
Abstract
The gene for β-agarase of an Agarivorans sp. JA-1 was expressed in Bacillus subtilis strain DB104 for efficient and economical mass-production of the enzyme. We isolated 360 mg protein with a specific activity of 201 U/mg from the culture broth. The efficiency of production was approximately 130-fold higher than that in E. coli. The enzyme produced neoagarohexaose, neoagarotetraose and neoagarobiose from agar. Neoagarooligosaccharides produced by the enzyme had a whitening effect and inhibited tyrosinase activity in the murine melanoma cell line, B16F10. Neoagarooligosaccharides were not cytotoxic to B16F10 or normal cells. β-Agarase could therefore be a good whitening, cosmetic additive.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
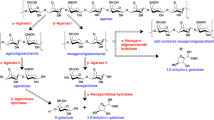
Agar is a cell wall component of some red algae and is composed of agarose and agaropectin. The two groups of agarases that degrade agarose, α-agarase and β-agarase, are divided based on their mode of action. While agarooligosaccharides are produced both by α-agarase and acids, neoagarooligosaccharides are produced only by β-agarase (Araki 1959).
Neoagarooligosaccharides can decrease the rate of degradation of starch and inhibit bacterial growth (Kohno et al. 1990, Kono and Hidaka 1989) and stimulate immune function (Yoshizawa et al. 1995). Furthermore, neoagarobiose moisturizes the skin and has a whitening (skin-lightening) effect on melanoma cells (Kobayashi et al. 1997). Hence, many attempts have been made to screen β-agarase producing bacteria such as Agarivorans sp. JA-1 (Lee et al. 2006); Pseudomonas sp. W7 (Kong et al. 1997); Bacillus cereus ASK202 (Kim et al. 1999); and Microbulbifer sp. JAMB-A94 (Ohta et al. 2004). Recently, we cloned and characterized the gene for a thermostable β-agarase from Agarivorans sp. JA-1 which was expressed in E. coli cells (Lee et al. 2006).
Melanins are the major pigments for human skin color and play a major role in the protection of the skin and underlying tissues from UV-induced skin injury. Melanin is produced by melanocyte cells in the basal layer of the epidermis (Hearing 2005). However, high melanin levels accumulate after chronic sun exposure, melasma, age spots, or other hyperpigmentation diseases (Briganti et al. 2003). Tyrosinase (EC:1.14.18.1), a copper-containing monooxygenase, participates in melanin biosynthesis (Sturn et al. 2001) by: the hydroxylation of monophenols to o-phenols (monophenolase activity), and the oxidation of o-phenols to o-quinones (diphenolase activity). These quinones are liable to polymerize to form melanins (Seo et al. 2003). Melanin biosynthesis can be reduced by inhibition of tyrosinase, avoiding UV exposure, or inhibition of melanocyte metabolism and proliferation (Seiberg et al. 2000). Tyrosinase inhibitors may be the least invasive method for maintaining white skin, and such agents are increasingly used in cosmetic products (Kadekaro et al. 2003) and medications (Seo et al. 2003) to prevent hyperpigmentation. Many natural tyrosinase inhibitors possess a phenol structure or are metal chelating agents (Mayer 1987; Passi and Nazzaro-Porro 1981; Pifferi et al. 1974). However, these inhibitors often have low activity, high toxicity, insufficient penetration, or undefined clinical efficiency (Briganti et al. 2003). Further work is needed to find tyrosinase inhibitors with high activity and low toxicity.
In this study, we over-produced the β-agarase of Agarivorans sp. JA-1 (Lee et al. 2006) in B. subtilis. We also produced neoagarooligosaccharides from agar with this recombinant enzyme and tested the degree of whitening, tyrosinase inhibition, and cytotoxicity, compared with other known tyrosinase inhibitors.
Materials and methods
Bacterial cells and culture conditions
Escherichia coli DH5α (F′ supE44 hsdS20 recA13 ara-14 proA2 lacY1 galK2 rpsL20 xyl-5 mtl-1 leuB6 thi-1) was used as the host for cloning. Bacillus subtilis DB104 (his, nprR2, nprE18, aprE A3) (BioLeaders, Daejeon, Korea) was used as the host for expression of β-agarase. Cells were routinely grown at 37°C in Luria–Bertani (LB) broth (BD, Sparks, MD, USA), supplemented with 100 μg ampicillin or 10 μg kanamycin per ml when required.
