
Abstract
The CRISPR–Cas system is the newest targeted nuclease for genome engineering. In less than 1 year, the ease, robustness and efficiency of this method have facilitated an immense range of genetic modifications in most model organisms. Full and conditional gene knock-outs, knock-ins, large chromosomal deletions and subtle mutations can be obtained using combinations of clustered regularly interspaced short palindromic repeats (CRISPRs) and DNA donors. In addition, with CRISPR–Cas compounds, multiple genetic modifications can be introduced seamlessly in a single step. CRISPR–Cas not only brings genome engineering capacities to species such as rodents and livestock in which the existing toolbox was already large, but has also enabled precise genetic engineering of organisms with difficult-to-edit genomes such as zebrafish, and of technically challenging species such as non-human primates. The CRISPR–Cas system allows generation of targeted mutations in mice, even in laboratories with limited or no access to the complex, time-consuming standard technology using mouse embryonic stem cells. Here we summarize the distinct applications of CRISPR–Cas technology for obtaining a variety of genetic modifications in different model organisms, underlining their advantages and limitations relative to other genome editing nucleases. We will guide the reader through the many publications that have seen the light in the first year of CRISPR–Cas technology.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Clustered regularly interspaced short palindromic repeats (CRISPRs) sequences and Cas (CRISPR-associated) proteins are the two elements of an ancient prokaryotic adaptive restriction system conserved in archaeal and other bacterial genomes (Jansen et al. 2002; Makarova et al. 2011; Jinek et al. 2012). CRISPRs represent the memory of the system; they form a repository of short, directly repeating nucleotide sequences that alternate with small unique DNA fragments (Ishino et al. 1987) acquired from previous infections (Bolotin et al. 2005). Cas proteins are the actual effectors (Haft et al. 2005). They are able to process CRISPR sequences into small RNAs (Haurwitz et al. 2010) and to cleave the infectious DNA molecules that match the CRISPR-derived RNA (Marraffini and Sontheimer 2008; Garneau et al. 2010; Gasiunas et al. 2012).
To translate a complex prokaryotic system into a simple genome editing tool, the crRNA (CRISPR RNA) and the tracrRNA (trans-activating crRNA) were fused into a synthetic, small guide RNA (sgRNA) comprised of a hairpin RNA structure that resembles the tracrRNA linked to a 20 bp sequence homologous to the target DNA (Jinek et al. 2012). Of all the Cas proteins (Chylinski et al. 2013), Cas9 is the final effector, able to complex (Nishimasu et al. 2014; Jinek et al. 2014) and to cleave both strands of a DNA molecule after detecting a typical Watson&Crick homologous base pair match with the sgRNA. Genome engineering with this RNA-programmable Cas9 nuclease has broad applications in biology, biomedicine and biotechnology (Charpentier and Doudna 2013). With their colleagues, microbiologist Emmanuelle Charpentier and structural biologist Jennifer Doudna co-authored the seminal publications on this subject and are to be credited for studying and bringing this prokaryotic tool to the attention and the benefit of the eukaryotic world (Jinek et al. 2012, 2014; Charpentier and Doudna 2013).
Different varieties of Cas9 protein are now available for transduction and expression in distinct organisms, and its prototype structure was recently reported (Jinek et al. 2014). Many additional Cas9 proteins from diverse bacteria are being characterized and will soon help to expand the CRISPR–Cas system toolkit (Fonfara et al. 2014).
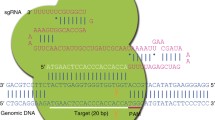
To begin using the CRISPR–Cas genome editing system, the researcher needs only to identify a 20 bp sequence from the target DNA that is followed by the protospacer adjacent motif (PAM) NGG (Fig. 1a), and clone it into the sgRNA expression vector appropriate for the organism of interest (Ran et al. 2013a, b).

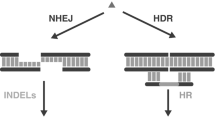
The principle of CRISPR–Cas genome editing. a Example of a CRISPR-binding site in mouse Tyr exon 1 (Giraldo and Montoliu 2002). The N(20) sequence is shown in bold, the PAM motif is underlined. b Typically, a DSB is produced 3 bp upstream of the PAM motif. The cell response to DNA damage includes an error-prone DNA repair mechanism (NHEJ), a HDR mechanism and might trigger apoptosis if the DNA damage cannot be repaired. The editing applications are listed beneath each repair route
As in the case of the zinc-finger nucleases (ZFN; Remy et al. 2010) and transcription activator-like effector nucleases (TALEN; Joung and Sander 2013), genome editing with CRISPR–Cas depends largely on the cell processes triggered by the DNA double strand break (DBS) at the targeted locus (Fig. 1b). The non-homologous end-joining (NHEJ) pathway repairs DNA damage in the absence of template DNA. Small insertions or deletions (indels) are introduced through NHEJ-mediated repair, rendering the resulting genetic modification unpredictable (Barnes 2001). When a DNA donor template is provided, DSB can also be repaired through homology-driven or homology-directed repair (HDR); DSB facilitate the frequency of homologous recombination (Liang et al. 1996; Urnov et al. 2005).
