
Abstract
Hemophagocytic lymphohistiocytosis (HLH) is a hyperinflammatory syndrome caused by defective lytic capability of cytotoxic T lymphocytes and NK cells, which results in proliferation of benign hemophagocytic histiocytes. A cytokine storm ensues, and a severe systemic inflammatory response syndrome, multiorgan dysfunction syndrome, and death frequently follow. It may occur as a primary (inherited) form, or be acquired secondary to malignancy, infection, rheumatologic disease, or immunosuppression. Cardinal manifestations include fever, cytopenias, hepatosplenomegaly, and dysfunction of liver, kidney, CNS, and/or lung. Additional laboratory findings include marked hyperferritinemia, hypofibrinogenemia, hypertriglyceridemia, abnormal LFTs, coagulopathy, and hyponatremia. Nephrologists need to be aware of this syndrome owing to the frequent occurrence of acute kidney injury in these severely ill patients. Glomerulopathy and nephrotic syndrome may develop. Kidney transplant recipients are at increased risk of HLH due to immunosuppression, and most such cases are triggered by infection with over 50 % mortality. Effective treatment of HLH usually requires chemoimmunotherapy to acutely suppress inflammation, specific treatment of underlying infection or malignancy, and in certain cases hematopoietic stem cell transplantation. The pathogenesis, clinical manifestations, diagnosis, and treatment of HLH are discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hemophagocytic lymphohistiocytosis (HLH) is a syndrome characterized by a hyperactive yet ineffective immune response to an antigenic challenge. HLH results from either an inherited (primary) or acquired (secondary HLH) inability of cytotoxic CD8+ T lymphocytes (CTLs) and natural killer cells (NKs) to lyse target cells [1–4]. These target cells include the initiators of the immune response, such as infected or malignant cells, and antigen-presenting cells upon resolution of the initial challenge. The consequent proliferation of CTLs results in a large production of interferon-γ (INF-γ) that causes a marked proliferation of benign histiocytes (macrophages). These macrophages and CTLs invade organs, such as liver, spleen, and lymph nodes, and release further inflammatory cytokines, including INF-γ, TNF-α, and interleukins (IL)-1, 6, and 18 [5]. The result is a so-called cytokine storm with severe systemic inflammatory response syndrome (SIRS), multiorgan dysfunction syndrome (MODS), and frequent death. The proliferating histiocytes engulf red cells, white cells, platelets, and their precursors and are called hemophagocytes (HPC), hence the alternative designation hemophagocytic syndrome.
The cardinal clinical manifestations of HLH include unremitting fever, hepatosplenomegaly, various cytopenias, and multiorgan dysfunction, including liver, CNS, lung, and kidney [1–4]. Characteristic laboratory findings in addition to the cytopenias include hyperferritinemia, hypertriglyceridemia, hypofibrinogenemia, abnormal LFTs, hyponatremia, elevated LDH, elevated soluble CD25 (sCD25), reduced or absent NK cell activity, and coagulopathy. Bone marrow aspiration typically, but not always, reveals HPCs, which may also be seen in the liver, spleen, or lymph nodes. Unfortunately, no pathognomonic finding or test is available to diagnose HLH, including the presence of HPCs, as these may be found in otherwise severely ill patients [6, 7].
Nephrologists need to be aware of HLH [8]. Acute kidney injury (AKI) can develop, especially in the critically ill [9], where it may be challenging to differentiate HLH from severe SIRS/MODS secondary to sepsis, trauma, or autoimmune/autoinflammatory disease. The distinction is critical, as immunomodulatory therapy may be required to dampen the hyperinflammatory state if HLH has developed, but may be harmful otherwise. In addition, various glomerulopathies are reported in HLH, often in the setting of nephrotic syndrome [10]. Thrombotic microangiopathy (TMA) can also occur [10]. Finally, HLH can develop post-kidney transplantation [11].
Pathophysiology: primary HLH
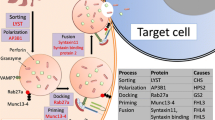
Primary or familial HLH (FHL) results from recessive mutations in genes involved in the function of the cytotoxic granules of CTLs and NKs. Normal degranulation requires several proteins for cytosolic vesicle transport, sorting, docking, priming, and finally fusion with the cell membrane [12]. Upon degranulation, perforin inserts into the target cell membrane, thereby allowing granzymes to enter and cause apoptosis. At least 5 types of FHL are described, termed FHL1-5. They are detailed in Table 1. FHL usually presents in the first months of life, often in response to a viral infection or immunization. However, some patients present later in adult life (see below).
Other inherited immune deficiency syndromes also can present with HLH, including oculocutaneous albinism syndromes (Chediak–Higashi syndrome, Griscelli syndrome type 2, and Hermansky–Pudlak syndrome type 2), which share defective cytotoxic degranulation similar to FHL3-5. The X-linked lymphoproliferative diseases (XLP), types 1 and 2, have normal degranulation but develop HLH in response to uncontrolled primary EBV infection [13]. Additional primary immunodeficiencies that may develop HLH include severe combined immunodeficiency (SCID), combined immunodeficiency, and chronic granulomatous disease [14]. Interestingly, HLH can develop in SCID patients with severe deficiencies of both CTLs and NKs. Nevertheless, macrophages can be activated with an associated cytokine storm [14].
Mice bearing analogous mutations to those found in human FHL and other primary immunodeficiencies have been developed [3, 15]. The best studied are perforin-deficient mice (Prf −/−), mimicking FHL2. These mice diseases do not develop spontaneously but require an infectious trigger, typically either the lymphocytic choriomeningitis virus (LCMV) or murine cytomegalovirus (MCMV). With LCMV, the full HLH syndrome develops, and the main pathogenic cytokine is INF-γ secreted by CD8+ CTLs. A somewhat more benign syndrome develops with MCMV. Although CTLs and NKs produce INF-γ to activate macrophages in this model, TNF-α secreted by macrophages and dendritic cells (DCs) is the main culprit, and IL-10 secreted by NKs tends to dampen the hyperinflammation. Analogous to FHL3 and FHL4, mice deficient in Munc13-4 (Unc13d jinx/jinx) and syntaxin 11 (Stx11 −/−), respectively, have been developed. Again, they are susceptible upon viral challenge to develop an HLH-like syndrome with variations in severity and cytokine mediators.
With FHL, the severity of the cytolytic defect of CTLs in patients with biallelic null mutations correlates with severity of disease, at least in terms of age of onset [15]. Perforin deficiency, the most severe defect, has the earliest age of onset (mean age 3 months). The age of onset increases with Griscelli syndrome type 2 (13 months), FHL4 (27 months), and Chediak–Higashi syndrome (38 months), although high variability exists within each group [15, 16]. Similar graded defects in cytotoxicity correlate with the severity of disease in the corresponding murine models: Perforin-deficient mice demonstrate the most severe HLH, followed by Rab27A deficiency (Griscelli syndrome type2), syntaxin 11 deficiency (FHL4), and Lyst deficiency (Chediak–Higashi syndrome). Of note, restoration of perforin expression to 10–20 % of normal with mixed chimerism in a mouse model (Prf −/−) reestablished normal immune regulation [17]. This is analogous to patients with hypomorphic missense mutations (as opposed to null mutations) who have reduced rather than absent protein expression. Such patients present later in life, with milder disease or atypical features [18–21].
Pathophysiology: secondary HLH
sHLH develops at any age without a detectable genetic defect. Inciting events include infection, malignancy, autoimmune/autoinflammatory disease, metabolic disease, and immunosuppression associated with HIV or solid organ transplantation [22–25]. An appropriate inflammatory response becomes exaggerated to produce a clinical syndrome similar to FHL. Polymorphisms or hypomorphic mutations in the same genes that cause FHL may underlie susceptibility to sHLH (see below). A predisposing condition and/or triggering event can be identified in the majority of sHLH cases. Multiple causes may coexist, such as malignancy or immunosuppression with an infectious trigger. Occasionally, no inciting event is found and these cases are considered idiopathic. Since primary HLH can be initiated by the same triggering agents as those causing sHLH, an apparent diagnosis of sHLH does not rule out an underlying genetic defect.
The hyperferritinemia and hemophagocytosis characteristic of HLH both contribute to its pathophysiology and, simultaneously, help to mitigate the hyperinflammation. Ferritin is an intracellular, iron-storage molecule composed of 24 subunits [26]. Ferritin is secreted by hepatocytes, Kupffer cells, and macrophages in an iron poor form. Secreted ferritin has pro-inflammatory effects by stimulating production of NF-κB in hepatic stellate cells. Alternatively, lymphocytes may be stimulated to produce the anti-inflammatory IL-10. In addition, ferritin inhibits CXC chemokine receptor 4, thereby reducing proliferation and migration.