Expression of recombinant β-agarase in B. subtilis
The methods used for molecular cloning were based on those of Sambrook et al. (1989). Plasmid DNA was isolated by the alkaline lysis method (Sambrook et al. 1989). The β-agarase gene of Agarivorans sp. JA-1 was generated by PCR using previously constructed pGEMTe-A_sp_b-agaE11 carrying the β-agarase gene of Agarivorans sp. JA-1 (Lee et al. 2006) as a template, with the sense primer A_sp_b-agaE8-F, 5′-GAATTCCATATGGCTGCTACCTTAG TCACCTC-3′ (inserted EcoRI and NdeI sites underlined) and antisense primer, A_sp_b-agaE7-R, 5′-GGATCCTACTCGAGCACTTTACGACGTCTTAG-3′ (inserted BamHI and XhoI sites underlined), and Pyrobest DNA polymerase (Takara Bio Inc., Otsu, Japan). The resultant fragment was ligated into the pGEM-T Easy vector (Promega, WI, USA), yielding pGEMTe-A_sp_b-agaE87. pGEMTe-A_sp_b-agaE87 was digested with EcoRI and BamHI, and a 2.9-kb DNA fragment was ligated to the corresponding sites of B. subtilis expression vector, pLip (BioLeaders), yielding pLip-A_sp_b-agaE87. The integrity of all constructs were verified by restriction analysis and sequencing. B. subtilis DB104 was transformed by pLip-A_sp_b-agaE87 and cultured in 1 l LB broth supplemented with 10 μg kanamycin/ml at 37°C for 9 h. The cells were removed by centrifugation at 5,000 × g for 5 min and the supernatant, which containing secreted β-agarase, was concentrated using Centriprep Ultracel YM-10 (Millipore, Billerica, MA, USA) and then dialyzed against 50 mM Tris/HCl (pH 7.4) buffer. The amount of protein was measured using a BCA protein assay reagent (Pierce Biotechnology, Rockford, IL, USA), utilizing bovine serum albumin as the standard protein.
Assay of β-agarase activity
Agarase activity was determined by the enzymatic production of reducing sugars from agarose (Somogyi 1952). The enzyme was incubated in 50 mM TAPS (Sigma) (pH 8.0) buffer containing 1 mM NaCl, 1 mM CaCl2, and 0.2% (w/v) molten agar at 40°C for 30 min. The enzyme reaction was ended by the addition of the Cu2+ reagent and used for the determination of reducing sugars. The mixture was boiled for 10 min and cooled, with arsenomolybdate reagent added afterwards. The amount of reducing sugar liberated was measured using d-galactose (Sigma) as a standard. One unit of the enzyme activity was defined as the amount of protein that produced 1 μmol reducing sugar per min under these assay conditions.
SDS-PAGE
SDS-PAGE was performed by the Laemmli method with an 11% (v/v) polyacrylamide gel. The enzyme solution was mixed with the sample buffer and boiled for 5 min before being placed on the gel. The gels were stained for protein with GelCode Blue Stain Reagent (Pierce Biotechnology).
Chromatographic analysis of the products of hydrolysis of agar
The hydrolyzed products of agar by β-agarase were identified using TLC. Enzymatic hydrolysis of agar was carried out at 50°C in 665 ml of 50 mM TAPS (pH 8.0) buffer containing 1 mM NaCl, 1 mM CaCl2 and 35 g agar. The agar was melted by heating at 95°C and was used as molten substrate at 50°C. The reaction mixture was applied to silica gel 60 TLC plates (Merck, Darmstadt, Germany) (Duckworth and Yaphe 1970; Groleau and Yaphe 1977). Neoagarohexaose (Sigma) and neoagarotetraose (V-Labs Inc., St. Covington, LA, USA) were used for size markers. The plates were developed using a solvent system composed of n-butanol/acetic acid/H2O (2:1:1, by vol.). The spots were visualized by spraying with 10% (v/v) H2SO4 and heating at 80°C. d-Galactose (Sigma), neoagarotetraose (V-Labs Inc.), and neoagarohexaose (Sigma) were used as standards. The products of the enzyme reaction were quantified by ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Assay of melanin production in B16F10 melanoma cell
The murine melanoma cell line B16F10 was obtained from the Korean Cell Line Bank (KCLB, Seoul, Korea) and cultured in Dulbecco’s modified Eagle’s medium (DMEM) (BioWhittaker, Walkersville, MD, USA) supplemented with 10% (v/v) fetal bovine serum (FBS) (BioWhittaker) at 37°C in a humidified atmosphere containing 5% CO2. B16F10 cells were cultured in 60 mm tissue culture dishes (Iwaki, Tokyo, Japan), and maintained in 2 ml DMEM containing 10% (v/v) FBS (BioWhittaker). After 48 h, the cells were washed twice with 2 ml each of phosphate-buffered saline (PBS) (BioWhittaker), fed 2 ml of fresh media, and treated with α-melanocyte stimulating hormone (MSH) (Sigma) at 200 nM. The samples were dissolved in PBS and added to dishes by serial dilution (final concentrations: 0.1, 1, 10, 100 μg/ml). After 48 h, the cells were harvested and washed twice with 2 ml each of PBS. The cells were collected by centrifugation (1,700 × g) at 4°C for 5 min and the supernatant discarded. Two hundred μl of 1 M NaOH (in 10% (v/v) DMSO) was added to the cells and incubated at 80°C for 1 h. After dissolving the melanin by vortexing, the melanin content was determined at 405 nm using a microplate reader.
