
Abstract
Diabetes mellitus (DM) is the leading cause of end stage renal disease. 40% of the patients worldwide will require replacement therapy after 20 years of DM worldwide. Early-stage diabetic nephropathy is characterized by hyperfiltration related to hypeglycemia-induced afferent artery vasodilatation with micro-and macroalbuminuria. Later on, proteinuria with arterial hypertension may appear, culminating in glomerular filtration rate (GFR) decline and end stage renal disease. Forty percent of diabetic patients develop microvascular and macrovascular complications, with increased risk among patients with genetic predisposition, such as Haptoglobin 2–2 phenotype. The most frequent complications in the daily clinical practice are diabetic kidney disease, diabetic retinopathy and vascular disease, such as coronary artery disease and stroke. Various pathways are involved in the pathogenesis of diabetic kidney disease. Chronic systemic inflammation and the inflammatory response, such as increased circulating cytokines (Interleukins), have been recognized as main players in the development and progression of diabetic kidney disease. DM is also associated with increased oxidative stress, and alterations in carbohydrate, lipid and protein metabolism. Overexpression of the renin-angiotensin-aldosterone system (RAAS) in the kidney, the vitamin D-Vitamin D receptor-klotho axis, and autophagy. Differences in the ATG5 protein levels or ATG5 gene expression involved in the autophagy process have been associated with diabetic complications such as diabetic kidney disease. Under normal blood glucose level, autophagy is an important protective mechanism in renal epithelial cells, including podocytes, proximal tubular, mesangial and endothelial cells. Down regulation of the autophagic mechanism, as in hyperglycemic condition, can contribute to the development and progression of diabetic kidney disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Diabetic kidney disease (DKD) is associated with decreased glomerular filtration rate (GFR) and ESRD with increased vascular complications [1,2,3,4,5] and increased cardiovascular morbidity and mortality worldwide in patients with type 2 diabetes mellitus (T2DM). T2DM is estimated to currently affect over 380 million people, with increasing prevalence in younger and more obese populations with metabolic syndrome and insulin resistance, especially in those with uncontrolled blood glucose levels and blood pressure, which eventually lead to a decline in renal function and proteinuria. The condition has become the most common cause of end stage renal disease (ESRD) in patients undergoing dialysis treatment or kidney transplantation, with approximately 40% of diabetic patients, either type 1 or type 2, eventually developing nephropathy 10–20 years after diagnosis [6]. Numerous risk factors are associated with the development and progression of DKD, such as the degree of hyperglycemia, extended duration of diabetes, hypertension, obesity and dyslipidemia, with most being modifiable by appropriate treatment. Other contributing factors, such as advanced age and genetic factors, such haptoglobun genotype, cannot be modified [7]. Among the risk factors mentioned above, hyperglycemia was identified as a factor of great importance in the development and progression of DKD [8,9,10].
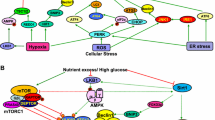
Hyperglycemia-mediated renal proximal tubule cells damage (proximal convolute tubules [PCT] and podocytes), is triggered by increased production of free radicals, accumulation of advanced glycation end products (AGEs), activation of protein kinase C (PKC), and the renin–angiotensin–aldosterone system (Fig. 1). Ligia Petrica and her group, in different studies showed that in patients with type 2 that in early diabetic kidney disease (DKD), PCT dysfunction may precede glomerular injury (Glomerular Theory). Patients with early DKD may display more abnormal tubular injury markers that show PCT dysfunction (Tubular Theory). MicroRNAs have been shown to play important role in gene expression that affect cellular function. miRNA-192 induces upregulation of fibrotic genes, thus promoting increased glomerular sclerosis and fibrosis. Upregulation of miRNA-192 expression in tubular cells treated with transforming growth factor (TGF)β1 increased fibrosis [11, 12].