Anemia in HLH develops rapidly, and hemophagocytosis per se has been ascribed a primary role. Zoller et al. [27] termed this “consumptive anemia of inflammation” and showed that interferon-γ signaling is required for both the anemia and hemophagocytosis by a process resembling apoptotic cell uptake. Hemophagocytosis preceded anemia and was considered the major proximate cause. In a different model, Behrens et al. [28] produced severe anemia without detectable hemophagocytosis. Subsequently, this group confirmed the prime importance of interferon-γ for development of severe anemia but not for hemophagocytosis, which was readily detectable in interferon null mice in the absence of anemia [29]. Hence, hemophagocytosis was neither necessary nor sufficient for developing severe anemia.
Recent evidence indicates that the driving force for MODS in HLH is the cytokine storm and not hemophagocytosis [30]. In fact, HPCs can release significant quantities of the anti-inflammatory IL-10, representing a mechanism to dampen the hyperinflammation [31]. In a mouse model, blocking either hemophagocytosis itself or the IL-10 released from HPCs enhanced virus-induced CTLs, liver damage, and mortality [31]. HPCs express markers of alternate activation [32] (M-2 macrophages), including the hemoglobin/haptoglobin scavenger receptor CD163 [32]. M-2 macrophages contribute to resolution of inflammation and tissue repair [33]. Both free and bound hemoglobin can be taken up by CD163+ macrophages and can activate heme oxygenase-1 (HO-1) [34]. Similarly, CD163+ HPCs have upregulated HO-1 in response to free heme liberated from phagocytosed erythrocytes [35]. The HO-1 catabolism of heme results in production of ferritin, bilirubin, and carbon monoxide, agents with potent anti-inflammatory effects [36]. Interestingly, postmortem bone marrow samples of patients dying from sepsis revealed abundant HPCs expressing HO-1, thereby indicating role for hemophagocytosis in hyperinflammatory states in general [35].
HLH in adults
Ramos-Casals et al. [22] reviewed MEDLINE and Embase databases supplemented with manual searches through 9/2011 for case series dealing with the clinical manifestations and treatment of HLH in adults and found 677 articles (2197 patients). Mortality was 41 % in a subset of 1109 patients. Infections were identified in 1108 (of the 2197), neoplasms in 1047, autoimmune disease in 276, transplantation in 95 (including 53 kidney), and other circumstances in 89. Only 81 were idiopathic. Nearly a third had multiple causes. The most common infections were viruses (762 of 1108), mainly EBV (330) and HIV (173). Other viruses included CMV (69), other herpes viruses (74), parvovirus, hepatitis viruses, and influenza. Bacteria were found in 206/1108, most commonly tuberculosis (78), but also staphylococcus and E coli. The most common parasite was leishmania and histoplasma the most common fungus. Of 1047 neoplasms, the majority were hematologic (981), including 369 with T cell or NK cell lymphoma, 333 with B cell lymphoma, 67 with leukemia, and 61 with Hodgkin’s lymphoma. Solid tumors were rare (32). The most common autoimmune diseases were SLE (133/276) and adult-onset Still’s disease (AOSD) (54).
Subsequently, 3 large series of HLH in adults were published. Riviere et al. identified 162 patients [23]. Hematologic malignancies were the most common triggers (92 patients). Infections were identified in 40, including 6 with concurrent malignancies. Autoimmune disease was found in 5. Parikh et al. [24] reported 62 adult patients. They also found the most common cause to be malignancy (32 patients). Infection was found in 21, autoimmune disease in 5, and 4 were idiopathic. Li et al. [25] reported 103 cases. Again, hematologic malignancies were most common (49 patients). Infections were found in 24, autoimmune diseases in 14, 24 with an unknown origin, and 8 with multiple causes.
When complicating rheumatic diseases, sHLH is termed macrophage activation syndrome (MAS). Most commonly, this occurs in systemic onset juvenile idiopathic arthritis (sJIA) [37], AOSD [38], or SLE (childhood [39] or adult onset [40]). HLH can also complicate Kawasaki’s disease or a vasculitis. With autoimmune or autoinflammatory conditions, HLH is typically triggered by a flare of the disease [37–39]. Less commonly, viruses such as EBV or antirheumatic medications are implicated.
HLH develops in about 10 % of cases of sJIA. Another 30–40 % evidence occult HLH [41–43]. Such cases have laboratory abnormalities consistent with HLH, but lack clinical manifestations [43]. When HLH complicates sJIA, 20 % of cases appear simultaneously with onset of the sJIA without frank arthritis and may be confused with FHL. Defective cytotoxic function of NKs occurs with HLH complicating sJIA, although a similar defect may be detectable in the absence of HLH [44].
Genetic considerations in adult HLH
The separation of primary HLH from sHLH is not clear-cut and should not be based simply on age of presentation. sHLH can present in childhood [45], and adults with sHLH may have genetic defects similar to the inherited conditions in Table 1. Realization of this overlap is important, because with an inherited mutation HLH may recur, and should the patient survive the initial episode prolonged therapy and/or hematopoietic stem cell transplantation (HSCT) need be considered.
Zhang et al. [21] studied 175 adult HLH patients referred for genetic testing. Missense and splice-site mutations/polymorphisms in the genes for perforin, MUNC13-4, and MUNC18-2 were found in 25 (14 %), including 12 (48 %) with the A91V polymorphism in both heterozygous and homozygous states. The A91V polymorphism reduces expression of perforin [46] resulting in susceptibility to HLH and is present in 3–17 % of the general population (1–4 % homozygous) [46]. Two patients were double heterozygotes with the A91V mutation and another mutation in one of the genes involved in degranulation. In a follow-up study, Zhang et al. [47] found an additional 21 patients with digenic heterozygous mutations involving perforin (10 of which had A91V) and one of the degranulation genes. Seven had digenic heterozygous mutations in 2 degranulation genes. These mutations/polymorphisms were considered hypomorphic, with adult HLH following a viral or other trigger. Sieni et al. [48] described 11 adult-onset patients with genetic defects underlying HLH in an Italian registry. These included 6 patients with biallelic perforin mutations, of which 4 included heterozygous A91V polymorphisms. Two also carried biallelic mutations consistent with FHL3, one FHL5, and 2 with XLP1. Wang et al. [49] studied 195 Chinese adults with HLH and found 3 with biallelic perforin mutations, 1 hemizygous SAP mutation (XLP1), and 6 monoallelic mutations (3 involving perforin, 3 syntaxin 11). None had the A91V polymorphism.
FHL mutations may also underly sHLH in the pediatric age range. In cases of HLH complicating sJIA, pathogenic biallelic MUNC13-4 mutations were found in 2 of 18 patients [50]. Another study found the A91V perforin polymorphism in 20 % of 15 sJIA patients with HLH compared to 10 % of 41 sJIA patients without HLH [51]. Similarly, whole-exome sequencing of 14 sJIA patients with HLH found 5 with protein-altering variants in FHL-related genes (MUNC13-4, STXBP2, and LYST) compared to 4 of 29 patients with sJIA without HLH [52]. In a study of 28 patients with HLH, 13 had 1 or more mutations in HLH-related genes, including 5 with STXPB2 and 5 with UNC13 mutations [53].
Clinical manifestations
The clinical manifestations of HLH result from tissue invasion by macrophages and CTLs, as well as the “cytokine storm” from the excessive release of inflammatory cytokines, especially IL-1, IL-6, IL-18, INF-γ, and TNF-α. A constellation of symptoms, signs, and laboratory abnormalities occurs that depends on the severity of the syndrome, the underlying predisposing conditions, and the presence of a triggering agent. Unremitting fever is nearly universal. Other constitutional symptoms include asthenia and weight loss. Splenomegaly, hepatomegaly, and adenopathy occur in a significant minority. Neurologic manifestations may develop in up to 70 % of sHLH cases [54–57] and may be obvious clinically or detectable only by imaging or CSF examination [56]. Clinical findings include altered mental status, seizures, hemiparesis, cranial nerve palsies, and meningitis. Permanent sequelae may result [54, 56, 57]. Coagulation abnormalities are common. Disseminated intravascular coagulation (DIC) may occur in 50 % HLH patients in the ICU, with over 20 % having severe bleeding [58]. Liver dysfunction is common and can progress to fulminant failure. Histologically, portal tract infiltration by CTLs and HPCs is present [59]. Cutaneous manifestations occur in up to 65 % of patients and include erythroderma, maculopapular rash, and morbilliform eruption [60]. HPCs may be found in skin biopsies [61]. A systemic inflammatory response syndrome (SIRS) with multiorgan dysfunction syndrome (MODS) may occur, including shock and acute lung or kidney injury. The clinical manifestations of HLH overlap with other causes of SIRS or MODS, such as bacterial sepsis or trauma, and such cases may be incorrectly labeled as “culture-negative sepsis [62]”. Patients dying from sepsis may have a marked proliferation of CD163+ HPCs predominantly ingesting RBCs and their precursors in the absence of frank HLH [35].