Assay of tyrosinase activity in B16F10 melanoma cell
B16F10 cells were cultured in 60 mm tissue culture dishes (Iwaki), and maintained in 2 ml DMEM containing 10% (v/v) FBS for 2 days. After 48 h, the cells were washed twice with 2 ml each of PBS, fed 2 ml of fresh media, and treated with α-MSH (Sigma) at of 200 nM. The samples were dissolved in PBS and added to the dishes by serial dilution (final concentrations: 0.1, 1, 10, 100 μg/ml). After 48 h, the cells were harvested and washed twice with 2 ml each of PBS. The cells were collected by centrifugation (1,700 × g) at 4°C for 5 min and the supernatant discarded. Fifty-five μl of lysis buffer (10 mM sodium phosphate (pH 7.0) buffer containing 1% Triton X-100 and 0.1 mM PMSF) was added to the cells and left on ice for 30 min. After 30 min, the cells were removed by centrifugation (1,700 × g) at 4°C for 5 min and 25 μl 10 mM l-tyrosine was added to the each supernatant. The reaction mixture was made to 200 μl with 50 mM sodium phosphate (pH 7.0) buffer and incubated at 37°C for 30 min. Tyrosinase activity was then measured at 475 nm using a microplate reader.
Cytotoxicity assay of melanoma cell line
B16F10 cells were cultured in a 96-well tissue culture plate and maintained in 200 μl DMEM containing 10% (v/v) FBS. After 48 h, the cells were washed twice with 1 ml each of PBS, fed 200 μl of fresh media, and treated with α-MSH (Sigma) at 200 nM. The samples were dissolved in PBS and added to the wells by serial dilution (final concentrations: 0.1, 1, 10, 100 μg/ml). After 48 h, the cells were washed twice with PBS and fed 200 μl fresh media. Twenty μl MTT (Sigma) solution (5 mg/ml in PBS) was then added to each well and the cells were incubated further for 12 h at 37°C. The media were removed and the cells were dissolved in 100 μl DMSO. The conversion of MTT to formazan was quantified from the absorbance at 570 nm using a microplate reader.
Cytotoxicity assay in primary cultures
Small pieces of spleens from naïve mice were incubated with collagenase type II (1 mg/ml) and DNase I (15 μg/ml) (Roche Applied Science, Mannheim, Germany) at 37°C for 40 min. Red blood cells were eliminated with red blood cell lysing buffer (Sigma). The cells were then washed and suspended in RPMI 1640 (BioWhittaker) supplemented with 10% (v/v) FBS, and were seeded in a 96-well cell culture plate at 4 × 106 cells/ml. After cells attached, the samples were dissolved in PBS and added to the wells by serial dilution (final concentrations: 0.1, 1, 10, 100 μg/ml). After 48 h, the cells were washed twice with PBS and fed 200 μl fresh media. Twenty μl MTT solution (5 mg/ml in PBS) was then added to each well and the cells were incubated further for 12 h at 37°C. The media were removed and the cells were dissolved in 100 μl DMSO. The conversion of MTT to formazan was quantified at 570 nm using a microplate reader.
Results
Secretory production of recombinant β-agarase using B. subtilis
Production of recombinant β-agarase was examined using B. subtilis DB104 as a host and pLip as a vector. B. subtilis DB104 cells harboring pLip-A_sp-b-agaE87 produced a high amount of β-agarase (Fig. 1). The amount of recombinant β-agarase was 360 mg/l and total activity was 72,360 U, with a specific activity of 201 U/mg protein in culture broth. SDS-PAGE of the enzyme exhibited a band with an apparent molecular mass of 109 kDa (Fig. 1). This value agreed with that produced in E. coli cells (Lee et al. 2006).