NHEJ-based CRISPR–Cas applications
NHEJ-mediated DSB repair leaves a footprint at the target site; depending on the nature of the two DNA ends, this can range from a single nucleotide exchange, deletion, or insertion to deletion of a few hundred base pairs. This error-prone DNA repair mechanism appears to be based on microhomologies between the two DNA ends. If the DSB overhangs do not allow immediate base pairing, cell exonucleases and polymerases will remove or add nucleotides until microhomology is suitable for DNA end joining (Lieber et al. 2003). This DNA scar, if located in the first coding exon of a gene, will probably result in a frame-shift mutation and almost complete loss of the protein encoded, hence a knockout (KO) allele will be produced simply by targeting the CRISPR–Cas mix to the desired DNA sequence.
Huang and colleagues first showed disruption of an EGFP transgene in mouse and in zebrafish transgenic lines (Shen et al. 2013). This breakthrough report was the first successful proof-of-principle of in vivo CRISPR–Cas use, although no information was provided regarding germ-line transmission of the mutations induced (often found as mosaics). In addition, the mice and zebrafish lines used in this study bore multiple copies of a GFP transgene, further complicating analysis of germ-line transmission.
One of the unique advantages of the CRISPR–Cas system is that NHEJ can be triggered simultaneously at various endogenous loci by co-injection of several sgRNA molecules with Cas9 mRNA. The first of these multiple-targeting experiments in mice, which included as many as five different sgRNA molecules, was reported just 1 year ago (Wang et al. 2013). CRISPR–Cas appears to be the editing tool of choice in mice compared to TALEN or ZFN, not only for its greater activity, but also because of the smaller RNA load needed to achieve such multiple gene targeting events (Wang et al. 2013). Multiplex genome editing using TALEN or ZFN is technically demanding, as the total amount of RNA involved would easily reach toxic concentrations before each individual nuclease reaches its working concentration. This might explain why no experiments involving multiple simultaneous gene targeting events have been reported for ZFN or TALEN.
CRISPR–Cas can be used to target two genes simultaneously on the same chromosome, as was shown for mice (Zhou et al. 2014); this would otherwise require several breeding rounds of two independent KO mouse lines, or two sequential rounds of gene targeting in embryonic stem (ES) cells using distinct, compatible selection approaches. Results were similar in rats, although it was observed that targeting two nearby sequences could lead to deletion of the intervening DNA (Li et al. 2013a). Ma et al. reported the simultaneous disruption of four genes in rats, using one or two sgRNA for each targeted gene (Ma et al. 2014a). Although illustrative of the unprecedented genome editing ability of CRISPR–Cas, these experiments also underline the fact that this approach can easily produce genetically difficult-to-handle animal models, due to the obvious segregation of the many simultaneous mutations in the founder animal.
Generation of full KO mouse lines by CRISPR–Cas embryo injection appears robust and reproducible, although key methodological details remain to be standardized, such as correct RNA concentration (or range) and recommended microinjection route (pronuclear-only, cytoplasmic-only, or a combination of the two). A recent publication provided a detailed technical note to guide microinjectionists in this task; after testing different microinjection routes, the authors recommended cytoplasmic-only microinjections (Horii et al. 2014); microinjection of some material into the pronucleus can be used to confirm positive delivery.
Genome-editing nucleases first allowed generation of targeted zebrafish KO lines (Meng et al. 2008; Bedell et al. 2012), and time-consuming, random ENU mutagenesis was no longer necessary (Kettleborough et al. 2011). Using CRISPR, biallelic KO of multiple targets can now be generated efficiently in zebrafish (Jao et al. 2013).
To mimic large chromosomal rearrangements, distal sequences on the same chromosome can be targeted with different sgRNA, to promote inversion or deletion of the intervening DNA region. Deletions as large as 10 kb were obtained using two distal sgRNA in mouse zygotes (Fujii et al. 2013) and haploid ES cells (Horii et al. 2013). In zebrafish, a 40 kb DNA region was deleted and inverted (Xiao et al. 2013). This strategy can be applied to study large non-coding regulatory elements that might need full deletion, rather than point inactivation, to observe the underlying phenotype. With the CRISPR–Cas system, functional analysis of intergenic/non-coding regulatory DNA sequences, recently annotated by the ENCODE project (Bernstein et al. 2012), is becoming a reality.