The most notable laboratory abnormalities in HLH include cytopenias, hyperferritinemia, hypofibrinogenemia due to consumption and liver injury, hypertriglyceridemia secondary to cytokine inhibition of lipoprotein lipase, and elevated transaminases and LDH. CRP may be markedly elevated, but the ESR is often normal or only minimally elevated because of hypofibrinogenemia. Elevated soluble CD25 and soluble CD163 reflect excessive CTL and macrophage activation, respectively. NK cells or their function is markedly reduced. Hyponatremia is common.
The most frequent renal manifestation is AKI. Aulagnon et al. [9] reported on 95 ICU patients with sHLH. Using current definitions, AKI occurred in 59 (62 %); 6-month survival was 37 % as compared to 56 % in those without AKI. Most (51 patients) reached stage 2 or 3 AKI, and dialysis was required in 59 %. AKI was attributed to acute tubular necrosis (49 %), hypoperfusion (46 %), tumor lysis (29 %), or glomerulopathy (17 %). Only 1 patient had a kidney biopsy. Nephrotic syndrome (NS) was present in 12, of whom 9 had AKI. Thirty-two percent of surviving patients had CKD at 6 months. The incidence of AKI was no different from that in a contemporaneous group of newly diagnosed, high-grade malignancy patients admitted to the same ICU (58 % of 202 patients). Direct interstitial infiltration by activated macrophages and T lymphocytes is reported to cause reversible AKI [63].
Glomerulopathy and NS complicating HLH result from primary podocyte pathology. Thaunat et al. [10] reported on 9 patients with NS and HLH that had kidney biopsies at 3 French hospitals and another 2 cases from the literature. AKI was present in 10/11, and 7/11 died. The underlying lesions included collapsing FSGS (5 patients, all of African descent), minimal change disease (4 Caucasian patients), and TMA (2 patients). A subsequent case report found minimal change disease in association with HLH [64].
HLH can complicate kidney transplantation. In 1979, Risdall et al. [65] described 19 patients with virus-associated HLH, and 13 were kidney transplant recipients. All cases were triggered by viruses, predominantly CMV. Karras et al. [66] studied 17 patients with HLH among 4230 renal transplants (prevalence of 0.4 %) collected from 8 Parisian transplant units. HLH developed from 10 days to 15 years after transplantation (median 52 days). All 17 were receiving corticosteroids, and 11 had received ATG within 3 months prior to developing HLH. Infections were detected in 14, most commonly herpes viruses (3 EBV, 3 CMV, 1 HHV 6, and 1 HHV8). Two patients had lymphoma, and 2 had no obvious trigger. Immunosuppression was reduced in all. Eight died, and 4 of the 9 survivors lost their allografts. Asci et al. [67] reported on 13 patients out of 403 renal transplant recipients (prevalence of 3.2 %) from a center in Turkey. HLH occurred from 2 weeks to 30 months post-transplantation (median 15 months). An infectious trigger was identified in 6 (tuberculosis in 4, CMV in 2, E. coli in 1). Hepatitis C was present in 8 of the 13. All patients had azathioprine discontinued and CNI reduced or discontinued. All 6 patients that received IVIg survived, as did 2 other patients responding to antimicrobial therapy.
In a 2009 Editorial Review, Ponticelli and Alberighi identified 76 cases of HLH in kidney transplant recipients with an overall 53 % mortality [11]. The majority of cases were triggered by infections, most commonly viral, but also bacterial and protozoal infections. HLH also occurred in patients with malignancy. Subsequently, additional cases of HLH in kidney transplant recipients have been reported with a variety of triggers, typically infections [68–74], including histoplasmosis, dengue, Bartonella, CMV, and BK virus. To date, 84 kidney transplant patients have been reported to have HLH [65–90]. Infectious triggers were identified in 76 % (Table 2).
Diagnosis of HLH
No pathognomonic finding or test confirms HLH. The Histiocyte Society proposed criteria for diagnosing pediatric FHL (Table 3). According to the most recent update (HLH-2004) [91], 5 of the following 8 criteria must be satisfied: (1) fever; (2) cytopenia in 2 lineages; (3) splenomegaly; (4) elevated ferritin; (5) elevated triglycerides and/or reduced fibrinogen; (6) hemophagocytosis in bone marrow, spleen, or lymph nodes; (7) low or absent NK cell cytotoxic activity; and (8) elevated soluble CD25. Supporting evidence not required for diagnosis includes abnormal liver function tests, CNS involvement (based on clinical examination, CSF findings, and/or CT or MRI scans), lymphadenopathy, rash, hyponatremia, and elevated LDH. Hence, a diagnosis is made by a constellation of clinicopathologic findings, familial history, or documentation of genetic mutations. The applicability of HLH-2004 criteria to adults with suspected HLH remains to be determined. Importantly, all of these features with the possible exception of splenomegaly can be found in severe SIRS secondary to trauma or sepsis [62]. Furthermore, the underlying predisposing conditions, such as rheumatologic disease or malignancy, may themselves affect baseline levels of some of these laboratory abnormalities. This clouds the issue of appropriate cutoffs and questions the use of specific criteria depending on the underlying disease.
A web-based, international Delphi study of 24 HLH experts identified 7 criteria as “absolutely required” or “important” for diagnosing sHLH in adults: cytopenia(s), demonstrable hemophagocytosis, fever, organomegaly, elevated ferritin, predisposing disease, and high LDH [92]. Four other criteria were of uncertain benefit: fibrinogen, triglycerides, elevated transaminases, and percentage of glycosylated ferritin. Fardet et al. [93] utilized the results of this Delphi study in a retrospective analysis of 312 patients to derive the HScore. This score included 6 of the “absolutely required” Delphi criteria, as well as 3 of 4 of uncertain benefit. Scores ranged from 0 to 337 with area under receiver operator curve of 0.97 in the original developmental data set and 0.95 in a separate validation set. The optimal cutoff was 169, accurately classifying 90 % of the patients. This scoring system is available online at http://saintantoine.aphp.fr/score/.
The use of HLH-2004 criteria to diagnose HLH (called MAS) in the setting of rheumatic disease is even more problematic. The cutoffs for thrombocytopenia, neutropenia, and fibrinogen in HLH-2004 may be too stringent for an autoinflammatory condition, such as sJIA where levels are typically high to start. More relevant may actually be a drop in these measurements. Ravelli et al. published guidelines for diagnosis of HLH in the setting of sJIA (Table 3) [94]. Laboratory criteria included (1) decreased platelet count (≤262 × 109/l), (2) elevated aspartate aminotransferase (>59 U/l), (3) decreased WBC count (≤4.0 × 109/l), and (4) hypofibrinogenemia (≤250 mg/dl). Clinical criteria included (1) CNS dysfunction, (2) hemorrhages, and (3) hepatomegaly. Any 2 or more laboratory criteria or 2 or more laboratory and clinical criteria would be sufficient for diagnosis. The higher cutoffs compared to HLH-2004 for platelets, WBC count, and fibrinogen were required because of the elevated baseline levels. These criteria were recently validated in a large, retrospective, multinational study, with better performance compared to modified HLH-2004 criteria [95]. In another study of 27 patients with sJIA diagnosed with MAS by these guidelines, 33 % did not satisfy HLH-2004 criteria [96]. Similar issues apply to diagnosing MAS in AOSD and SLE, the 2 most common triggering autoimmune diseases in adults. In this situation, no specific diagnostic criteria have been published for adults.