SDS-PAGE of β-agarase from B. subtilis cells harboring pLip-A_sp-b-agaE87. Lane M, size marker, lane P, produced enzyme. The arrow indicates the position of β-agarase.
Production of neoagarooligosaccharides from agar using recombinant β-agarase
Thirty-five g agar was hydrolyzed at 50°C for 48 h using recombinant β-agarase to generate neoagarooligosaccharides. The reaction mixture was lyophilized and 32 g hydrolyzed product was obtained. Neoagarooligosaccharides produced by the enzyme reaction were analyzed by TLC (Fig. 2). The enzyme could produce neoagarohexaose, neoagarotetraose, and neoagarobiose from agar (Fig 2). The main products were neoagarotetraose (48% total products) and neoagarobiose (29% total products) (Fig. 2).

TLC of the products of agar hydrolysis by recombinant β-agarase. The reactions were carried out at 50°C in 665 ml of 50 mM TAPS (pH 8.0) buffer containing 1 mM NaCl, 1 mM CaCl2, and 35 g agar with 700 U enzyme for 48 h. The reaction mixture was developed by TLC. Gal, d-galatose; NA2, neoagarobiose; NA4, neoagarotetraose; NA6, neoagarohexaose; P, neoagarooligosaccharides.
Whitening effect of neoagarooligosaccharides
The whitening effects of neoagarooligosaccharides, neoagarohexaose, arbutin, and kojic acid were evaluated by measuring melanin content in melanocytes (Fig. 3). Murine melanoma B16F10 cells were whitened by neoagarooligosaccharides treatment in a dose-dependent manner (Fig. 3). Kobayashi et al. (1997) reported that neoagarobiose had whitening effect at 100 μg/ml, and kojic acid and arbutin showed a greater whitening effect than neoagarobiose at the same concentration. In this study, however, neoagarooligosaccharides showed whitening effects from 0.1 μg/ml, and kojic acid and arbutin, well-known whitening compounds, had similar whitening effects at the same concentration (Fig. 3). On the other hand, neoagarohexaose showed whitening only at 100 μg/ml. These observations suggest that whitening by neoagarooligosaccharides below 10 μg/ml may be influenced by neoagarotetraose and neoagarobiose that included in neoagarooligosaccharides. While Kobayashi et al. (1997) tested only neoagarobiose for whitening, we used neoagarooligosaccharides.

Whitening effects of neoagarohexaose, neoagarooligosaccharides, kojic acid and arbutin at various concentrations in B16F10 cells. Means ± SEM for three wells are shown as fold-increase compared with no treatment. Factorial ANOVA with Fisher’s PLSD post-hoc test *P < 0.0001 compared with no treatment. This experiment was repeated at least twice yielding reproducible results.
Effect of neoagarooligosaccharides on tyrosinase activity
Neoagarooligosaccharides inhibited tyrosinase activity in a dose-dependent manner (Fig. 4). Neoagarohexaose also inhibited tyrosinase activity, but at an 100-fold higher dose than neoagarooligosaccharides (Fig. 4). These results suggest that inhibition of tyrosinase activity by neoagarooligosaccharides below 10 μg/ml may be affected by the neoagarotetraose and neoagarobiose included in neoagarooligosaccharides. Kojic acid and arbutin also inhibited tyrosinase activity (Fig. 4).

Tyrosinase inhibition of neoagarohexaose, neoagarooligosaccharides, kojic acid, and arbutin at various concentrations in B16F10 cells. Means ± SEM for three wells are shown as fold-increase compared with no treatment. Factorial ANOVA with Fisher’s PLSD post-hoc test *P < 0.0001 compared with no treatment. This experiment was repeated at least twice yielding reproducible results.
Effect of neoagarooligosaccharides on cell viability
Neoagarooligosaccharides and neoagarohexaose were not cytotoxic to B16F10 melanoma cells at concentrations up to 100 μg/ml, whereas kojic acid and arbutin showed dose-dependent cytotoxicity (Fig. 5a). To test for cytotoxicity in normal cells, spleen cells of naïve mice were treated with neoagarooligosaccharides at the same concentrations that showed whitening effects (Fig. 5b). Neoagarooligosaccharides were not cytotoxic to normal cells also (Fig. 5b).