Although it is apparently counterintuitive, the error-prone NHEJ repair pathway can promote efficiently targeted transgene integration, with no need for a targeting vector with large homology arms (Auer et al. 2014). The knock-in (KI) vector must bear the same target sequence as the endogenous target region, and must be provided as a circular plasmid. This new method greatly simplifies the complex genotyping strategies, based on laborious Southern blot and long-range PCR assays, needed to identify correctly targeted alleles in the presence of large homology arms (Lay et al. 1998). A homology-independent KI approach was tested in cultured cells with TALEN and ZFN (Maresca et al. 2013), but has not been reported in vivo. Although efficient and straightforward, these KI are thus still largely based on error-prone NHEJ-mediated DNA repair, and the targeted insertion will be accompanied by small indels at the insertion site. For gene-replacement or in-frame reporter gene insertion, one should thus recall that only one-third of the integrants will retain the insertion site open reading frame in the inserted cassette.
Generation and genetic modification of haploid ES cells is a promising tool that will expedite the generation of biallelic complex modified alleles (Li et al. 2012). CRISPR–Cas was used successfully to engineer mouse (Horii et al. 2013) and rat haploid stem cells, generating triple-mutant clones in a single step (Li et al. 2013b).
The CRISPR–Cas system was recently used to generate a double knockout in the Cynomolgus monkey (Niu et al. 2014), a technically challenging species. Cas9 mRNA and an sgRNA mix [directed against Nr0b1 (two sgRNA), Ppar-γ (two sgRNA) and Rag1 (one sgRNA)] were co-injected in intracytoplasmic sperm injection-fertilized eggs. Although some pregnancies were still in gestation at the time of publication, the first two newborn monkeys analysed showed genomic modification of two of the three target genes. Several mutated genotypes were found in the same individual, suggesting distinct CRISPR–Cas cleavage events at different embryonic stages (Niu et al. 2014).
HDR-based CRISPR–Cas applications
Standard ubiquitous targeted gene disruption, although valuable, might not satisfy all the geneticist’s wishes. Conditional mutagenesis allows interrogation of specific tissues, conditions or developmental stages, and is often used to rescue mutated alleles that would otherwise be lethal (i.e., Mastracci et al. 2013). In mice, conditional mutants are generated in a routine but complex process based on traditional gene targeting in ES cells (i.e., using the Cre-loxP system); the resulting mice bearing the genetically-altered loxP-tagged alleles are then bred with the Cre-driver line of choice (Rossant and McMahon 1999). The expression pattern of the Cre lines must be compatible with the organ, tissue or developmental stage in which gene inactivation is to be studied, and requires considerable expertise and resources. For this reason, the task of systematic generation of conditional KOs across all mouse genes has been delegated to specialized large-scale consortia such as the International Knockout Mouse Consortium (IKMC) (Skarnes et al. 2011). Even so, these large consortia often cannot fulfil all researcher interests for inactivation/deletion of a specific exon or introduction of a specific mutation. Targeted nucleases, and particularly the CRISPR–Cas system, might fill this gap, providing a simpler strategy for conditional mutagenesis that does not require ES cell work (Shen et al. 2014).
As commented above, a targeted DSB facilitates homologous recombination by activating the HDR pathway (Rouet et al. 1994). Wang et al. used a combination of an sgRNA and a single-stranded (ss)DNA donor to introduce precise point mutations by homologous recombination into mouse zygotes (Wang et al. 2013). Using two oligonucleotides bearing loxP sites and two sgRNA that targeted the desired insertion sites around a critical exon, they generated a conditional allele directly in a single step by embryo injection (Yang et al. 2013a). This impressive result unequivocally illustrates the power of CRISPR–Cas technology, as this new approach can be used to obtain mouse founders genotypically confirmed to carry a floxed allele in only about 2 months. In contrast, the traditional process (ES cell route) for producing a conditional (floxed) mouse mutant allele might require a month to build the targeting construct, 3 months for transfection, screening and validation of positive recombinant ES cell clones, and 4 to 6 additional months to obtain and confirm a germline-transmitting chimera.
Conditional alleles have been also generated in the rat by embryo injection of one or two sgRNA and a circular targeting vector bearing a floxed exon and short homology arms (Ma et al. 2014b). In this case, the use of two sgRNA appeared to be the most efficient strategy to target loxP sites at the correct location.