If HLH is a consideration, the majority of criteria in either HLH-2004 or HScore are readily obtainable, with the exception of NK cell function and soluble CD25 levels. Of special note are serum ferritin and bone marrow aspiration (BMA). Unfortunately, neither is specific, and BMA lacks sensitivity. The pediatric HLH-2004 criteria use a ferritin cutoff of 500 ng/ml. Although quite sensitive, it is not specific, even in childhood. Allen et al. studied 330 consecutive children with maximum ferritin levels above 500 ng/ml. Only 10 were diagnosed with HLH. The optimal cutoff for diagnosing HLH in this retrospective pediatric cohort was 10,000 ng/ml with a sensitivity of 90 % and specificity of 96 % [97]. Adult series show even less specificity. Moore et al. studied 627 adult patients with maximum ferritin levels above 1000 ng/ml and found only 4 with HLH [98]. Beer et al. [99] studied 405 adult patients with ferritin levels above 5000 ng/ml and found only 3 cases had HLH. Schram et al. [100] evaluated 113 adult patients with ferritin levels above 50,000 ng/ml. HLH was found in only 19 (17 %), even at these extraordinarily high levels. Major contributing disorders to such hyperferritinemia in these series included renal failure, iron overload, hepatocellular injury, infection, and malignancy.
A bone marrow aspiration is mandatory to determine HPCs are present, as well as to rule out hematologic malignancy. The presence of HPCs on bone marrow aspiration is not required for diagnosis, however, as the sensitivity is only 60–85 % [101, 102]. Hence, a negative aspiration does not rule out HLH and should not delay specific treatment, if otherwise indicated [101]. Furthermore, finding HPCs is clearly not specific for HLH, as they can often be found following transfusions or surgery [103] and in the critically ill [7]. Suster et al. [104] studied 230 consecutive, autopsied adults and found moderate-to-severe HPCs in the bone marrow of 102 cases (44 %). This result was strongly associated with the number of recent RBC transfusions. In those with ≥5 units transfused, the adjusted odds ratio was nearly 60. Strauss et al. [7] studied 107 consecutive autopsied medical ICU patients and found hemophagocytosis in the bone marrow of 69 (64.5 %).
In all cases diagnosed as HLH, screening for genetic defects is recommended. If present, a decision regarding aggressive therapy and possible HSCT is simplified. Formal genetic testing is labor intensive and takes weeks to complete. Flow cytometric (FC) assays are available with results in several days. Normal degranulation of NK cells and CTLs results in surface expression of CD107a. Such expression is abnormal in FHL3-5 and the oculocutaneous albinism syndromes, but normal in FHL2 and the XLP syndromes. FC staining for intracellular perforin is absent or greatly reduced in FHL2, and intracellular SAP and XIAP are deficient in XLP1 and 2, respectively. All patients should have CD107a and perforin assayed, and all male patients SAP and XIAP assayed as well [105]. Using this protocol, Bryceson et al. [106] evaluated 494 patients by FC with suspected HLH and found a sensitivity of 96 % and specificity of 88 % for differentiating genetic degranulation defects (FHL3-5 and oculocutaneous albinism syndromes) from FHL2, XLP1 and 2, and sHLH. Directed, formal genetic testing can then follow.
Identification of triggering agent
After establishing a diagnosis of HLH, it is imperative to search for a triggering agent that may require specific therapy [2, 105]. Malignancy and infection are the 2 most common triggers in adult HLH, with autoimmune disease a distant third [22–25]. The most common triggering infections are viral, especially EBV [45, 107] and other herpes viruses. Blood for PCR analysis should be obtained for EBV, CMV, VZV, herpes simplex, HHV6, HHV8, parvovirus B19, adenovirus, hepatitis, and influenza. Many other infections have been identified as triggering agents, especially intracellular pathogens, but also pyogenic bacteria [108]. If suspected, PCR of a bone marrow aspirate for leishmania should be performed. A malignancy evaluation is indicated in sHLH, especially in the absence of an identified infection or auto-inflammatory condition, and should include CT or MRI of chest and abdomen [109]. A bone marrow evaluation is mandatory, and a PET scan may also be considered.
Treatment of HLH
Treatment of HLH depends on the severity of hyperinflammation, underlying disease, the specific trigger, and whether or not an underlying genetic predisposition exists. No randomized, controlled treatment trials have been published, and only observational data exist. FHL in the pediatric age range is nearly uniformly fatal, with 1-year survival in early reports of less than 5 % [110]. The HLH-94 protocol of 8 weeks of dexamethasone and etoposide with intrathecal methotrexate in selected cases dramatically improved outcomes. In patients with persistent, familial, or relapsing disease, continued dexamethasone pulses, daily cyclosporine, and intermittent etoposide were used as a bridge to HSCT [55]. In a multinational series of 249 pediatric patients using this protocol, the estimated 5-year survival was 54 %, and this improved to 66 % in the 124 able to undergo HSCT [55]. Of note, 49 children were alive and well >1 year after completion of therapy that had a median duration of 4 months without HSCT. Presumably, these patients had sHLH. The HLH-2004 protocol added cyclosporine during the 8-week induction phase [91]. As an alternative regimen, a single-center series of 38 pediatric FHL patients received antithymocyte globulin (ATG) and methylprednisolone, along with intrathecal methotrexate and corticosteroids. Maintenance therapy then included cyclosporine and intermittent intravenous immunoglobulins until 26 eventually underwent HSCT [111]. The complete response rate to ATG was 73 % with another 24 % attaining a partial response. HSCT is indicated in patients with documented genetic mutations, as well as in those with familial, relapsing, or refractory disease. Reduced intensity conditioning appears to be better tolerated than myoablative conditioning.
The optimal treatment of sHLH in adults remains undefined. Although some cases resolve with just supportive therapy and treatment of the trigger, the most immediate issue is usually to quell the intense hyperinflammatory state. At a minimum for cases requiring urgent treatment, high-dose corticosteroids are indicated. In severe, familial, or relapsing disease, HLH-2004 should be considered. Etoposide appears to be especially suited for HLH, as it selectively deletes activated CD8+ CTLs in LCMV-infected Prf −/− mice and alleviates all manifestations of HLH [112]. Cyclophosphamide and methotrexate had similar effects, although other chemotherapeutic agents did not. In a retrospective analysis of 162 adults with sHLH, first-line use of etoposide was associated with significantly improved 30-day survival by multivariable analysis [113].
Treatment of an identified infectious trigger is mandatory, such as ganciclovir for CMV or amphotericin for leishmania. In the latter circumstance, antimicrobial therapy alone may suffice. In cases triggered by EBV, observational data support the use of etoposide in both pediatric [114] and young adult [115] patients. Treatment was most effective when instituted within 4 weeks of onset of disease [114, 115]. Theoretical support for use of etoposide derives from studies demonstrating EBV infection of CD8+ CTLs in EBV-HLH [116, 117]. Etoposide was also shown to have direct antiviral effects by inhibiting EBNA synthesis and EBV-induced transformation of mononuclear cells in vitro [118]. B cells may also be infected in EBV-HLH [119], and rituximab combined with traditional HLH therapy significantly reduced ferritin levels and EBV viral titers [120]. HSCT has also been effective in EBV-associated HLH [121].
Hyperferritinemic MODS in the ICU patient is not uncommon and merits consideration of sHLH. Such patients have severe SIRS caused by suspected/confirmed sepsis or a noninfectious illness, such as active rheumatologic disease, catastrophic antiphospholipid syndrome, or trauma [26, 62]. Malignancy is also common [122, 123]. If sHLH is deemed present, the use of chemoimmunotherapy in a potentially septic patient, however, poses a dilemma. A family history of HLH or possibly consanguineous parents would necessitate the HLH-2004 protocol [124]. Rapid flow cytometric screening for genetic defects as outlined above should be performed, and if positive would also support HLH-2004 protocol, as would significant CNS involvement. Active malignancy would necessitate either HLH-2004 protocol or specific therapy.
Two recent series describe mortality and treatment of adult HLH cases admitted to the ICU, one based on HLH-2004 criteria [122] and one based on the HScore [123]. Hospital mortality ranged from 52 to 68 %, respectively. Steroids were used in 55 and 66 %, etoposide in 80 and 40 %, and intravenous immunoglobulin (IVIG) in 5 and 27 %, respectively. Some authors favor methylprednisolone over dexamethasone in ICU cases [125]. Plasma exchange (PE) and anakinra have also been employed. In a multicenter, retrospective cohort study of 23 critically ill children with hyperferritinemic syndrome, suspected sHLH was treated with PE and either IVIG or methylprednisolone (n = 17) and compared to PE and IVIG with dexamethasone, cyclosporine, or etoposide (n = 6). Despite documented infections in 15 patients, only 3 died, all receiving the more aggressive HLH-like agents [125]. Other data support the use of PE and IVIG [26, 62]. Anakinra appeared effective as initial therapy in a retrospective case series of 8 pediatric sHLH cases admitted to the ICU. However, 6 also received high-dose steroids, and 5 received IVIG [126]. Therapy was well tolerated, and anakinra is safe in patients with severe sepsis [127].