Cell viability after exposure to neoagarohexaose, neoagarooligosaccharides, kojic acid, and arbutin in B16F10 cells (a) and to neoagarooligosaccharides in mouse spleen cells (b). Means ± SEM for three wells are shown as fold-change compared with no treatment. Factorial ANOVA with Fisher’s PLSD post-hoc test *P < 0.0001 compared with no treatment. These experiments were repeated at least twice yielding reproducible results.
Discussion
We heterologously produced 360 mg β-agarase from the marine bacterium, Agarivorans sp. JA-1 with a specific activity of 201 U/mg using B. subtilis DB104 as the host. β-Agarase from the Agarivorans sp. JA-1 is a member of GH family-50, which can only produce neoagarobiose, and can be heterologously expressed in E. coli cells at the level of 1.9 U/mg in culture broth (Lee et al. 2006). We produced approximately 130-fold higher expression levels in this study than in E. coli (Lee et al. 2006). Ohta et al. (2004) expressed β-agarase of Agarivarans sp. JAMB-A11 in B. subtilis, and total protein and specific activity was 1,662 mg and 0.8 U/mg, respectively, in 72 ml of culture supernatant. Protein amount produced by Ohta et al. (2004) was higher than ours; however, the total activity and specific activity of the enzyme were greater in our work. Ohta et al. (2004) commented that the biohazard risks of members of genus Agarivorans have not been fully evaluated, and therefore heterologous β-agarase production in B. subtilis is safer and efficient.
We previously described the initial cloning and characterization of β-agarase gene from the Agarivorans sp. JA-1 (Lee et al. 2006), and demonstrated that the gene product could produce a mixture of neoagarobiose (58%) and neoagarotetraose (42%) from agarose as a substrate (Lee et al. 2006). In this study, we used agar instead of expensive agarose as a substrate and successfully produced a mixture of neoagarobiose (29%), neoagarotetraose (48%), and neoagarohexaose (23%) (Fig. 2). Neoagarobiose is the biologically active substance produced by several β-agarases and Kobayashi et al. (1997) used neoagarobiose for the study of whitening effects. However we obtained the better whitening effect than that of Kobayashi et al. (1997) by using the neoagarooligosaccharides produced from agar.
Tyrosinase is a key enzyme in melanin biosynthesis, and avoiding UV exposure and using tyrosinase inhibitors are the best methods for skin whitening (Kadekaro et al. 2003). Tyrosinase inhibitors have clinical applications in melanin hyperpigmentation, cosmetics for whitening, and depigmentation after sunburn, and many attempts have been made to screen tyrosinase inhibitory agents. Tyrosinase (EC:1.14.18.1) is a copper-containing monooxygenase (Sturn et al. 2001) and many natural tyrosinase inhibitors have a phenol structure or the activity of metal chelating (Mayer 1987; Passi and Nazzaro-Porro 1981; Pifferi et al. 1974). Kojic acid and arbutin are strong natural tyrosinase inhibitors, but they represent cytotoxicity (Briganti et al. 2003). They have been used as the standard materials for the degree of tyrosinase inhibition and whitening effect. Kojic acid [5-hydroxy-2-(hydroxymethyl)-4-pyrone] is a chelating agent produced by several species of fungi, especially Aspergillus oryzae (Yabuta 1924) and arbutin [2-hydroxymethyl-6-(4-hydroxyphenoxy)oxane-3,4,5-triol] is a glycosylated hydroquinone extracted from bearberry plant in the genus Arctostaphylos (O’Donoghue 2006).