Pathways involved in hyperglycemia-induced alteration of intracellular metabolism with consequent diabetic kidney disease
Low molecular weight AGEs, especially glycated peptides and carboxymethyllysine, reabsorbed and metabolized by PCT cells and severely modified albumin molecules become tubulotoxic component by activating NF-kappaB and IL-6 antigen [11, 12]. These structural changes in the PCT and glomerular cells (podocytes), including thickening of glomerular and tubular basement membranes, glomerular hypertrophy, mesangial expansion and accumulation of extracellular matrix components (ECM) resulting in tubulointerstitial fibrosis, tubular cell apoptosis and glomerulosclerosis, all of which lead to a decline in renal function and ESRD [11].
The soluble klotho anti-aging factor is known to be involved in the pathogenesis of DKD with decreased expression measured in podocytes of mice in early stages of DKD [14]. Various works, including those of our laboratory, have shown that the main protective effect of klotho on renal epithelial cells is via upregulation of the VDR in PCT. Others had shown that klotho inhibits intrarenal renin-angiotensin–aldosterone system (RAAS) or exerts a direct inhibitory effect on aldosterone synthesis in adrenal glands [15]. In line with these reports, our group has demonstrated the beneficial effects of the selective active vitamin D 1,25 (OH)2 (data not published) (Paricalcitol) in diabetic mice, especially on VDR and fibronectin expression. Several clinical studies have shown the renoprotective effect of vitamin D treatment in patients with diabetic kidney disease [16], and is manifested by reduced albuminuria or proteinuria and renal failure progression, primarily mediated via RAAS down regulation (Fig. 2).

Different pathways involved in the pathogenesis of diabetic kidney disease
RAAS inhibitors are the first line of care to be used in DKD treatment, particularly in the early stages of the disease. However, there has been an enormous increase in the number of DM patients with ESRD treated chronically with hemodialysis in spite of maximum dose of angiotensin converting enzyme inhibitors (ACEIs) or angiotensin receptor blockers (ARBs) [17, 18]. Clinical trials performed until 2009 failed to show any success with different ACEIs in delaying DKD progression. In contrast, several recent studies have shown an impressive protective effect of sodium-glucose cotransporter 2 (SGLT2) inhibitors (EMPA study) in slowing the progression of DKD [19], possibly through the restoration of glomerular hypertension or hyperfiltration, and upregulation of the autophagy pathway [19, 20]. Regarding hyperglycemia and diabetic retinopathy, hyperglycemia decreases retinal blood flow with consequent hypoxia with increased vascular endothelial growth factor (VEGF) and neovascularization, and macular damage. SGLT2Is can be of benefit in the future regarding diabetic retinopathy and blindness.
Autophagy
Autophagy is a mechanism required to maintain homeostasis of glomeruli and tubules, and plays important roles in human health and diseases. Impairment of autophagy is implicated in various inflammatory diseases, and particularly in the pathogenesis of DKD. Hyperglycemia-induced alterations in intracellular metabolism and cellular events, including accumulation of advanced glycation end-products (AGEs), increased oxidative stress, endoplasmic reticulum stress, and activation of the renin angiotensin system [21,22,23,24].
Autophagy is a cellular self-protection mechanism by which cells degrade damaged proteins and macromolecules, through a lysosome-dependent mechanism classified as macro-autophagy. The process involves several steps, including initiation, elongation, maturation, fusion and degradation. At initiation, a phagophore forms around cytoplasmic components that are then sequestered by a double membrane, which forms the autophagosome. The autophagosome subsequently fuses with the lysosome to form an autolysosome; the enclosed contents are then degraded. Autophagosome formation is regulated by several autophagy-related gene (Atg) proteins [24,25,26,27,28,29].