Recent series of adult HLH implicate malignancy as the most common trigger, usually lymphomas. HLH can complicate the active phase of malignancy or occur following chemotherapy-induced remission, where it is typically triggered by an infection [109, 128]. With active malignancy, it remains unclear whether first-line therapy should be HLH-directed (e.g., HLH-2004 protocol) or targeted to the specific malignancy. If HLH directed, specific malignancy therapy should immediately follow resolution of the hyperinflammation. Infection can also coexist with active malignancy, most notably EBV [129], and in such cases anti-B cell therapy is probably additionally indicated [109]. Active malignancy, usually lymphoma, is also found in over 50 % of HIV-associated HLH cases [130]. Chemotherapy-induced HLH results from infection and necessitates either reduction in intensity or interruption of further chemotherapy [109].
Mortality rates with HLH complicating auto-inflammatory/autoimmune diseases are generally much lower than with FHL or other causes of sHLH. Hence, initial therapy is less intense than HLH-2004 and usually does not include etoposide. For example, mortality in a large series of 362 HLH cases complicating sJIA was 8 % [131]. Nearly all (98 %) received corticosteroids, 61 % received cyclosporine, and 36 % received IVIG. Biologic agents were given to 15 %, most commonly anakinra (10 %), but also etanercept, rituximab, tocilizumab, infliximab, and canakinumab in a handful. Etoposide was only used in 12 %. Interestingly, HLH can develop in patients with sJIA undergoing treatment with biologic agents, including tocilizumab [132, 133], canakinumab [134], and anakinra [135]. In the latter case, dose escalation was effective in treatment [135]. In juvenile lupus-associated HLH, mortality is around 10 % [39], with steroids (100 %), cyclosporine (38 %), and IVIG (32 %) being the mainstays of therapy in a multicenter series and literature review of 38 patients [39]. The largest series of adult autoimmune/autoinflammatory HLH in the absence of coexisting active infection or malignancy reported 116 patients, including 61 with SLE and 31 with AOSD. Overall mortality was 13 %. Corticosteroids were used in 98 %, with 53 % of 87 patients responding to steroid monotherapy [38]; however, IVIG was used in 24 %, cyclosporine in 21 %, IV cyclophosphamide in 15 %, and etoposide in only 3 %. In the presence of an infectious trigger, and in the absence of an underlying disease flare, reduction in immunosuppression may be preferred in SLE-associated HLH [40].
Little data exist to guide therapy of HLH in kidney transplant recipients. As shown above, the vast majority of cases are triggered by infections, which should be specifically treated whenever possible. We believe calcineurin inhibitor therapy should be continued given the role of cyclosporine in HLH-2004. High-dose steroids are indicated, and antimetabolite therapy should be discontinued to reduce possible over-immunosuppression. If rejection develops, IVIG is a consideration, with PE if antibody mediated. If EBV is detected, dexamethasone, etoposide, and rituximab seem justifiable.
Conclusion
Nephrologists need to be aware of the clinical manifestations, diagnosis, and treatment of HLH in its various settings. In the acutely ill ICU patient with AKI in the setting of MODS, HLH may have supervened, a situation necessitating specific treatment. Similarly, patients with autoimmune and autoinflammatory diseases may develop HLH and present with glomerulopathy associated with the either the underlying disease or HLH. Finally, in immunosuppressed kidney transplant patients, when clinical conditions suggest it, HLH must be recognized as mortality is over 50 %.
References
Chandrakasan S, Filipovich AH (2013) Hemophagocytic lymphohistiocytosis: advances in pathophysiology, diagnosis, and treatment. J Pediatr 163(5):1253–1259
Janka GE, Lehmberg K (2014) Hemophagocytic syndromes—an update. Blood Rev 28(4):135–142
Brisse E, Wouters CH, Matthys P (2015) Hemophagocytic lymphohistiocytosis (HLH): a heterogeneous spectrum of cytokine-driven immune disorders. Cytokine Growth Factor Rev 26(3):263–280
Schram AM, Berliner N (2015) How I treat hemophagocytic lymphohistiocytosis in the adult patient. Blood 125(19):2908–2914
Fujiwara FMD, Hibi SMD, Imashuku SMD (1993) Hypercytokinemia in hemophagocytic syndrome. Am J Pediatr Hematol 15(1):92–98
François B, Trimoreau F, Vignon P, Fixe P, Praloran V, Gastinne H (1997) Thrombocytopenia in the sepsis syndrome: role of hemophagocytosis and macrophage colony-stimulating factor. Am J Med 103(2):114–120
Strauss Richard Neureiter, Westenburger Daniel, Wehler Bert, Kirchner Markus, Hahn Thomas, Eckhart G, Facp (2004) Multifactorial risk analysis of bone marrow histiocytic hyperplasia with hemophagocytosis in critically ill medical patients-A postmortem clinicopathologic analysis. Crit Care Med 32(6):1316–1321
Karras A (2009) What nephrologists need to know about hemophagocytic syndrome. Nat Rev Nephrol 5(6):329–336
Aulagnon F, Lapidus N, Canet E et al (2015) Acute kidney injury in adults with hemophagocytic lymphohistiocytosis. Am J Kidney Dis 65(6):851–859
Thaunat O, Delahousse M, Fakhouri F et al (2006) Nephrotic syndrome associated with hemophagocytic syndrome. Kidney Int 69(10):1892–1898
Ponticelli C, Alberighi ODC (2009) Haemophagocytic syndrome—a life-threatening complication of renal transplantation. Nephrol Dial Transplant 24(9):2623–2627
de Saint Basile G, Ménasché G, Fischer A (2010) Molecular mechanisms of biogenesis and exocytosis of cytotoxic granules. Nat Rev Immunol 10(8):568–579
Parvaneh N, Filipovich AH, Borkhardt A (2013) Primary immunodeficiencies predisposed to epstein–barr virus-driven haematological diseases. Br J Haematol 162(5):573–586
Bode SFN, Ammann S, Al-Herz W et al (2015) The syndrome of hemophagocytic lymphohistiocytosis in primary immunodeficiencies: implications for differential diagnosis and pathogenesis. Haematologica 100(7):978–988
Jessen B, Kögl T, Sepulveda FE, de Saint Basile G, Aichele P, Ehl S (2013) Graded defects in cytotoxicity determine severity of hemophagocytic lymphohistiocytosis in humans and mice. Front Immunol 4:448-1–448-13
Sepulveda FE, Debeurme F, Ménasché G et al (2012) Distinct severity of HLH in both human and murine mutants with complete loss of cytotoxic effector PRF1, RAB27A, and STX11. Blood 121(4):595–603
Terrell CE, Jordan MB (2013) Mixed hematopoietic or T-cell chimerism above a minimal threshold restores perforin-dependent immune regulation in perforin-deficient mice. Blood 122(15):2618–2621
Nagafuji K, Nonami A, Kumano T et al (2007) Perforin gene mutations in adult-onset hemophagocytic lymphohistiocytosis. Haematologica 92(7):978–981
Ueda I, Kurokawa Y, Koike K et al (2007) Late-onset cases of familial hemophagocytic lymphohistiocytosis with missense perforin gene mutations. Am J Hematol 82(6):427–432
Tesi B, Chiang SC, El-Ghoneimy D et al (2015) Spectrum of atypical clinical presentations in patients with biallelic PRF1 missense mutations. Pediatr Blood Cancer 62(12):2094–2100
Zhang K, Jordan MB, Marsh RA et al (2011) Hypomorphic mutations in PRF1, MUNC13-4, and STXBP2 are associated with adult-onset familial HLH. Blood 118(22):5794–5798
Ramos-Casals M, Brito-Zerón P, López-Guillermo A, Khamashta MA, Bosch X (2014) Adult haemophagocytic syndrome. Lancet 383(9927):1503–1516
Rivière S, Galicier L, Coppo P et al (2014) Reactive hemophagocytic syndrome in adults: a retrospective analysis of 162 patients. Am J Med 127(11):1118–1125
