
Abstract
In recent years, diabetic kidney disease has been the main cause of end-stage renal disease; more and more people have faced this serious public health problem worldwide. Autophagy is a conserved multistep pathway that degrades and recycles damaged organelles and macromolecules to maintain intracellular homeostasis. Autophagy plays key roles in several diseases, including kidney diseases. It has been suggested that dysregulated autophagy plays a vital role in both glomerular and tubulointerstitial pathologies in kidneys under diabetic conditions. The advances in our understanding of autophagy in diabetic kidney disease will be helpful for us to discover a new therapeutic target for the prevention and treatment of this life-threatening diabetes complication.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
1 The Pathogenesis of Diabetic Kidney Disease
Diabetic kidney disease (DKD) is a serious complication of diabetes mellitus and one of the most significant contributing factors to end-stage renal disease (ESRD). It is reported that around 35–40% of diabetes patients eventually develop DKD, which accounts for a significant increase in mortality in these patients and exacerbates a grave threat to the clinical outcome of diabetic patients. The rapidly increasing prevalence of diabetes and its complications will further increase an already high cost of therapies and economic burden on patients.
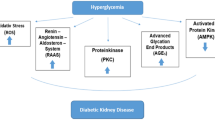
The development of DKD in diabetic patients is caused by multifactorial interactions between hemodynamic and metabolic pathways. It is well known that hyperglycemia, hypertension, and genetic pre-disposition are the main risk factors besides elevated serum lipids, smoking, overweight or obesity, physical inactivity, and the amount of dietary proteins (Yang et al. 2018).
There are many factors involved in the pathogenesis of DKD, including activation of protein kinase C (PKC), accumulation of advanced glycation end-products (AGEs). In addition, intracellular stress caused by endoplasmic reticulum (ER) stress, oxidative stress, and hypoxia are also the pathological factors in DKD. Meanwhile, hemodynamic changes such as systemic and glomerular hypertension associated with a hyperactive renin-angiotensin system (RAS) are also involved in the progression of DKD. In addition, it has been shown that the metabolic and hemodynamic abnormalities lead to inflammation and kidney fibrosis associated with DKD via the increased production of a variety of cytokines, chemokines, and growth factors.
The prevention and management of DKD have been multi-targeted, advocating a healthy lifestyle and targeting cellular and molecular factors involved in the pathogenesis of this disease. It has been shown that intensive interventions such as glycemic control, blood pressure control, and inhibition of the RAS can delay or decrease the risks for the onset and progression of albuminuria. Unfortunately, these treatments are unable to prevent loss of GFR or progression to ESRD as some patients develop treatment-resistant albuminuria.
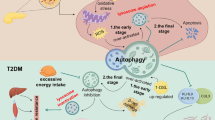
Therefore, in order to improve the prognosis of DKD, we need to explore effective therapeutic options. In diabetic kidneys, it has been shown that the level of autophagy is decreased. According to these findings, autophagy deficiency in diabetic kidneys may enhance the susceptibility of kidney cells to diabetes-associated damage, which worse albuminuria and renal function.
In recent years, it has been shown that inflammation is a risk factor for the progression of diabetes nephritis (Najafian et al. 2011). Diabetes is associated with increased intracellular ROS, an activator of the inflammasome. Advanced glycation end-products (AGEs) are irreversibly glycated proteins generated under hyperglycemia and can be endocytosed by kidney proximal tubules for lysosomal degradation, which finally suppress AGEs-induced inflammation. The increased level of AGEs via both overproduction of AGEs and impairment of degradation of AGEs can activate inflammation in diabetic nephropathy. In addition, IL1B (the product of inflammasome activation) pivotally aggravates glucose tolerance. Therefore, inflammasome activation is recognized as a key pathophysiology of diabetic nephropathy.
In lots of studies, it has been indicated that AGEs cause obvious alternations of structure and function in kidney (Ahmed 2005). The formation of AGEs can induce cross-linking of extracellular matrices such as collagen and generalize cellular dysfunction. The level of AGEs production is increased in the hyperglycemic state. Conversely, the clearance of AGEs in the kidneys is decreased, which are responsible for the accumulation of AGEs in DKD patients. It is reported that both the endogenous AGEs in kidneys are degraded in the lysosomes of the proximal tubular epithelial cells.
2 The Role of Autophagy in the Development of Diabetic Nephropathy
In DKD, autophagy plays a vital role in maintaining lysosomal homeostasis in podocytes. In addition, it has been reported that AGE overload disrupts lysosomal membrane permeabilization and damages autophagy in DKD.