In biomedicine, animal disease modelling using sophisticated genome engineering techniques enables customised editing of the same mutations found in patients, rather than full disruption of the causative gene, which is rarely observed in man. The CRISPR–Cas technology could provide innovative, more efficient ways to generate precise animal models of human disease. Use of long, single-stranded oligonucleotides as DNA donor templates to repair the DSB created by these nucleases is the most desirable simplification of the process. Current in vitro studies nonetheless indicate that many parameters require additional optimisation (Yang et al. 2013b), including length of the DNA oligonucleotide, relative position between the cleavage site and the mutation and finally, which DNA strand (or strands) to provide. Quality, purity and concentration of the oligonucleotide DNA donor still lack the necessary standardisation.
In zebrafish, CRISPR–Cas and oligonucleotides were used to introduce an HA tag in two different genes (Hruscha et al. 2013). Accurate small insertions and single nucleotide substitutions were introduced with ssDNA donors; in some cases, additional mutations were found (Hwang et al. 2013), indicating a second NHEJ-repair event subsequent to the first homologous recombination event. Introduction of mutations in the PAM sequence, if compatible with the result desired, will of course prevent re-cutting the edited allele. In mice, correction of a mutation in the Crygc gene rescued a cataract phenotype (Wu et al. 2013). This correction could be driven by the endogenous wild type allele or by an oligonucleotide donor DNA; in this example, dominant-negative mutations with a pathological phenotype were reversed in the absence of a DNA donor, by simply designing an sgRNA specific for the mutated allele (Wu et al. 2013).
The CRISPR–Cas technology has also been welcomed in the field of large animal/livestock transgenesis. The first successful experiments for genetic alteration of pig and cow genomes using CRISPR–Cas systems were reported recently (Tan et al. 2013). In livestock, selective breeding (classical genetics) is traditionally used to transfer valuable traits to the production strain. This process is time-consuming, however, and desired traits are often genetically linked to undesired ones, resulting in lower quality breeds. Targeted nucleases and oligonucleotide donors can be used to introduce allelic variants of agricultural interest directly into the breed of choice. For example, bulls could be genetically dehorned by introducing the Angus POLLED allelic variant (Tan et al. 2013); interestingly, in this report CRISPR–Cas yielded fewer recombinant clones than TALEN.
The CRISPR–Cas system compared with other targeted nuclease platforms
Whereas ZFN design requires complex algorithms and extensive experimental validation to match the target site with a ZF array (Maeder et al. 2008), the TALEN platform benefits from direct correspondence between a set of protein modules (repeat variable di-residue; RVD) and each base pair of the target site (Moscou and Bogdanove 2009) with fewer sequence constraints (Doyle et al. 2012). This modular design allows application of simple, iterative assembly techniques such as Golden Gate (GG) assembly (Engler et al. 2009), which can be completed within a week (Fig. 2a). Solid protocols are reported using TALEN for mouse genome engineering (Hermann et al. 2014). In the case of ZFN, although there are open platforms (Hermann et al. 2012), most molecules are produced commercially and cannot be prepared easily by the researcher.

Comparison of steps for assembly of TALEN and CRISPR reagents in the laboratory. a TALEN assembly needs two consecutive cloning steps, which involve pipetting of more than 40 different compounds. The full process, including assembly and validation in cultured cells, requires at least 7 days. b CRISPR nucleases can be assembled and tested in cultured cells within 5 days. Short annealed oligonucleotides are cloned in an sgRNA plasmid and co-transfected with a Cas9 expression vector. RE restriction enzyme, GG Golden Gate cloning system (Engler et al. 2009), NLS nuclear localisation signal
With CRISPR–Cas, the nuclease recognises its target by Watson&Crick base pairing with the RNA guide molecule. The only sequence constraint is the presence at the 3′ end of the target site of the PAM, an NGG trinucleotide, which occurs on average once every 8 bp in the mammalian genome (Cong et al. 2013).
In a comparative study using human pluripotent stem cells, TALEN mutagenesis efficiency ranged from 0 to 34 %, which reached 51–79 % when CRISPR–Cas was used to target the same set of genes (Ding et al. 2013).
TALEN assembly requires a week’s work by an experienced molecular biologist (Cermak et al. 2011), and the cell must translate and correctly fold two 110 kDa proteins to scan and bind the correct DNA target site. In contrast, the laboratory workload for CRISPR–Cas production is minimal (Fig. 2b) and from the cellular point of view, a 100 bp-long sgRNA is delivered and Cas9 mRNA is translated and folded into a single 150 kDa protein.
The rapid optimisation and wide application of the CRISPR–Cas tools in genome editing are also due to uncomplicated access to inexpensive reagents for academic use from plasmid repositories such as Addgene (Baker 2014). These distribution portals explain the rapid universalisation of CRISPR–Cas reagents.