Parikh SA, Kapoor P, Letendre L, Kumar S, Wolanskyj AP (2014) Prognostic factors and outcomes of adults with hemophagocytic lymphohistiocytosis. Mayo Clin Proc 89(4):484–492
Li J, Wang Q, Zheng W et al (2014) Hemophagocytic lymphohistiocytosis: clinical analysis of 103 adult patients. Medicine 93(2):100–105
Rosario C, Zandman-Goddard G, Meyron-Holtz EG, D’Cruz DP, Shoenfeld Y (2013) The hyperferritinemic syndrome: macrophage activation syndrome, still’s disease, septic shock and catastrophic antiphospholipid syndrome. BMC Med 11:185-7015-11-185
Zoller EE, Lykens JE, Terrell CE et al (2011) Hemophagocytosis causes a consumptive anemia of inflammation. J Exp Med 208(6):1203–1214
Behrens EM, Canna SW, Slade K et al (2011) Repeated TLR9 stimulation results in macrophage activation syndrome—like disease in mice. J Clin Invest 121(6):2264–2277
Canna SW, Wrobel J, Chu N, Kreiger PA, Paessler M, Behrens EM (2013) Interferon-γ mediates anemia but is dispensable for fulminant toll-like receptor 9-induced macrophage activation syndrome and hemophagocytosis in mice. Arthritis Rheum 65(7):1764–1775
Weaver LK, Behrens EM (2014) Hyperinflammation, rather than hemophagocytosis, is the common link between macrophage activation syndrome and hemophagocytic lymphohistiocytosis. Curr Opin Rheumatol 26(5):562–569
Ohyagi H, Onai N, Sato T et al (2013) Monocyte-derived dendritic cells perform hemophagocytosis to fine-tune excessive immune responses. Immunity 39(3):584–598
Canna SW, Costa-Reis P, Bernal WE et al (2014) Brief report: alternative activation of laser-captured murine hemophagocytes. Arthritis Rheumatol 66(6):1666–1671
Sica A, Mantovani A (2012) Macrophage plasticity and polarization: in vivo veritas. J Clin Invest 122(3):787–795
Schaer CA, Schoedon G, Imhof A, Kurrer MO, Schaer DJ (2006) Constitutive endocytosis of CD163 mediates hemoglobin-heme uptake and determines the noninflammatory and protective transcriptional response of macrophages to hemoglobin. Circ Res 99(9):943–950
Schaer DJ, Schaer CA, Schoedon G, Imhof A, Kurrer MO (2006) Hemophagocytic macrophages constitute a major compartment of heme oxygenase expression in sepsis. Eur J Haematol 77(5):432–436
Otterbein LE, Soares MP, Yamashita K, Bach FH (2003) Heme oxygenase-1: unleashing the protective properties of heme. Trends Immunol 24(8):449–455
Minoia F, Davì S, Horne A et al (2014) Clinical features, treatment, and outcome of macrophage activation syndrome complicating systemic juvenile idiopathic arthritis: a multinational, multicenter study of 362 patients. Arthritis Rheumatol 66(11):3160–3169
Kumakura S, Murakawa Y (2014) Clinical characteristics and treatment outcomes of autoimmune-associated hemophagocytic syndrome in adults. Arthritis Rheum 66(8):2297–2307
Parodi A, Davì S, Pringe AB et al (2009) Macrophage activation syndrome in juvenile systemic lupus erythematosus: a multinational multicenter study of thirty-eight patients. Arthritis Rheum 60(11):3388–3399
Lambotte O, Khellaf M, Harmouche H et al (2006) Characteristics and long-term outcome of 15 episodes of systemic lupus erythematosus-associated hemophagocytic syndrome. Medicine 85(3):169–182. Accessed 6 Jan 2016
Behrens EM, Beukelman T, Paessler M, Cron RQ (2007) Occult macrophage activation syndrome in patients with systemic juvenile idiopathic arthritis. J Rheumatol 34(5):1133–1138
Bleesing J, Prada A, Siegel DM et al (2007) The diagnostic significance of soluble CD163 and soluble interleukin-2 receptor α-chain in macrophage activation syndrome and untreated new-onset systemic juvenile idiopathic arthritis. Arthritis Rheum 56(3):965–971
Reddy VV, Myles A, Cheekatla SS, Singh S, Aggarwal A (2014) Soluble CD25 in serum: a potential marker for subclinical macrophage activation syndrome in patients with active systemic onset juvenile idiopathic arthritis. Int J Rheum Dis 17(3):261–267
Villanueva J, Lee S, Giannini EH et al (2005) Natural killer cell dysfunction is a distinguishing feature of systemic onset juvenile rheumatoid arthritis and macrophage activation syndrome. Arthritis Res Ther 7(1):R30–R37
Ishii E, Ohga S, Imashuku S et al (2007) Nationwide survey of hemophagocytic lymphohistiocytosis in Japan. Int J Hematol 86(1):58–65
Voskoboinik I, Thia M, Trapani JA (2005) A functional analysis of the putative polymorphisms A91V and N252S and 22 missense perforin mutations associated with familial hemophagocytic lymphohistiocytosis. Blood 105(12):4700–4706
Zhang K, Chandrakasan S, Chapman H et al (2014) Synergistic defects of different molecules in the cytotoxic pathway lead to clinical familial hemophagocytic lymphohistiocytosis. Blood 124(8):1331–1334
Sieni E, Cetica V, Piccin A et al (2012) Familial hemophagocytic lymphohistiocytosis may present during adulthood: clinical and genetic features of a small series. PLoS ONE 7(9):e44649
Wang Y, Wang Z, Zhang J et al (2014) Genetic features of late onset primary hemophagocytic lymphohistiocytosis in adolescence or adulthood. PLoS ONE 9(9):e107386
Zhang K, Biroschak J, Glass DN et al (2008) Macrophage activation syndrome in patients with systemic juvenile idiopathic arthritis is associated with MUNC13-4 polymorphisms. Arthritis Rheum 58(9):2892–2896
Vastert SJ, van Wijk R, D’Urbano LE et al (2010) Mutations in the perforin gene can be linked to macrophage activation syndrome in patients with systemic onset juvenile idiopathic arthritis. Rheumatology 49(3):441–449
Kaufman KM, Linghu B, Szustakowski JD et al (2014) Whole-exome sequencing reveals overlap between macrophage activation syndrome in systemic juvenile idiopathic arthritis and familial hemophagocytic lymphohistiocytosis. Arthritis Rheumatol 66(12):3486–3495
Zhang M, Behrens E, Atkinson TP, Shakoory B, Grom A, Cron R (2014) Genetic defects in cytolysis in macrophage activation syndrome. Curr Rheumatol Rep 16(9):1–8
Horne A, Trottestam H, Aricò M et al (2008) Frequency and spectrum of central nervous system involvement in 193 children with haemophagocytic lymphohistiocytosis. Br J Haematol 140(3):327–335. doi:10.1111/j.1365-2141.2007.06922.x
Trottestam H, Horne A, Aricò M et al (2011) Chemoimmunotherapy for hemophagocytic lymphohistiocytosis: long-term results of the HLH-94 treatment protocol. Blood 118(17):4577–4584
Deiva K, Mahlaoui N, Beaudonnet F et al (2012) CNS involvement at the onset of primary hemophagocytic lymphohistiocytosis. Neurology 78(15):1150–1156
Gratton SM, Powell TR, Theeler BJ, Hawley JS, Amjad FS, Tornatore C (2015) Neurological involvement and characterization in acquired hemophagocytic lymphohistiocytosis in adulthood. J Neurol Sci 357(1–2):136–142
Valade S, Azoulay E, Galicier L et al (2015) Coagulation disorders and bleedings in critically ill patients with hemophagocytic lymphohistiocytosis. Medicine 94(40):e1692
Chen J, Fleming MD, Pinkus GS et al (2010) Pathology of the liver in familial hemophagocytic lymphohistiocytosis. Am J Surg Pathol 34(6):852–867
Morrell DS, Pepping MA, Scott JP, Esterly NB, Drolet BA (2002) Cutaneous manifestations of hemophagocytic lymphohistiocytosis. Arch Dermatol 138(9):1208–1212
Kerl K, Wolf IH, Cerroni L, Wolf P, French LE, Kerl H (2015) Hemophagocytosis in cutaneous autoimmune disease. Am J Dermatopathol 37(7):539–543
Castillo L, Carcillo J (2009) Secondary hemophagocytic lymphohistiocytosis and severe sepsis/systemic inflammatory response syndrome/multiorgan dysfunction syndrome/macrophage activation syndrome share common intermediate phenotypes on a spectrum of inflammation. Pediatr Crit Care Med 10(3):387–392
Malaga-Dieguez L, Ming W, Trachtman H (2015) Direct reversible kidney injury in familial hemophagocytic lymphohistiocytosis type 3. J Am Soc Nephrol 26(8):1777–1780