The initiation of hyperglycemia-induced autophagy is regulated by the phosphorylation of Atg1 via the intracellular mammalian target of rapamycin (mTORC1) and adenosine monophosphate (AMP)-activated protein kinase (AMPK) [30,31,32,33]. The conjugation of microtubule-associated protein 1 light chain 3 (LC3I), the mammalian homolog of Atg8, toAtg7 and Atg3, to form LC3II, is a critical step in autophagosome formation [34]. This LC3 conjugation reaction is regulated positively by the Atg12-Atg5-Atg16 complex [33, 34]. Several studies have suggested that targeted these complexes, and restoration of autophagy activity may have reno-protective effects.
Regulation of autophagy
The discovery of over 32 Atgs that are essential for the regulation and achievement of autophagy, has raised great interest in the field of DM and its complications. Numerous regulators are involved in the autophagy regulation in basal and under stimulation. One of these regulators is nitrogen-dependent regulation. The primary sensor of amino acid and nitrogen change is the mammalian homolog mTOR [mechanistic target of rapamycin (serine/threonine kinase)], which is the main negative regulator of autophagy. In nutrient-rich conditions, TORC1 directly phosphorylates Atg13, Atg1 and Atg14, which prevents the formation and/or activation of the Atg1-Atg13-Atg17-Atg31-Atg29 complex and suppresses the autophagy-specific PtdIns3K, thus inhibiting autophagy induction. In the presence of glucose, protein kinase A (PKA) is activated by binding with cAMP. PKA then phosphorylates Atg1 and Atg13, which prevents the localization of Atg13 to the PAS. In addition, PKA can inhibit autophagy by direct phosphorylation of TORC1 or in mammalian cells by indirect activation of MTORC1 through inhibition of AMPK [11,12,13, 35].
Other regulator is a lipid kinase signaling complexes that mediate vesicle nucleation (Atg6, Atg14). Atg12, Atg5, Atg16, Atg7, Atg8 and Atg10 have a significant role in the autophagy process, specifically in autophagosome formation. Free fatty acids stimulate autophagy through EIF2AK2/PKR-dependent activation of EIF2S1/eIF2α, MAPK8 (mitogen-activated protein kinase eight) or inhibition of MTORC1. These genes operate through two regulatory systems [11,12,13] as we will discuss in the following paragraph.
Atg12-Atg5 conjugation system
The first system covalently conjugates Atg12 to Atg5, via an isopeptide bond. The mode of conjugation is similar to the ubiquitin conjugation system and depends on the activity of two other proteins, Atg7 and Atg10. During autophagy, Atg12 is first activated by Atg7 before being transferred to the active cysteine 133 of Atg10, to form an Atg12-Atg10 complex. Next, Atg12 is next conjugated to its target protein Atg5, forming an Atg12-Atg5 complex. This complex interacts further with Atg16, to form the multimeric protein complex Atg12-Atg5-Atg16, which is actively involved in the expansion of the autophagosome membranes (Fig. 3). Atg16 is required for the stability of this complex [34,35,36,37,38,39].

Proposed mechanism of action of sodium-glucose transporters inhibitors and Rapamycin. Negative color indicates suppressing effect, while green color indicates activation of autophagy
The LC3 conjugation system
The Atg8/LC3 conjugation system is another ubiquitin-like system involving LC3, a homologue of yeast protein Atg8. Immediately after synthesis, LC3 is proteolytically cleaved by Atg4, to generate the cytosolic LC3-I, which is then activated by Atg7 which binds it via a thoiester bond. The activated LC3-I is then transferred to Atg3. Finally, LC3-I is conjugated to phosphatidiletanolamine to form LC3-II. LC3-II is then connected to an autophagosome, where it serves as an important player in the elongation and expansion of the autophagosome membrane. Recruitment and integration of LC3-II into the expanding autophagosome is dependent on the Atg5–Atg12 complex, indicating a cooperative interplay between the two LC3/ATG8 and Atg5–Atg12 conjugation systems in autophagosome maturation. LC3-II remains attached to the internal and external membranes until completion of autophagosome formation. In contrast, the Arg12-Atg5-Atg16 complex is generally localized at the outer membrane of the expanding autophagosome and is disconnected before or immediately after autophagosome formation is completed. Thus, LC3-II is the best marker for estimating the level of autophagy activity [40, 41].