Cas9 and sgRNA variants (activity and off-targeting)
After codon optimisation and addition of a suitable nuclear localisation signal, Streptococcus pyogenes Cas9 is able to mediate RNA–guided dsDNA cleavage in vertebrates. Last year, four independent groups nonetheless reported that the RNA-guiding process tolerates several mismatches between the RNA guide and the target DNA sequence (Fu et al. 2013; Mali et al. 2013; Pattanayak et al. 2013; Hsu et al. 2013), questioning the specificity of the CRISPR–Cas system. These studies were conducted mainly using cell lines, in which undesired genetic modifications cannot be easily segregated by breeding, as they can in animal models such as mice. These reports nevertheless pushed the scientific community into a search for new, safer strategies. Zhang and co-workers proposed the use of two offset sgRNA together with a new Cas9 variant, D10A, a mutant version that lacks one of the two nuclease domains (Jinek et al. 2012; Fig. 3a). This procedure generates two nearby single-strand breaks (nicks) (Fig. 3b) that are still able to trigger both NHEJ and HDR responses. With this approach, two sgRNA–DNA interactions are necessary to mediate a DSB, while undesired contacts of a single sgRNA with off-target sequences will result in non-mutagenic, easily repairable nicks on just one DNA strand (Ran et al. 2013a, b). Combined use of D10A, known as “nickase”, with duplicated sets of sgRNA boosts the efficiency and nearly abolish the off-target sequences observed (Ran et al. 2013a, b). This double-nicking strategy is effective in mice; Skarnes and colleagues obtained gene KO in the near-absence of off-target mutations (Shen et al. 2014), and Fujii et al. produced a 1 kb deletion by combining four sgRNA (that produced two distal DSB) and Cas9/nickase (Fujii et al. 2014). Use of the Cas9/nickase variant will greatly benefit multiplex editing, as simultaneous targeting of several genes by standard Cas9 protein exponentially increases the risk of off-targeting.

DNA cleavage using wild type and mutant D10A Cas9 proteins. a Above using original Cas9, a double-stranded blunt DNA cleavage is typically observed 3 bp upstream of the PAM motif. Below using the mutant D10A Cas9 variant (nickase), a single-stranded DNA nick is produced in the DNA strand homologous to the sgRNA used. b Two DNA nicks, correctly ordered and spaced, can trigger the same DNA repair pathway and associated mutagenesis as a DSB
Alternative design of guide RNA increased specificity in genome editing; a recent report showed that truncated sgRNA can be more specific on-target than full-length sgRNA (Fu et al. 2014). In the presence of mismatches, shorter sgRNA will interact weakly with DNA. Reducing mismatch tolerance increases specificity. It is tempting to speculate that combined double-nicking strategy and truncated sgRNA design could further improve the specificity of genome engineering.
The relationship between binding and cleavage efficiencies was further studied using a non-functional Cas9 variant combined with sgRNA targeted for a specific locus (Wu et al. 2014). The authors propose a two-state model for Cas9 binding and cleavage, in which a seed match would trigger binding, but extensive additional pairing with target DNA would be necessary for effective cleavage (Wu et al. 2014).
In summary, the widespread success of the CRISPR–Cas technology in less than a year suggests that we are witnessing the birth of a new era in genome engineering. Similar to the revolution in genetic engineering caused by restriction enzymes in the 1970s, the CRISPR–Cas approach (another restriction system from bacteria) appears to offer a new paradigm for genome manipulation. This will enable the production of improved animal models of human disease, the generation of safer and more precise biotechnological products, and possibly a new way to devise innovative gene therapy approaches.