Landau D, Gurevich E, Kapelushnik J, Tamary H, Shelef I, Lazar I (2013) Association between childhood nephrotic syndrome and hemophagocytic lymphohistiocytosis. Pediatr Nephrol 28(12):2389–2392
Risdall RJ, McKenna RW, Nesbit ME et al (1979) Virus-associated hemophagocytic syndrome A benign histiocytic proliferation distinct from malignant histiocytosis. Cancer 44(3):993–1002
Karras A, Thervet E, Legendre C, for the Groupe Cooperatif de transplantation d’Ile de France (2004) Hemophagocytic syndrome in renal transplant recipients: report of 17 cases and review of literature. Transplantation 77(2):238–243
Asci G, Toz H, Ozkahya M et al (2006) High-dose immunoglobulin therapy in renal transplant recipients with hemophagocytic histiocytic syndrome. J Nephrol 19(3):322–326
Lo MM, Mo JQ, Dixon BP, Czech KA (2010) Disseminated histoplasmosis associated with hemophagocytic lymphohistiocytosis in kidney transplant recipients. Am J Transplant 10(3):687–691
Raffray L, Couzi L, Viallard JF et al (2010) Mycophenolate mofetil: a possible cause of hemophagocytic syndrome following renal transplantation? Am J Transplant 10(10):2378–2379
Tangnararatchakit K, Tirapanich W, Tapaneya-Olarn W et al (2012) Severe nonfebrile dengue infection in an adolescent after postoperative kidney transplantation: a case report. Transplant Proc 44(1):303–306
Grabas M, Darrieux L, Potier J, Safa G (2012) Hemophagocytic syndrome as the presenting manifestation of bacillary angiomatosis in a renal transplant recipient. J Am Acad Dermatol 67(5):e236–e237
Yaich S, Charfeddine K, Hsairi D et al (2014) BK virus-associated hemophagocytic syndrome in a renal transplant recipient. Saudi J Kidney Dis Transplant 25(3):610–614
Wisanuyotin S, Jiravuttipong A, Puapairoj A (2014) De novo lupus nephritis in a renal transplanted child: a case report. Transplant Proc 46(2):648–650
Poudel A, Lew J, Slayton W, Dharnidharka VR (2014) Bartonella henselae infection inducing hemophagocytic lymphohistiocytosis in a kidney transplant recipient. Pediatr Transplant 18(3):E83–E87
Broeckaert-van Orshoven A, Michielsen P, Vandepitte J (1979) Fatal leishmaniasis in renal-transplant patient. Lancet 314(8145):740–741
Reiner AP, Spivak JL, Dietz N (1988) Hematophagic histiocytosis: a report of 23 new patients and a review of the literature. Medicine 67(6):369–388
Calonge VM, Glotz D, Bouscary D et al (1995) Hemophagocytic histiocytosis (HH) in renal transplant recipients under cyclosporin therapy: report of the first two cases. Clin Transplant 9(2):88–91
Slovut DP, Benedetti E, Matas AJ (1996) Babesiosis and hemophagocytic syndrome in an asplenic renal transplant recipient. Transplantation 62(4):537–539
Kursat S, Cagirgan S, Ok E et al (1997) Haemophagocytic–histiocytic syndrome in renal transplantation. Nephrol Dial Transplant 12(5):1058–1060
Dargent J, Vermylen P, Abramowicz D et al (1997) Disseminated angiosarcoma presenting as a hemophagocytic syndrome in a renal allograft recipient. Transplant Int 10(1):61–64
Peeters P, Sennesael J, De Raeve H, De Waele M, Verbeelen D (1997) Hemophagocytic syndrome and T-cell lymphoma after kidney transplantation: a case report. Transplant Int 10(6):471–474
Rossi C, Delforge M-, Jacobs F et al (2001) Fatal primary infection due to human herpesvirus 6 variant A in a renal transplant recipient. Transplantation 71(2):288–292
Luppi M, Barozzi P, Rasini V et al (2002) Severe pancytopenia and hemophagocytosis after HHV-8 primary infection in a renal transplant patient successfully treated with foscarnet. Transplantation 74(1):131–133
Gurkan A, Yakupoglu U, Yavuz A et al (2006) Hemophagocytic syndrome in kidney transplant recipients: report of four cases from a single center. Acta Haematol 116(2):108–113. Accessed 29 Nov 2015
Hot A, Madoux MHG, Viard JP, Coppéré B, Ninet J (2008) Successful treatment of cytomegalovirus-associated hemophagocytic syndrome by intravenous immunoglobulins. Am J Hematol 83(2):159–162
Bossini N, Sandrini S, Setti G et al (2005) Successful treatment with liposomal doxorubicin and Foscarnet in a patient with widespread Kaposi’s sarcoma and human herpes virus 8-related, serious hemophagocytic syndrome, after renal transplantation. G Ital Nefrol 22(3):281–286
Esposito L, Hirsch H, Basse G, Fillola G, Kamar N, Rostaing L (2007) BK virus-related hemophagocytic syndrome in a renal transplant patient [3]. Transplantation 83(3):365
Segall L, Moal M, Doucet L, Kergoat N, Bourbigot B (2006) Toxoplasmosis-associated hemophagocytic syndrome in renal transplantation. Transplant Int 19(1):78–80
González-Posada JM, Hernández D, Martin A et al (2008) Hemophagocytic lymphohistiocytosis in a pancreas-kidney transplant recipient: response to dexamethasone and cyclosporine. Clin Nephrol 70(1):82–86
Ardalan MR, Shoja MM, Tubbs RS, Esmaili H, Keyvani H (2008) Postrenal transplant hemophagocytic lymphohistiocytosis and thrombotic microangiopathy associated with parvovirus B19 infection. Am J Transplant 8(6):1340–1344
Henter J, Horne A, Aricó M et al (2007) HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 48(2):124–131
Hejblum G, Lambotte O, Galicier L et al (2014) A web-based delphi study for eliciting helpful criteria in the positive diagnosis of hemophagocytic syndrome in adult patients. PLoS One 9(4):e94024-1–e94024-6
Fardet L, Galicier L, Lambotte O et al (2014) Development and validation of the hscore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol 66(9):2613–2620
Ravelli A, Magni-Manzoni S, Pistorio A et al (2005) Preliminary diagnostic guidelines for macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. J Pediatr 146(5):598–604
Davì S, Minoia F, Pistorio A et al (2014) Performance of current guidelines for diagnosis of macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. Arthritis Rheumatol 66(10):2871–2880
Lehmberg K, Pink I, Eulenburg C, Beutel K, Maul-Pavicic A, Janka G (2013) Differentiating macrophage activation syndrome in systemic juvenile idiopathic arthritis from other forms of hemophagocytic lymphohistiocytosis. J Pediatr 162(6):1245–1251
Allen CE, Yu X, Kozinetz CA, McClain KL (2008) Highly elevated ferritin levels and the diagnosis of hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 50(6):1227–1235
Moore C Jr, Ormseth M, Fuchs H (2013) Causes and significance of markedly elevated serum ferritin levels in an academic medical center. JCR: J Clin Rheumatol 19(6):324–328
Beer T, Vadakara J (2015) Etiologies and short-term mortality in patients with ultraelevated serum ferritin. South Med J 108(9):574–578
Schram AM, Campigotto F, Mullally A et al (2015) Marked hyperferritinemia does not predict for HLH in the adult population. Blood 125(10):1548–1552
Gupta A, Tyrrell P, Valani R, Benseler S, Weitzman S, Abdelhaleem M (2008) The role of the initial bone marrow aspirate in the diagnosis of hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 51(3):402–404
Goel S, Polski JM, Imran H (2012) Sensitivity and specificity of bone marrow hemophagocytosis in hemophagocytic lymphohistiocytosis. Ann Clin Lab Sci 42(1):21–25
Listinsky CM (1988) Common reactive erythrophagocytosis in axillary lymph nodes. Am J Clin Pathol 90(2):189–192
Suster S, Hilsenbeck S, Rywlin AM (1988) Reactive histiocytic hyperplasia with hemophagocytosis in hematopoietic organs: a reevaluation of the benign hemophagocytic proliferations. Hum Pathol 19(6):705–712
Lehmberg K, Ehl S (2013) Diagnostic evaluation of patients with suspected haemophagocytic lymphohistiocytosis. Br J Haematol 160(3):275–287
