoagarohexaose; NA8, neoagarooctaose. b NAOS degradation of rHZ2. Fifty μl solution of rHZ2 (7.4 U/ml) was added into the equivalent volume of NAOS substrate (prepared in 20 mM Tris/HCl, pH 8 buffer). Reactions were carried out at 40°C for 24 h. Lane 1, rHZ2 + NA4 (7.5 mM); Lane 2, NA4; Lane 3, rHZ2 + NAOS mixture 46 containing NA4 (4.6 mM) and NA6 (4.6 mM); Lane 4, self-made NAOS mixture 46; Lane 5, rHZ2 + NAOS mixture 814 containing each 2.5 mM of NA8, NA10 (neoagarodecaose), NA12 (neoagarododecaose) and NA14 (neoagarotetradecaose); Lane 6, self-made NAOS mixture 814; Lane 7 and lane 8, self-made standards of NAOS
Most of the reported agarases belonging to glycoside hydrolase family 50 degraded agarose to produce NA2 as the dominant end product (Table 3; Sugano et al. 1993; Ohta et al. 2005; Lee et al. 2006). While agarase rHZ2 differed from those GH50 agarases to yield NA4 as the dominant end product.
Agarase rHZ2 hydrolyzed NAOS with DP above 4 to generate NA4 with only a low amount of NA2 but could not degrade NA4 further (Fig. 4b), which was coincident with the result that the main product of agarose degradation by rHZ2 was NA4.
The study by Jang et al. (2009) suggested that the NA4 generated from agar by recombinant β-agarase might be a good candidate as a cosmetic additive for the skin whitening effect. In this study the recombinant β-agarase rHZ2 with a high K cat/K m value toward agarose, which degraded agarose to yield NA4 as the dominant product, might also be useful to produce NA4 in the cosmetic industry.
References
Allouch J, Jam M, Helbert W, Barbeyron T, Kloareg B, Henrissat B, Czjzek M (2003) The three-dimensional structures of two β-agarases. J Biol Chem 278:47171–47180
Du Z, Wang J, Yang L, Chen G (2011) Identification of a marine agarolytic bacterium Agarivorans albus QM38 and cloning and sequencing its beta-agarase genes. Acta Oceanol Sin 30:118–124
Fu XT, Kim SM (2010) Agarase: review of major sources, categories, purification method, enzyme characteristics and applications. Mar Drugs 8:200–218
Fu XT, Lin H, Kim SM (2008) Purification and characterization of a novel beta-agarase, AgaA34, from Agarivorans albus YKW-34. Appl Microbiol Biotechnol 78:265–273
Fu XT, Pan C-H, Lin H, Kim SM (2009) Gene cloning, expression, and characterization of a β-agarase, AgaB34, from Agarivorans albus YKW-34. J Microbiol Biotechnol 19:257–264
Hu B, Gong Q, Wang Y, Ma Y, Li J, Yu W (2006) Prebiotic effects of neoagaro-oligosaccharides prepared by enzymatic hydrolysis of agarose. Anaerobe 12:260–266
Hu Z, Lin BK, Xu Y, Zhong MQ, Liu GM (2009) Production of agarase from a marine agarolytic bacterium Agarivorans sp. HZ105. J Appl Microbiol 106:181–190
Jam M, Flament D, Allouch J, Potin P, Thion L, Kloareg B, Czjzek M, Helbert W, Michel G, Barbeyron T (2005) The endo-beta-agarases AgaA and AgaB from the marine bacterium Zobellia galactanivorans: two paralogue enzymes with different molecular organizations and catalytic behaviours. Biochem J 385:703–713
Jang MK, Lee DG, Kim NY, Yu KH, Jang HJ, Lee SW, Jang HJ, Lee Y, Lee S (2009) Purification and characterization of neoagarotetraose from hydrolyzed agar. J Microbiol Biotechnol 19:1197–1200
Kim HT, Lee S, Lee D, Kim HS, Bang WG, Kim KH, Choi IG (2010) Overexpression and molecular characterization of Aga50D from Saccharophagus degradans 2–40: an exo-type β-agarase producing neo- agarobiose. Appl Microbiol Biotechnol 86:227–234
Kobayashi R, Takisada M, Suzuki T, Kirimura K, Usami S (1997) Neoagarobiose as a novel moisturizer with whitening effect. Biosci Biotechnol Biochem 61:162–163
Lee DG, Park GT, Kim NY, Lee EJ, Jang MK, Shin YG, Park GS, Kim TM, Lee JH, Lee JH, Kim SJ, Lee SH (2006) Cloning, expression, and characterization of a glycoside hydrolase family 50 β-agarase from a marine Agarivorans isolate. Biotechnol Lett 28:1925–1932
Lee DG, Jang MK, Lee OH, Kim NY, Ju SA, Lee SH (2008) Over-production of a glycoside hydrolase family 50 β-agarase from Agarivorans sp. JA-1 in Bacillus subtilis and the whitening effect of its product. Biotechnol Lett 30:911–918
Long M, Yu Z, Xu X (2010) A Novel β-Agarase with high pH stability from marine Agarivorans sp. LQ48. Mar Biotechnol 12:62–69
Miller GL (1959) Use of dinitrosalicylic acid reagent for determination of reducing sugar. Anal Chem 31:426–428
Nikapitiya C, Oh C, Lee Y, Lee S, Whang I, Lee J (2010) Characterization of a glycoside hydrolase family 50 thermostable beta agarase AgrA from marine bacteria Agarivorans sp. AG17. Fish Aquat Sci 13:36–48
Ohta Y, Hatada Y, Nogi Y, Li Z, Ito S, Horikoshi K (2004) Cloning, expression, and characterization of a glycoside hydrolase family 86 -agarase from a deep-sea Microbulbifer-like isolate. Appl Microbiol Biotechnol 66:266–275
Ohta Y, Hatada Y, Ito S, Horikoshi K (2005) High-level expression of a neoagarobiose-producing β-agarase gene from Agarivorans sp. JAMB-A11 in Bacillus subtilis and enzymic properties of the recombinant enzyme. Biotechnol Appl Biochem 41:183–191
Potin P, Richard C, Rochas C, Kloareg B (1993) Purification and characterization of the α-agarase from Atteromonas agarlyticus (Cataldi) comb.nov., strain GJ1B. Eur J Biochem 214:599–607
Sugano Y, Matsumoto T, Kodama H, Noma M (1993) Cloning and sequencing of agaA, a unique agarase 0107 gene from a marine bacterium, Vibrio sp. strain JT0107. Appl Environ Microbiol 59:3750–3756
Sugano Y, Matsumoto T, Noma M (1994) Sequence analysis of the agaB gene encoding a new beta-agarase from Vibrio sp. strain JT0107. Biochim Biophys Acta 1218:105–108
Wang J, Jiang X, Mou H, Guan H (2004) Anti-oxidation of agar oligosaccharides produced by agarase from a marine bacterium. J Appl Phycol 16:333–340
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 41076106), Guangdong Natural Science Foundation (No. S2011030005257), the Science & Technology Project of Guangdong Province (No. 2009B030803051) and the Key Science and Technology Innovation Project for University by the Department of Education of Guangdong Province (No.CXZD1124).
Author information
Authors and Affiliations
Corresponding author
Additional information
Bokun Lin and Guoyong Lu contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Lin, B., Lu, G., Zheng, Y. et al. Gene cloning, expression and characterization of a neoagarotetraose-producing β-agarase from the marine bacterium Agarivorans sp. HZ105. World J Microbiol Biotechnol 28, 1691–1697 (2012). https://doi.org/10.1007/s11274-011-0977-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11274-011-0977-y