References
Auer TO, Duroure K, De Cian A, Concordet JP, Del Bene F (2014) Highly efficient CRISPR/Cas9-mediated knock-in in zebrafish by homology-independent DNA repair. Genome Res 24:142–153
Baker M (2014) Repositories share key research tools. Nature 505:272
Barnes DE (2001) Non-homologous end joining as a mechanism of DNA repair. Curr Biol 11:R455–R457
Bedell VM, Wang Y, Campbell JM, Poshusta TL, Starker CG, Krung RG II, Tan W, Penheiter SG, Ma AC, Leung AY, Fahrenkrug SC, Carlson DF, Voytas DF, Clark KJ, Essner JJ, Ekker SC (2012) In vivo genome editing using a high-efficiency TALEN system. Nature 491:114–118
Bernstein BE, Birney E, Dunham I, Green ED, Gunter C, Snyder M, ENCODE Project Consortium (2012) An integrated encyclopedia of DNA elements in the human genome. Nature 489:57–74
Bolotin A, Quinquis B, Sorokin A, Ehrlich SD (2005) Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology 151:2551–2561
Cermak T, Doyle EL, Christian M, Wang L, Zhang Y, Schmidt C, Baller JA, Somia NV, Bogdanove AJ, Voytas DF (2011) Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res 39:e82
Charpentier E, Doudna JA (2013) Biotechnology: rewriting a genome. Nature 495:50–51
Chylinski K, Le Rhun A, Charpentier E (2013) The tracrRNA and Cas9 families of type II CRISPR–Cas immunity systems. RNA Biol 10:726–737
Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339:819–823
Ding Q, Regan SN, Xia Y, Oostrom LA, Cowan CA, Musunuru K (2013) Enhanced efficiency of human pluripotent stem cell genome editing through replacing TALENs with CRISPRs. Cell Stem Cell 12:393–394
Doyle EL, Booher NJ, Standage DS, Voytas DF, Brendel VP, Vandyk JK, Bogdanove AJ (2012) TAL effector-nucleotide targeter (TALE-NT) 2.0: tools for TAL effector design and target prediction. Nucleic Acids Res 40:W117–W122
Engler C, Gruetzner R, Kandzia R, Marillonnet S (2009) Golden gate shuffling: a one-pot DNA shuffling method based on type IIs restriction enzymes. PLoS One 4:e5553
Fonfara I, Le Rhun A, Chylinski K, Makarova KS, Lécrivain AL, Bzdrenga J, Koonin EV, Charpentier E (2014) Phylogeny of Cas9 determines functional exchangeability of dual-RNA and Cas9 among orthologous type II CRISPR–Cas systems. Nucleic Acids Res 42:2577–2590
Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, Sander JD (2013) High-frequency off-target mutagenesis induced by CRISPR–Cas nucleases in human cells. Nat Biotechnol 31:822–826
Fu Y, Sander JD, Reyon D, Cascio VM, Joung JK (2014) Improving CRISPR–Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol 32(3):279–284
Fujii W, Kawasaki K, Sugiura K, Naito K (2013) Efficient generation of large-scale genome-modified mice using gRNA and CAS9 endonuclease. Nucleic Acids Res 41:e187
Fujii W, Onuma A, Sugiura K, Naito K (2014) Efficient generation of genome-modified mice via offset-nicking by CRISPR/Cas system. Biochem Biophys Res Commun 445:791–794
Garneau JE, Dupuis MÈ, Villion M, Romero DA, Barrangou R, Boyaval P, Fremaux C, Horvath P, Magadán AH, Moineau S (2010) The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 468:67–71
Gasiunas G, Barrangou R, Horvath P, Siksnys V (2012) Cas9–crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci USA 109:E2579–E2586
Giraldo P, Montoliu L (2002) Artificial chromosome transgenesis in pigmentary research. Pigment Cell Res 15:258–264
Haft DH, Selengut J, Mongodin EF, Nelson KE (2005) A guild of 45 CRISPR-associated (Cas) protein families and multiple CRISPR/Cas subtypes exist in prokaryotic genomes. PLoS Comput Biol 1:e60
Haurwitz RE, Jinek M, Wiedenheft B, Zhou K, Doudna JA (2010) Sequence- and structure-specific RNA processing by a CRISPR endonuclease. Science 329:1355–1358
Hermann M, Maeder ML, Rector K, Ruiz J, Becher B, Bürki K, Khayter C, Aguzzi A, Joung JK, Buch T, Pelczar P (2012) Evaluation of OPEN zinc finger nucleases for direct gene targeting of the ROSA26 locus in mouse embryos. PLoS One 7:e41796
Hermann M, Cermak T, Voytas DF, Pelczar P (2014) Mouse genome engineering using designer nucleases. J Vis Exp (86):e50930. doi:10.3791/50930