Considering that the blood glucose levels were comparable, it is possible that AGE accumulation because of autophagy deficiency leads to tubular injury, inflammation, and interstitial fibrosis. In addition, it has been reported that AGEs induce extracellular matrix expansion and epithelial mesenchymal transition and inflammation. After AGE-BSA exposure in the autophagy-deficient PTECs, the level of MCP-1 was increased than in autophagy-competent PTECs. In fact, the mRNA levels of NLRP3, ASC, and caspase-1 and the protein level of IL-1β in the diabetic proximal tubule are increased in vivo. The fact indicates that autophagy plays a vital role in preventing kidney fibrosis and inflammation in DKD by upregulation of lysosomal degradation of AGEs (Takahashi et al. 2017).
Because AGEs can block autophagic flux in cultured PTECs, people speculate that AGEs should accumulate even in autophagy-competent cells treated with AGEs, which has not been the case. In the previous reports, AGEs were directly endocytosed to lysosomes for degradation. In one study, autophagic flux has been measured in vivo by immunostaining for the substrate of autophagy SQSTM1/p62. The results of the above study were the numbers of SQSTM1/p62-positive dots in STZ-treated Atg5F/F:KAP mice compared to vehicle-treated Atg5F/F:KAP mice which were significantly increased, whereas they were comparable between STZ- and vehicle-treated control mice. Therefore, autophagic flux is not completely inhibited in diabetic control mice.
STZ can induce various physiological changes including lipid metabolism besides hyperglycemia which may affect autophagic activity. It is speculated that lysosomal dysfunction and accumulation of AGEs may be result from autophagy deficiency (Chowdhury et al. 2019).
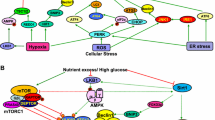
In another streptozotocin-induced diabetes model (type I diabetes model), endothelial-specific autophagy deficiency worsens the diabetic phenotype, which demonstrates that the deficiency of autophagy worsens diabetic nephropathy in vivo. In streptozotocin-induced autophagy-deficient mice, there are severe microalbuminuria, endothelial lesions, and damage in podocytes. High-fat diet challenge induces hyperglycemia with proteinuria, and finally causes damages to podocytes in podocyte-specific autophagy-deficient mice. Autophagy also results in the degradation of AGEs, and thus suppresses inflammation in kidneys (Fig. 36.1).

It is illustrated that possible mechanisms by which autophagy protects PTECs from the accumulation of AGEs under diabetic conditions. Autophagy inhibits the accumulation of endogenous AGEs by degrading intracellular proteins and exogenous AGEs by upregulating lysosomal functions, resulting in inhibiting inflammation and fibrosis in DKD (Takahashi et al. 2017)
Currently, the overall activity of autophagy, either activated or inactivated, in diabetic nephropathy is still controversial. It is reported that hyperinsulinemia due to hyperglycemia may suppress autophagic activity through MTOR activation. Importantly, this suppressive function of insulin seems to have a tissue-specificity, i.e., insulin suppresses autophagy in muscles whereas amino acids suppress autophagy in liver.
Conversely, it is also suggested that autophagy can be induced under hyperglycemia due to the production of ROS or direct cytotoxicity of hyperglycemia. In future, we still need to be determined whether over-nutrition status and hyperinsulinemia could suppress autophagy in kidney.
Moreover, what further modifies the autophagic activity under diabetic condition is the presence of diabetic byproducts and resulting complications. AGEs suppress autophagic/lysosomal degradation activity. Similar phenomena were reported under hyperlipidemia (high-fat diet challenge), a common metabolic complication seen in diabetes. Mechanistically, it is suggested that AGEs impair lysosomal membrane permeability and function. Conversely, autophagy upregulates lysosomal biogenesis and function via nuclear translocation of TFEB (transcription factor EB), a key regulator of lysosomal biogenesis.
In autophagy-deficient kidney tubular cells, it is shown that neither TFEB nuclear translocation nor lysosomal biogenesis upon AGEs challenge. The facts suggest a critical role of the autophagy-TFEB axis against AGEs. The scientists speculate that these byproducts and complications of diabetes may be associated with autophagic regulation of inflammation of the kidney in diabetes.
In conclusion, autophagy indeed plays a protective role in diabetic nephropathy. However, it is not clear whether autophagy is active or inactive in DKD. It is acknowledged that inflammation is the vital factor in the DKD pathogenesis. Importantly, autophagy may play a protective role against kidney inflammation in DKD, which may help us to explore the mechanism of the DKD.