Autophagy in renal disease
Autophagy plays a central role in DKD. A large number of studies in human kidney tissues and experimental animal models have shown that autophagy is activated to maintain homeostasis of renal glomeruli and tubules under stress conditions, such as oxidative stress and hypoxia. When this process is impaired, it can lead to glomerular diseases, tubular injuries, and DKD [42,43,44,45].
Podocytes are the glomerular epithelial cells and play a crucial role in maintaining the structure and function of the glomerular filtration barrier, the slit. Under normal physiological conditions, basal autophagy activity in podocytes is very high, suggesting that the autophagylysosome system plays an essential role in maintaining podocyte homeostasis. Dysfunction of podocytes has proven to be a major risk factor for glomerular diseases that induce proteinuria and subsequent development of DKD [45]. Proximal tubular epithelial cells, the most prevalent cell type in the kidney, are responsible for the reabsorption of substances such as glucose and amino acids. Proximal tubule cell dysfunction is closely related to the progression of kidney diseases, such as tubulointerstitial injury, interstitial nephritis, and diabetic nephropathy [35, 46,47,48].
Recent studies have demonstrated that diabetic conditions can significantly suppress the protective effect of autophagy in kidney cells, particularly in podocytes and proximal tubular cells, with both cell types showing lower levels of autophagy in hyperglycemia, which may contribute to diabetes-related podocyte injury. This emphasizes the key homeostatic role of autophagy in maintaining podocyte integrity [49, 50].
Some reports suggest that hyperglycemia can suppress autophagy activity in podocytes and proximal tubular cells by inducing hyper-activation of mammalian target of rapamycin complex 1 (mTORc1). mTORc1 is a nutritional status sensor involved in cell growth, development and proliferation. Under high glucose concentrations, mTORC1 is highly activated and promotes the synthesis of cell components such as proteins, lipids and nucleotides, and at the same time, represses autophagy [41]. Hyperglycemia can also inhibit the expression of beclin-1, Atg12-5, and LC3, resulting in the inhibition of autophagosome membrane formation [32, 37].
A decrease in the number of podocytes with effusion can be caused by increased oxidative stress triggered by high glucose and endoplasmic reticulum stress (ER) stress, leading to slit loss, with consequent albuminuria, characteristic of the early stages of DKD. Increased podocyte damage and reduced levels of autophagy-related protein expression, including beclin-1, LC3- II and the Atg5, have been observed in a type I DM mouse model, characterized by high glucose levels.
Therapeutic targeting of autophagy
New compounds have been designed to target different genes involved in the autophagy process, especially the mTOR system, ATG5, the Atg12-Atg5 complex, and LC3I–LC3II proteins, and have demonstrated therapeutic value in the treatment of DKD [37, 40, 50]. It is well acknowledged that ATG5 plays a significant role in the formation of isolation membranes and autophagosomes, by participating in both Atg12-Atg5 conjugation and LC3 conjugation. LC3-II is a gold-standard marker used to trace changes in autophagic process; earlier reports have implicated that there is a close relationship between the LC3-II and autophagosome formation [40, 41].
Sodium glucose co-transporter 2 inhibitors (SGLT2I) and rapamycin, a well-known inhibitor of mTORC1 and a potent activator of autophagy, are promising candidates for treatment of DKD [51].
Sodium-glucose cotransporter 2 inhibitors (SGLT2I)
DM is characterized by hyperfiltration with increased intraglomerular pressure, podocyte damage with mesangial expansion resulting in fibrosis, and ESRD. High blood glucose concentrations induce increased reactive oxygen species and oxidative stress with podocyte injury. SGLT2Is are a newer class of antihyperglycemic agents that exert glucose-lowering effects via inhibition of the SGL2T in the proximal tubule, with consequent natriuresis and glucosuria [51, 52]. Aside from lowering blood glucose, SGL2Tis reduce the hyper filtration induced by hyperglycemia via restoration of afferent artery diameter. Preclinical studies and clinical trials of SGLT2I treatment have consistently demonstrated reduction of albuminuria and preservation of kidney function. Although decreased blood glucose levels by SGLT2 inhibition are likely to contribute to these favorable changes in the glomeruli, the possibility that SGLT2 inhibition can directly affect basal autophagy in podocytes and PCT cells via Atg5 protein is under investigation (ongoing project in our lab) (Fig. 3).