Horii T, Morita S, Kimura M, Kobayashi R, Tamura D, Takahashi RU, Kimura H, Suetake I, Ohata H, Okamoto K, Tajima S, Ochiya T, Abe Y, Hatada I (2013) Genome engineering of mammalian haploid embryonic stem cells using the Cas9/RNA system. PeerJ 1:e230
Horii T, Arai Y, Yamazaki M, Morita S, Kimura M, Itoh M, Abe Y, Hatada I (2014) Validation of microinjection methods for generating knockout mice by CRISPR/Cas-mediated genome engineering. Sci Rep 4:4513
Hruscha A, Krawitz P, Rechenberg A, Heinrich V, Hecht J, Haass C, Schmid B (2013) Efficient CRISPR/Cas9 genome editing with low off-target effects in zebrafish. Development 140:4982–4987
Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O, Cradick TJ, Marraffini LA, Bao G, Zhang F (2013) DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol 31:827–832
Hwang WY, Fu Y, Reyon D, Maeder ML, Kaini P, Sander JD, Joung JK, Peterson RT, Yeh JR (2013) Heritable and precise zebrafish genome editing using a CRISPR–Cas system. PLoS One 8:e68708
Ishino Y, Shinagawa H, Makino K, Amemura M, Nakata A (1987) Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J Bacteriol 169:5429–5433
Jansen R, Embden JD, Gaastra W, Schouls LM (2002) Identification of genes that are associated with DNA repeats in prokaryotes. Mol Microbiol 43:1565–1575
Jao LE, Wente SR, Chen W (2013) Efficient multiplex biallelic zebrafish genome editing using a CRISPR nuclease system. Proc Natl Acad Sci USA 110:13904–13909
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E (2012) A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science 337:816–821
Jinek M, Jiang F, Taylor DW, Sternberg SH, Kaya E, Ma E, Anders C, Hauer M, Zhou K, Lin S, Kaplan M, Iavarone AT, Charpentier E, Nogales E, Doudna JA (2014) Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science 343:1247997
Joung JK, Sander JD (2013) TALENs: a widely applicable technology for targeted genome editing. Nat Rev Mol Cell Biol 14:49–55
Kettleborough RN, Ed Bruijn, Fv Eeden, Cuppen E, Stemple DL (2011) High-throughput target-selected gene inactivation in zebrafish. Methods Cell Biol 104:121–127
Lay JM, Friis-Hansen L, Gillespie OJ, Samuelson LC (1998) Rapid confirmation of gene targeting in embryonic stem cells using two long-range PCR techniques. Transgenic Res 7:135–140
Li W, Shuai L, Wan H, Dong M, Wang M, Sang L, Feng C, Luo GZ, Li T, Li X, Wang L, Zheng QY, Sheng C, Wu HJ, Liu Z, Liu L, Wang L, Wang XJ, Zhao XY, Zhou Q (2012) Androgenetic haploid embryonic stem cells produce live transgenic mice. Nature 490:407–411
Li D, Qiu Z, Shao Y, Chen Y, Guan Y, Liu M, Li Y, Gao N, Wang L, Lu X, Zhao Y, Liu M (2013a) Heritable gene targeting in the mouse and rat using a CRISPR–Cas system. Nat Biotechnol 31:681–683
Li W, Li X, Li T, Jiang MG, Wan H, Luo GZ, Feng C, Cui X, Teng F, Yuan Y, Zhou Q, Gu Q, Shuai L, Sha J, Xiao Y, Wang L, Liu Z, Wang XJ, Zhao XY, Zhou Q (2013b) Genetic modification and screening in rat using haploid embryonic stem cells. Cell Stem Cell 14:404–414
Liang F, Romanienko PJ, Weaver DT, Jeggo PA, Jasin M (1996) Chromosomal double-strand break repair in Ku80-deficient cells. Proc Natl Acad Sci USA 93:8929–8933
Lieber MR, Ma Y, Pannicke U, Schwarz K (2003) Mechanism and regulation of human non-homologous DNA end-joining. Nat Rev Mol Cell 4(9):712–720
Ma Y, Shen B, Zhang X, Lu Y, Chen W, Ma J, Huang X, Zhang L (2014a) heritable multiplex genetic engineering in rats using CRISPR/Cas9. PLoS One 9:e89413
Ma Y, Zhang X, Shen B, Lu Y, Chen W, Ma J, Bai L, Huang X, Zhang L (2014b) Generating rats with conditional alleles using CRISPR/Cas9. Cell Res 24:122–125
Maeder ML, Thibodeau-Beganny S, Osiak A, Wright DA, Anthony RM, Eichtinger M, Jiang T, Foley JE, Winfrey RJ, Townsend JA, Unger-Wallace E, Sander JD, Müller-Lerch F, Fu F, Pearlberg J, Göbel C, Dassie JP, Pruett-Miller SM, Porteus MH, Sgroi DC, Iafrate AJ, Dobbs D, McCray PB Jr, Cathomen T, Voytas DF, Joung JK (2008) Rapid “open-source” engineering of customized zinc-finger nucleases for highly efficient gene modification. Mol Cell 31:294–301
Makarova KS, Haft DH, Barrangou R, Brouns SJ, Charpentier E, Horvath P, Moineau S, Mojica FJ, Wolf YI, Yakunin AF, van der Oost J, Koonin EV (2011) Evolution and classification of the CRISPR–Cas systems. Nat Rev Microbiol 9:467–477