It is the fact that in diabetic nephropathy models, the activation of mTOR in the proximal and distal tubules and the downregulation of the AMP-activated protein kinase pathway result in autophagy inhibition. In addition, the scientists also found that prolonged exposure to high glucose levels induced apoptosis and autophagy in tubular epithelial cells in vitro. Meanwhile, in the diabetic nephropathy, elevated ROS production and ER stress both can upregulate autophagy. In addition, autophagy is also induced by lipotoxicity in tubular cells via modulating fatty acid β-oxidation.
3 Interventional Treatment of Diabetic Nephropathy by Regulating Autophagy Activity
It is reported that a phytochemical ferulic acid protects against varied diseased conditions. In one study, the scientists explored the ameliorative role and mechanisms of ferulic acid in averting STZ-mediated nephrotoxicity. In vivo, they administered a single intraperitoneal injection of streptozotocin (50 mg/kg) in experimental rats to induce diabetes. In diabetic rats, the results were the increased level of blood glucose, as well as kidney to body weight ratio, a decrease in serum insulin level, severe kidney tissue damage and dysfunction. The participation of oxidative stress in hyperglycemia-triggered renal injury was demonstrated by elevation of intracellular ROS level, altered mitochondrial membrane potential, and cellular redox balance impairment. In the kidneys of ferulic acid group, kidney damage, apoptosis, inflammation, and defective autophagy were markedly ameliorated, probably via the underlying mechanism for such protection, such as AGEs, MAPKs, NF-κB-mediated inflammatory pathways, autophagy induction, and so on. In vitro, ferulic acid could decrease excessive ROS generation, active autophagy, and prevent apoptotic death of cells under high glucose environment. When autophagy is inhibited, the protective effect of ferulic acid is significantly eradicated in high glucose-mediated cell death. In conclusion, it has been confirmed that ferulic acid play antioxidant, anti-inflammatory, anti-apoptotic activities and role in autophagy, which could avoid oxidative stress-mediated renal cell damage (Chowdhury et al. 2019).
In another study, the role of spironolactone in podocyte loss and autophagy has been investigated. It is reported that spironolactone decreased the urinary albumin excretion, fasting glucose levels and lipids, and alleviated kidney damage. Spironolactone also increased the expression of the podocyte-specific markers WT1 and NPHS2, as well as the autophagic markers Beclin1 and LC3B. In addition, spironolactone not entirely blocked the renin–angiotensin–aldosterone system (RAAS) by regulating the ACE1, ACE2, and aldosterone levels. In one word, spironolactone promoted autophagy in podocytes and further alleviated DN through partially impeding the RAAS (Dong et al. 2019).
It is the fact that podocyte loss and apoptosis play a crucial role in the progression of DKD. One widely used Chinese herb called tripterygium glycoside (TG) has exerted comprehensive protective effects on preventing DKD progression. In a new study, people assessed the podocyte protective effect of tripterygium glycoside on DKD by the potential role of activation of autophagy and downregulating β-arrestin-1. In vitro, tripterygium glycoside and small interfering RNA (siRNA) of β-arrestin-1 were added to 10% db/db mice high-glucose serum to induce podocytes. In DKD mouse serum treated podocytes, pretreatment of tripterygium glycoside significantly ameliorated podocyte apoptosis, increased nephrin and podocin levels and inhibited expression of β-arrestin-1. Meanwhile, levels of autophagic activity were significantly higher. They demonstrated that silencing β-arrestin-1 upregulated autophagic activity and ameliorated podocyte apoptosis. In addition, silencing β-arrestin-1 in combination with tripterygium glycoside enhanced the levels of LC3-II and LC3-II/LC3-I ratios and reduced the expression of p62. Finally, in podocyte apoptotic rate was reduced notably in DKD serum + siRNA-β-arrestin-1 + TG group compared to DKD serum + siRNA-β-arrestin-1 group, and the levels of nephrin and podocin were increased compared to treatment with siRNA-β-arrestin-1 only. They demonstrated that tripterygium glycoside provided protection against podocyte injury induced by high-glucose serum, and that this effect was mediated by the concomitant activation of autophagy and downregulation of β-arrestin-1 (Zhan et al. 2019).
Generally, autophagy activity is inhibited under diabetic conditions, thus new therapeutic strategy has been focusing on restoring autophagy. In clinic, diet or calorie restriction is an important glycemic control therapy in diabetic patients and plays a renoprotective role in several metabolic or age-related kidney diseases. It is essential that activation of autophagy plays an indispensable role in calorie restriction-mediated anti-aging effects. Therefore, we believe that a calorie restriction regimen can activate autophagy which should be a potent therapeutic strategy to prevent DKD. In fact, researchers demonstrated that calorie restriction can restore autophagy activity in PTECs and attenuate renal damage in type 2 diabetic Wistar fatty rats. However, in recent human clinical studies, the quality of diet affects the development of insulin resistance and new onset of diabetes. In addition, under diabetic conditions, it is reported that a high energy/nutrient intake along with a low-protein diet may cause an insufficiency of diet restriction. Finally, we also need to identify the types of carbohydrates, amino acids, and fatty acids in different diet regimens that can result in a better prognosis.