References
Araki C (1959) Seaweed polysaccharides. In: Wolfrom ML (eds) Carbohydrate chemistry of substances of biological interest. Pergamon Press, London, pp 15–30
Briganti S, Camera E, Picardo M (2003) Chemical and instrumental approaches to treat hyperpigmentation. Pigment Cell Res 16:101–110
Duckworth M, Yaphe W (1970) Thin-layer chromatographic analysis of enzymic hydrolysate of agar. J Chromatogr 49:482–487
Groleau D, Yaphe W (1977) Enzymatic hydrolysis of agar: purification and characterization of β-neoagarotetraose hydrolase from Peudomonas atlantica. Can J Microbiol 23:672–679
Hearing VJ (2005) Biogenesis of pigment granules: a sensitive way to regulate melanocyte function. J Dermatol Sci 37:3–14
Kadekaro AL, Kanto H, Kavanagh R, Abdel-Malek ZA (2003) Significance of the melanocortin 1 receptor in regulating human melanocyte pigmentation, proliferation, and survival. Ann NY Acad Sci 994:359–365
Kang NY, Choi YR, Cho YS, Kim BK, Jeon BS, Cha JY, Kim CH, Lee YC (2003) Cloning, expression and characterization of a β-agarase gene from a marine bacterium, Pseudomonas sp. SK38. Biotechnol Lett 23:1165–1170
Kim BJ, Kim HJ, Ha SD, Hwang SH, Byun DS, Lee TH, Kong JY (1999) Purification and characterization of β-agarase from marine bacterium Bacillus cereus ASK202. Biotechnol Lett 21:1011–1105
Kobayashi R, Takisada M, Suzuki T, Kirimura K, Usami S (1997) Neoagarobiose as a novel moisturizer with whitening effect. Biosci Biotechnol Biochem 61:162–163
Kohno T, Kitagawa H, Hiraga T (1990) Production of hetero-oligosaccharides. In: Gijutsu Kenkyu Kukami (eds) Shokuhin sangyo bioreactor system, Jissen bioreactor. Shokuhin Kagaku Shimbunsa, Tokyo, Japan, pp 87–105
Kong JY, Hwang SH, Kim BJ, Bae SK, Kim JD (1997) Cloning and expression of an agarase gene from a marine bacterium Pseudomonas sp. w7. Biotechnol Lett 19:23–26
Kono T, Hidaka H (1989) Properties and production of neoagarooligosaccharide. Nippon Nogeikagaku Kaishi 63:1126–1129
Lee DG, Park GT, Kim NY, Lee EJ, Jang MK, Shin YG, Park GS, Kim TM, Lee JH, Lee JH, Kim SJ, Lee SH (2006) Cloning, expression, and characterization of a glycoside hydrolase family 50 beta-agarase from a marine Agarivorans isolate. Biotechnol Lett 28:1925–1932
Mayer AM (1987) Polyphenol oxidases in plants. Recent progress. Phytochemistry 26:11–20
O’Donoghue JL (2006) Hydroquinone and its analogues in dermatology – a risk-benefit viewpoint. J Cosmet Dermatol 5:196–203
Ohta Y, Nogi Y, Miyazaki M, Li Z, Hatada Y, Ito S, Horikoshi K (2004) Enzymatic properties and nucleotide and amino acid sequences of a thermostable beta-agarase from the novel marine isolate, JAMB-A94. Biosci Biotechnol Biochem 68:1073–1081
Passi S, Nazzaro-Porro M (1981) Molecular basis of substrate and inhibitory specificity of tyrosinase: phenolic compounds. Br J Dermatol 104:659–665
Pifferi PG, Baldassari L, Cultrera R (1974) Inhibition by carboxylic acids of an o-diphenol oxidase from Prunus avium fruits. J Sci Food Agric 25:263–270
Sambrook E, Fritsch F, Maniatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Labarotory, Cold Spring Harbor, New York
Seo SY, Sharma VK, Sharma N (2003) Mushroom tyrosinase: recent prospects. J Agric Food Chem 51:2837–2853
Seiberg M, Paine C, Sharlow E, Andrade-Gordon P, Costanzo M, Eisinger M, Shapiro SS (2000) Inhibition of melanosome transfer results in skin lightening. J Invest Dermatol 115:162–167
Somogyi M (1952) Notes on sugar determination. J Biol Chem 195:19–23
Sturm RA, Teasdale RD, Box NF (2001) Human pigmentation genes: identification, structure and consequences of polymorphic variation. Gene 277:49–62
Yabuta T (1924) The constitution of kojic acid, a gamma-pyrone derivative formed by Aspergillus oryzae from carbohydrates. J Chem Soc 125:575–587
Yoshizawa Y, Ametani A, Tsunehiro J, Nomura K, Itoh M, Fukui F, Kaminogawa S (1995) Macrophage stimulation activity of the polysaccharide fraction from a marine alga (Porphyra yezoensis): structure-function relationships and improved solubility. Biosci Biotechnol Biochem 59:1933–1937
Acknowledgement
This work was supported by the Marine and Extreme Genome Research Center Program, Ministry of Maritime Affairs & Fisheries, Republic of Korea.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lee, DG., Jang, M.K., Lee, OH. et al. Over-production of a glycoside hydrolase family 50 β-agarase from Agarivorans sp. JA-1 in Bacillus subtilis and the whitening effect of its product. Biotechnol Lett 30, 911–918 (2008). https://doi.org/10.1007/s10529-008-9634-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-008-9634-4