Preclinical studies showing that SGLT2 inhibition elicits beneficial metabolic effects in diabetic kidneys are in accordance with recent clinical trials demonstrating that SGLT2 inhibition not only lowers blood glucose levels, but also reduces renal events with GFR amelioration and stabilization with time. SGLT2 is known as a biological indicator of nutrient excess, During DM, increased tubular concentration of glucose can increase the expression of SGLT2, but reduce the expression of SIRT1. SGLT2 in the PCT appears to promote autophagic flux, and that SGLT2 in the PCT can be an entry point for therapeutic interventions that aim to stimulate nutrient deprivation signaling such as SIRT1 and AMPK. Up-regulation of AMPK and SIRT1 signalling can increase the autophagic flux in the PCT and podocytes. Additionally, SGLt2i may influence HIF-1α in a manner that favorably affects the course of DKD.
SGLT2 inhibition may have additional anti-inflammatory and ant fibrotic actions that protect the kidney. In primary proximal tubular cells, SGLT2 inhibition suppressed the generation of a hyperglycemia mediated increase in ROS, which likely upregulated the autophagic process [53,54,55,56].
VitaminD/vitamin D receptor
The vitamin D receptor (VDR) is a nuclear receptor superfamily, which, once activated, enters the nucleus, where it regulates target gene transcription by interacting with their promoters. We have shown that VDR expression is reduced in the podocytes of a diabetic mouse model and in patients with diabetic kidney disease (data not published yet). The active vitamin D, via VDR, can exert a renoprotective effect through multiple mechanisms, such as inhibiting inflammation, and preventing the production of transforming growth factor (TGF) and fibrosis by inhibiting RAAS [57, 58]. Basal autophagy can have an effective homeostatic role in renal tubules and glomeruli, via cross talk between autophagy and apoptosis in the pathogenesis of DKD.
New data from Magda Hamzawy and her group [58] showed that cell loss in renal cortex of diabetic rats is associated with significant decrease in autophagy and anti-apoptotic gene expression (LC3II, LC31, beclin-1 and Bcl2), and significant increase of pro-apoptotic gene expression (Caspase-3). The same group report the reciprocal effects of vitamin D analoge, 22-oxa-1,25-dihydroxyvitamin D3 on both autophagy and apoptosis during DM. Vitamin D analoge (OCT) can reduce cell loss and renal protection. The upregulation of both antu-apoptoticv Bcl-2and Pro-autophagy beclin-1, could be involved in vitamin D regulation of cell selection between apoptosis and autophagy.
Several studies had demonstrated that LC3 is a negative regulator of high glucose-induced fibrogenic gene expression through its ability to promote VDR signaling. Li and his group reported that the levels of both nuclear VDR and LC3 were decreased in a high-glucose environment, while overexpression of LC3 by new molecules, such as Rapamun, can restore the nuclear VDR levels. Therefore, LC3 is abundant in the nucleus, but its functions in autophagy is primarily conducted in the cytoplasm after the nuclear translocation of the VDR [58, 59].
Rapamycin
Under normal conditions, podocytes with nephrin-podocin protein that build the slit structure, play an important role as a barrier and can effectively prevent plasma protein leakage. Rapamycin (RAPA), is an anti-solid organ rejection agent, and can specifically bind mTOR kinase and inhibit its activity, thereby regulating pathological autophagy. RAPA has been found to increase the autophagic activity of kidney podocytes in diabetic mice and to inhibit podocyte apoptosis [60]. Taken together, impaired autophagy is involved in the pathogenesis of DKD, suggesting that autophagy activation by new therapeutic molecules could be a potential therapy in the near future for combatting DKD.