In addition, it is the fact that drugs that can activate autophagy which affect mTORC1, AMPK, and SIRT1 may have therapeutic potency in DKD treatment. Rapamycin can induce autophagy and improve glomerular and tubular damage in DKD. However, it has been well-recognized that the side effects of long-term or complete mTORC1 inhibition.
Currently, it is still under debate whether mTOR inhibition by rapamycin or other mTOR inhibitors is safe and effective for treating DKD. In addition, it is also being explored that targeting AMPK and SIRT1 with activators such as resveratrol, metformin, and AICAR. It has been demonstrated that the above agents activate autophagy in different animal models of diabetes and protect the kidney from diabetic injury, making them be attractive therapeutic options for DKD. Moreover, the use of antioxidants to counter oxidative stress or chemical chaperons to reduce ER stress and to restore autophagy activity may also be a hopeful therapeutic approach to treat the diabetic kidney disease.
4 Conclusion
It is no doubt that autophagy would be a key therapeutic option in kidney diseases. However, modulation of autophagy in cancer therapy can worsen kidney function, which is demonstrated that general modulation of autophagy might result in unexpected side effects on the kidney. It is the fact that rough and bulk modulation of autophagy, therefore, may result in unwanted side effects in the kidney.
We still need to explore in future what the targets of autophagy are and how autophagy recognizes these specific targets in kidney diseases. For example, the mechanism and the proteins that are degraded in kidney diseases by autophagy are scarcely known. It is obvious that mitochondria are the targets of autophagy in kidney. However, how autophagy selectively recognizes damaged or depolarized mitochondria in unhealthy ones awaits determination. So far, it has not been identified as receptors of mitophagy for kidney diseases. Of course, specific targeting of autophagic targets would promote precision medicine. In other words, precise degradation of autophagic targets by precision autophagy would benefit kidney disease patients.
It has been reported that autophagy has dual roles (both activating and suppressing) in the kidney diseases, so the precise mechanistic study of these regulatory processes would also benefit kidney disease patients. Because autophagy is a metabolic process, one potential regulatory mechanism may be via metabolism. The modulations of autophagy may have a vital role in immunometabolic regulations in kidney diseases.
In conclusion, autophagic regulation protects most kidney diseases. In future, we need to clear the precise role of autophagy in kidney diseases.
References
Ahmed N (2005) Advanced glycation endproducts–role in pathology of diabetic complications. Diabetes Res Clin Pract 67:3–21
Chowdhury S, Ghosh S, Das AK, Sil PC (2019) Ferulic acid protects hyperglycemia-induced kidney damage by regulating oxidative insult inflammation and autophagy. Front Pharmacol 10:27
Dong D, Fan TT, Ji YS, Yu JY, Wu S, Zhang L (2019) Spironolactone alleviates diabetic nephropathy through promoting autophagy in podocytes. Int Urol Nephrol
Najafian B, Alpers CE, Fogo AB (2011) Pathology of human diabetic nephropathy. Contrib Nephrol 170:36–47
Takahashi A, Takabatake Y, Kimura T, Maejima I, Namba T, Yamamoto T, Matsuda J, Minami S, Kaimori JY, Matsui I, Matsusaka T, Niimura F, Yoshimori T, Isaka Y (2017) Autophagy inhibits the accumulation of advanced glycation end products by promoting lysosomal biogenesis and function in the kidney proximal tubules. Diabetes 66:1359–1372
Yang D, Livingston MJ, Liu Z, Dong G, Zhang M, Chen JK, Dong Z (2018) Autophagy in diabetic kidney disease: regulation, pathological role and therapeutic potential. Cell Mol Life Sci 75:669–688
Zhan H, Jin J, Liang S, Zhao L, Gong J, He Q (2019) Tripterygium glycoside protects diabetic kidney disease mouse serum-induced podocyte injury by upregulating autophagy and downregulating beta-arrestin-1. Histol Histopathol 18097
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Science Press and Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Cui, J., Bai, X., Chen, X. (2020). Autophagy and Diabetic Nephropathy. In: Le, W. (eds) Autophagy: Biology and Diseases. Advances in Experimental Medicine and Biology, vol 1207. Springer, Singapore. https://doi.org/10.1007/978-981-15-4272-5_36
Download citation
DOI: https://doi.org/10.1007/978-981-15-4272-5_36
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-15-4271-8
Online ISBN: 978-981-15-4272-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)