Bryceson YT, Pende D, Maul-Pavicic A et al (2012) A prospective evaluation of degranulation assays in the rapid diagnosis of familial hemophagocytic syndromes. Blood 119(12):2754–2763
Koh K, Im HJ, Chung N et al (2015) Clinical features, genetics, and outcome of pediatric patients with hemophagocytic lymphohistiocytosis in Korea: report of a nationwide survey from Korea histiocytosis working party. Eur J Haematol 94(1):51–59
Rouphael NG, Talati NJ, Vaughan C, Cunningham K, Moreira R, Gould C (2007) Infections associated with haemophagocytic syndrome. Lancet Infect Dis 7(12):814–822
Lehmberg K, Nichols KE, Henter J- et al (2015) Consensus recommendations for the diagnosis and management of hemophagocytic lymphohistiocytosis associated with malignancies. Haematologica 100(8):997–1004
Janka GE (1983) Familial hemophagocytic lymphohistiocytosis. Eur J Pediatr 140(3):221–230. Accessed 4 Jan 2016
Mahlaoui N, Ouachée-Chardin M, de Saint Basile G et al (2007) Immunotherapy of familial hemophagocytic lymphohistiocytosis with antithymocyte globulins: a single-center retrospective report of 38 patients. Pediatrics 120(3):e622–e628
Johnson TS, Terrell CE, Millen SH, Katz JD, Hildeman DA, Jordan MB (2014) Etoposide selectively ablates activated T cells to control the immunoregulatory disorder hemophagocytic lymphohistiocytosis. J Immunol 192(1):84–91
Arca M, Fardet L, Galicier L et al (2015) Prognostic factors of early death in a cohort of 162 adult haemophagocytic syndrome: impact of triggering disease and early treatment with etoposide. Br J Haematol 168(1):63–68
Imashuku S, Kuriyama K, Teramura T et al (2001) Requirement for etoposide in the treatment of epstein–barr virus—associated hemophagocytic lymphohistiocytosis. J Clin Oncol 19(10):2665–2673
Imashuku S, Kuriyama K, Sakai R et al (2003) Treatment of epstein–barr virus-associated hemophagocytic lymphohistiocytosis (EBV-HLH) in young adults: a report from the HLH studyl center. Med Pediatr Oncol 41(2):103–109
Su IJ, Chen RL, Lin DT, Lin KS, Chen CC (1994) Epstein–barr virus (EBV) infects T lymphocytes in childhood EBV-associated hemophagocytic syndrome in taiwan. Am J Pathol 144(6):1219–1225
Beutel K, Gross-Wieltsch U, Wiesel T, Stadt UZ, Janka G, Wagner H (2009) Infection of T lymphocytes in epstein–barr virus-associated hemophagocytic lymphohistiocytosis in children of non-asian origin. Pediatr Blood Cancer 53(2):184–190
Kikuta H, Sakiyama Y (1995) Etoposide (VP-16) inhibits epstein–barr virus determined nuclear antigen (EBNA) synthesis. Br J Haematol 90(4):971–972
Kasahara Y, Yachie A (2002) Cell type specific infection of epstein–barr virus (EBV) in EBV-associated hemophagocytic lymphohistiocytosis and chronic active EBV infection. Crit Rev Oncol 44(3):283–294
Chellapandian D, Das R, Zelley K et al (2013) Treatment of epstein barr virus-induced haemophagocytic lymphohistiocytosis with rituximab-containing chemo-immunotherapeutic regimens. Br J Haematol 162(3):376–382
Ohga S, Kudo K, Ishii E et al (2010) Hematopoietic stem cell transplantation for familial hemophagocytic lymphohistiocytosis and epstein–barr virus-associated hemophagocytic lymphohistiocytosis in japan. Pediatr Blood Cancer 54(2):299–306
Buyse S, Teixeira L, Galicier L et al (2010) Critical care management of patients with hemophagocytic lymphohistiocytosis. Intensive Care Med 36(10):1695–1702
Barba T, Maucort-Boulch D, Iwaz J et al (2015) Hemophagocytic lymphohistiocytosis in intensive care unit: a 71-case strobe-compliant retrospective study. Medicine 94(51):e2318
Carcillo JA, Simon DW, Podd BS (2015) How we manage hyperferritinemic sepsis-related multiple organ dysfunction syndrome/macrophage activation syndrome/secondary hemophagocytic lymphohistiocytosis histiocytosis. Pediatr Crit Care Med 16(6):598–600
Demirkol D, Yildizdas D, Bayrakci B et al (2012) Hyperferritinemia in the critically ill child with secondary hemophagocytic lymphohistiocytosis/sepsis/multiple organ dysfunction syndrome/macrophage activation syndrome: what is the treatment? Crit Care 16(2):R52
Rajasekaran S, Kruse K, Kovey K et al (2014) Therapeutic role of anakinra, an interleukin-1 receptor antagonist, in the management of secondary hemophagocytic lymphohistiocytosis/sepsis/multiple organ dysfunction/macrophage activating syndrome in critically ill children. Pediatr Crit Care Med 15(5):401–408
Opal SM, Fisher CJ Jr, Dhainaut J-A et al (1997) Confirmatory interleukin-1 receptor antagonist trial in severe sepsis: a phase III, randomized, double-blind, placebo-controlled, multicenter trial. Crit Care Med 25(7):1115–1124
Lehmberg K, Sprekels B, Nichols KE et al (2015) Malignancy-associated haemophagocytic lymphohistiocytosis in children and adolescents. Br J Haematol 170(4):539–549
Mánard F, Besson C, Rincá P et al (2008) Hodgkin lymphoma—associated hemophagocytic syndrome: a disorder strongly correlated with epstein–barr virus. Clin Infect Dis 47(4):531–534
Fardet L, Lambotte O, Meynard J et al (2010) Reactive haemophagocytic syndrome in 58 HIV-1-infected patients: clinical features, underlying diseases and prognosis. AIDS 24(9):1299–1306
Minoia F, Davì S, Horne A et al (2014) Clinical features, treatment, and outcome of macrophage activation syndrome complicating systemic juvenile idiopathic arthritis: a multinational, multicenter study of 362 patients. Arthritis Rheumatol 66(11):3160–3169
De Benedetti F, Brunner HI, Ruperto N et al (2012) Randomized trial of tocilizumab in systemic juvenile idiopathic arthritis. New Engl J Med 367(25):2385–2395
Yokota S, Itoh Y, Morio T, Sumitomo N, Daimaru K, Minota S (2015) Macrophage activation syndrome in patients with systemic juvenile idiopathic arthritis under treatment with tocilizumab. J Rheumatol 42(4):712–722
Ruperto N, Brunner HI, Quartier P et al (2012) Two randomized trials of canakinumab in systemic juvenile idiopathic arthritis. New Engl J Med 367(25):2396–2406
Nigrovic PA, Mannion M, Prince FHM et al (2011) Anakinra as first-line disease-modifying therapy in systemic juvenile idiopathic arthritis: report of forty-six patients from an international multicenter series. Arthritis Rheum 63(2):545–555
Boehler A, Schaffner A, Salomon F, Keusch G (1994) Cytomegalovirus disease of late onset following renal transplantation: a potentially fatal entity. Scand J Infect Dis 26(4):369–373. Accessed 29 Nov 2015
Rostaing L, Fillola G, Baron E, Cisterne JM, Durand D (1995) Course of hemophagocytic histiocytic syndrome in renal transplant patients. Transplantation 60(5):506–509
Drut R, Drut RM (1994) EBV-associated kaposi’s sarcoma in a pediatric renal transplant recipient. Fetal Pediatr Pathol 14(5):863–872
Gupta P, Hurley RW, Helseth PH, Goodman JL, Hammerschmidt DE (1995) Pancytopenia due to hemophagocytic syndrome as the presenting manifestation of babesiosis. Am J Hematol 50(1):60–62
Ravelli A, Minoia F, Davi S et al (2016) 2016 Classification criteria for macrophage activation syndrome complicating systemic juvenile idiopathic arthritis: a European league against Rheumatism/American college of Rheumatology/Paediatric rheumatology international trials organisation collaborative initiative. Ann Rheum Dis 75 (3):481–489
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
EJF: speaker’s bureau for Mallinckrodt Pharmaceuticals; JLF: none.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Rights and permissions
About this article

Cite this article
Filippone, E.J., Farber, J.L. Hemophagocytic lymphohistiocytosis: an update for nephrologists. Int Urol Nephrol 48, 1291–1304 (2016). https://doi.org/10.1007/s11255-016-1294-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11255-016-1294-z