Abbreviations
- DM:
-
Diabetes Mellitus
- DKD:
-
Diabetic Kidney Disease
- VDR:
-
Vitamin D Receptor
- SGLT2I:
-
Sodium-Glucose-Transport 2 Inhibitor
- GFR:
-
Glomerular Filtration rate
- ESRD:
-
End Stage Renal Disease
- PCT:
-
Proximal Convolute Tubule
References
Zimmet P, Alberti KG, Magliano DJ et al (2016) Diabetes mellitus statistics on prevalence and mortality: facts and fallacies. Nat Rev Endocrinol 12:616–622
Polonsky KS (2012) The past 200 years in diabetes. N Engl J Med 367:1332–1340
Genuth S (1982) Classification and diagnosis of diabetes mellitus. Med Clin North Am 66:1191–1207
Doshi SM, Friedman AN (2017) Diagnosis and management of type 2 diabetic kidney disease. CJASN 12(8):1366–1373
Fowler MJ (2008) Microvascular and macrovascular complications of diabetes. Clin Diabetes 26(2):77–82
Lim AKH (2014) Diabetic nephropathy–complications and treatment. Int J Nephrol Renovasc Dis 12:361–381
Levy AP, Asleh R, Blum S, Nakhoul F et al (2010) Haptoglobin: basic and clinical aspects. Antioxid Redox Signal 12(2):293–304
Mora-Fernández C, Domínguez-Pimentel V, de Fuentes MM et al (2014) Diabetic kidney disease: from physiology to therapeutics. J Physiol 592:3997–4012
Liang S, Cai GY, Chen XM (2017) Clinical and pathological factors associated with progression of diabetic nephropathy. Nephrology 22:14–19
Petrica L, Vlad A, Gluhovschi G et al (2015) Glycated peptides are associated with proximal tubule dysfunction in type 2 diabetes mellitus. Iny J Clin Exp Med 8(2):2516–2525
Petrica L, Petrica M, Vlad A et al (2011) Proximal tubule dysfunction is dissociated from endothelial dysfunction in normoalbuminuric patients with type 2 diabetes mellitus: a cross-sectional study. Nephron Clin Pract 118:155–164
Milas O, Petrica GF et al (2018) Deregulated profiles of urinary microRNAs may explain podocyte injury and proximal tubule dysfunction in normoalbuminuric patients with type 2 diabetes mellitus. J Invest Med 2:18
Petrica L, Milas O, Mihaela VladInterleukins M et al (2019) Interleukines and miRNAs intervene in the early stages of diabetic kidney disease in Type 2 diabetes mellitus patient. Biomarkers Med 13:18
Dahan I, Thawho N, Farber E et al (2018) The Iron-Klotho-VDR axis is a major determinant of proximal convoluted tubule Injury in Haptoglobin 2–2 Genotype diabetic nephropathy patients and mice. J Diabetes Res 71:63–652
Nakhoul F, Nakhoul N, Thaucho N et al (2015) The Non Mineral Axis Klotho-Vitamin D in diabetic nephropathy:review. J Diabetes Metab 6:7
Eltablawy N, Ashour H, Rashed LA, Hamza W (2018) Vitamin D protection from rat diabetic nephropathy is partly mediated through Klotho expression and renin-angiotensin inhibition. Arch Physiol Biochem 124(5):461–467
Nakhoul R, Nakhoul F, Nakhoul N (2017) Diabetic Nephropathy from RAAS to Autophagy: The Era for New Players. J Clin Exp Nephrol 2:43
Kim MK (2017) Treatment of diabetic kidney disease : current and future targets. Korean J Intern Med 32(4):622–630
Lin YC, Chang YH, Yang SY, Wu KD, Chu TS (2018) Update of pathophysiology and management of diabetic kidney disease. J Formos Med Assoc 117(8):662–675
Nakhoul R, Koch E, Nakhoul F et al (2018) Sodium-glucose transporter inhibitors and diabetic nephropathy in humans and animal model. J Clini Experim Nephrol 12:6
Ding Y, Choi ME (2015) Autophagy in diabetic nephropathy. J Endocrinol 224(1):R15–R30
Kume S, Yamahara K, Yasuda M et al (2014) Autophagy: emerging therapeutic target for diabetic nephropathy. Semin Nephrol 34(1):9–16
Takabatake Y, Kimura T, Takahashi A et al (2014) Autophagy and the kidney: health and disease. Nephrol Dial Transplant 29:1639–1647
Yang D, Livingston MJ, Liu Z et al (2018) Autophagy in diabetic kidney disease: regulation, pathological role and therapeutic potential. Cell Mol Life Sci 75(4):669–688
Choi AM, Ryter SW, Levine B (2013) Autophagy in human health and disease. New Engl J Med 368(7):651–662
Ding Y, Choi ME (2015) Autophagy in diabetic nephropathy. J Endocrinol 224:15–23
Glick D, Barth S, Macleod KF (2010) Autophagy: cellular and molecular mechanisms. J Pathol 221:3–12
Yamahara K, Yasuda M, Kume S et al (2013) The role of autophagy in the pathogenesis of diabetic nephropathy. J Diabetes Res 2013:193757
Kim YC, Guan KL (2015) mTOR: a pharmacologic target for autophagy regulation. J Clin Investig 125(1):25–32
Kim J, Kundu M, Viollet B et al (2011) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13(2):132–141
Inoki K (2014) mTOR signaling in autophagy. Sem Nephrol 34:2–8
Tanida I, Ueno T, Kominam E (2008) LC3 and autophagy - methods in molecular biology. Methods Mol Biol 445:77–88
Arakawa S, Honda S, Yamaguchi H et al (2017) Molecular mechanisms and physiological roles of Atg5/Atg7-independent alternative autophagy. Proc Acad Ser B Phys Biol Sci 93(6):378–385
Geng J, Klionsky D (2008) The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. Protein Rev EMBO Rep 9:859–864
Xu Y, Liu L, Xin W et al (2015) The renoprotective role of autophagy activation in proximal tubular epithelial cells in diabetic nephropathy. J Diabetes Complications 29:976–983
Walczak M, Martens S (2013) Dissecting the role of the Atg12–Atg5-Atg16 complex during autophagosome formation. Autophagy 9(3):424–425
Hanada T, Noda NN, Satomi Y et al (2007) The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem 282:37298–37302
Rabanal-Ruiz Y, Otten EG, Korolchuk V (2017) MTORC1 as the main gateway to autophagy. Essays Biochem 61:565–584
Ye X, Zhou XJ, Zhange H (2018) Exploring the role of autophagy-related gene 5 (ATG5ATG5) yields important insights into autophagy in autoimmune/autoinflammatory diseases. Front Immunol 9:1–15
Li A, Zhang H, Han H, Wang WZS, Hwang Z et al (2019) LC3 promotes the nuclear translocation of the vitamin D receptor and decreases fibrogenic gene expression in proximal renal tubules. Metabolism 98:95–103
Liu L, Yang L, Changc B, Zhang J, Guo Y, Yang X (2018) The protective effects of rapamycin on cell autophagy in the renal tissues of rats with diabetic nephropathy via mTOR-S6K1-LC3II signaling pathway. Ren Fail 40(1):492–497
Kitada M, Ogura Y, Itaru Monno I et al (2017) Regulating autophagy as a therapeutic target for diabetic nephropathy. Curr Diab Rep 17:53
Warren AM, Knudsen ST, Cooper ME (2019) Diabetic nephropathy: an insight into molecular mechanisms and emerging therapies. Expert Opin Ther Targets 23(7):579–591
Lenoir O, Tharaux PL, Huber TB (2016) Autophagy in kidney disease and aging: lessons from rodent models. Kidney Int 90:950–964
Kimura T, Isaka Y (2017) Yoshimori T autophagy and kidney inflammation. Autophagy 13(6):997–1003
Sakai S, Yamamoto T, Takabatake Y, Takahashi A, Namba-Hamano T, Minami S et al (2019) Proximal tubule autophagy differs in type 1 and 2 diabetes. J Am Soc Nephrol 30(6):929–945
Xu Y, Liub L, Wei Xin W et al (2015) The renoprotective role of autophagy activation in proximal tubular epithelial cells in diabetic nephropathy. J Diabetes Compl 29:976–983
Fang L, Zhou Y, Cao H et al (2013) Autophagy attenuates diabetic glomerular damage through protection of hyperglycemia- induced podocyte injury. PLoS ONE 8(4):e60546
Tanaka Y, Kume S, Kitada M et al (2012) Autophagy as a therapeutic target in diabetic nephropathy. Exp Diabetes Res 12:4
Kume S, Koya D (2015) Autophagy: a novel therapeutic target for diabetic nephropathy. Diabetes Metab J 39(6):451–460
Fioretto P, Zambon A, Rossato M et al (2016) SGLT2 Inhibitors and the diabetic kidney. Diabetes Care 39(2):S165–S171
Alicic AZ, Neumiller JJ, Johnson EJ et al (2019) Sodium-glucose cotransporter 2 inhibition and diabetic kidney disease. Diabetes 68(2):248–257
Wanner C, Inzucchi SE, Lachin JM et al (2016) Empagliflozin and progression of kidney disease in type 2 diabetes. New Engl J Med 375(4):323–334
Xu C, Wang W, Zhong J et al (2018) Canagliflozin exerts anti-inflammatory effects by inhibiting intracellular glucose metabolism and promoting autophagy in immune cells. Biochem Pharmacol 152:45–59
Liu WJ, Huang WF, Ye L (2018) The activity and role of autophagy in the pathogenesis of diabetic nephropathy. Eur Rev Med Pharmacol Sci 22:3182–3189
Yamahara K, Yasuda M, Kume S (2013) The role of autophagy in the pathogenesis of diabetic nephropathy. J Diabetes Res 20:13
Schuster A, Al-Makki A, Shepler B (2019) Use of paricalcitol as adjunctive therapy to renin-angiotensin-aldosterone system inhibition for diabetic nephropathy: a systematic review of the literature. Clin Ther 41(11):2416–2423
Hamzawy M, Gouda SAA, Rashid L, Attia Morcos M, Shoukry H, Sharawy N (2017) The cellular selection between apoptosis and autophagy: roles of vitamin D, glucose and immune response in diabetic nephropathy. Endocrine 58(1):66–80
Wang H, Wang J, Qu H et al (2016) In vitro and in vivo inhibition of mTOR by 1, 25-dihydroxyvitamin D 3 to improve early diabetic nephropathy via the DDIT4/TSC2/mTOR pathway. Endocrine 1:348–359
Xiao T, Guan X, Nie L et al (2014) Rapamycin promotes podocyte autophagy and ameliorates renal injury in diabetic mice. Mol Cell Biochem 394:145–154
Acknowledgments
This work was supported by Karim Family in memory of their son Hasan karim Magd Alkoroum, ISRAEL. MIGAL The Internal ministry for the development of Galilee-North Israel
Author information
Authors and Affiliations
Contributions
All of the authors contributed to the formation of overall concept. Nn, Nf and KE wrote the manuscript and NR, EF, Ht edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
All authors have no conflicts of interest to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Koch, E.A.T., Nakhoul, R., Nakhoul, F. et al. Autophagy in diabetic nephropathy: a review. Int Urol Nephrol 52, 1705–1712 (2020). https://doi.org/10.1007/s11255-020-02545-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11255-020-02545-4