Mali P, Aach J, Stranges PB, Esvelt KM, Moosburner M, Kosuri S, Yang L, Church GM (2013) CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat Biotechnol 31:833–838
Maresca M, Lin VG, Guo N, Yang Y (2013) Obligate ligation-gated recombination (ObLiGaRe): custom-designed nuclease-mediated targeted integration through nonhomologous end joining. Genome Res 23:539–546
Marraffini LA, Sontheimer EJ (2008) CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science 322:1843–1845
Mastracci TL, Lin CS, Sussel L (2013) Generation of mice encoding a conditional allele of Nkx2.2. Transgenic Res 22:965–972
Meng X, Noyes MB, Zhu LJ, Lawson ND, Wolfe SA (2008) Targeted gene inactivation in zebrafish using engineered zinc-finger nucleases. Nat Biotechnol 26:695–701
Moscou MJ, Bogdanove AJ (2009) A simple cipher governs DNA recognition by TAL effectors. Science 326:1501
Nishimasu H, Ran FA, Hsu PD, Konermann S, Shehata SI, Dohmae N, Ishitani R, Zhang F, Nureki O (2014) Crystal structure of Cas9 in complex with Guide RNA and target DNA. Cell 156:935–949
Niu Y, Shen B, Cui Y, Chen Y, Wang J, Wang L, Kang Y, Zhao X, Si W, Li W, Xiang AP, Zhou J, Guo X, Bi Y, Si C, Hu B, Dong G, Wang H, Zhou Z, Li T, Tan T, Pu X, Wang F, Ji S, Zhou Q, Huang X, Ji W, Sha J (2014) Generation of gene-modified cynomolgus monkey via Cas9/RNA-mediated gene targeting in one-cell embryos. Cell 156:836–843
Pattanayak V, Lin S, Guilinger JP, Ma E, Doudna JA, Liu DR (2013) High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat Biotechnol 31:839–843
Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, Scott DA, Inoue A, Matoba S, Zhang Y, Zhang F (2013a) Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154:1380–1389
Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F (2013b) Genome engineering using the CRISPR–Cas9 system. Nat Protoc 8:2281–2308
Remy S, Tesson L, Menoret S, Usal C, Scharenberg AM, Anegon I (2010) Zinc-finger nucleases: a powerful tool for genetic engineering of animals. Transgenic Res 19:363–371
Rossant J, McMahon A (1999) ”Cre”-ating mouse mutants-a meeting review on conditional mouse genetics. Genes Dev 13:142–145
Rouet P, Smih F, Jasin M (1994) Expression of a site-specific endonuclease stimulates homologous recombination in mammalian cells. Proc Natl Acad Sci USA 91:6064–6068
Shen B, Zhang J, Wu H, Wang J, Ma K, Li Z, Zhang X, Zhang P, Huang X (2013) Generation of gene-modified mice via Cas9/RNA-mediated gene targeting. Cell Res 23:720–723
Shen B, Zhang W, Zhang J, Zhou J, Wang J, Chen L, Wang L, Hodgkins A, Iyer V, Huang X, Skarnes WC (2014) Efficient genome modification by CRISPR–Cas9 nickase with minimal off-target effects. Nat Methods 11:399–402
Skarnes WC, Rosen B, West AP, Koutsourakis M, Bushell W, Iyer V, Mujica AO, Thomas M, Harrow J, Cox T, Jackson D, Severin J, Biggs P, Fu J, Nefedov M, de Jong PJ, Stewart AF, Bradley A (2011) A conditional knockout resource for the genome-wide study of mouse gene function. Nature 474:337–342
Tan W, Carlson DF, Lancto CA, Garbe JR, Webster DA, Hackett PB, Fahrenkrug SC (2013) Efficient nonmeiotic allele introgression in livestock using custom endonucleases. Proc Natl Acad Sci USA 110:16526–16531
Urnov FD, Miller JC, Lee YL, Beausejour CM, Rock JM, Augustus S, Jamieson AC, Porteus MH, Gregory PD, Holmes MC (2005) Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature 435:646–651
Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, Jaenisch R (2013) One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153:910–918
Wu Y, Liang D, Wang Y, Bai M, Tang W, Bao S, Yan Z, Li D, Li J (2013) Correction of a genetic disease in mouse via use of CRISPR–Cas9. Cell Stem Cell 13:659–662
Wu X, Scott DA, Kriz AJ, Chiu AC, Hsu PD, Dadon DB, Cheng AW, Trevino AE, Konermann S, Chen S, Jaenisch R, Zhang F, Sharp PA (2014) Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nat Biotechnol. doi:10.1038/nbt.2889
Xiao A, Wang Z, Hu Y, Wu Y, Luo Z, Yang Z, Zu Y, Li W, Huang P, Tong X, Zhu Z, Lin S, Zhang B (2013) Chromosomal deletions and inversions mediated by TALENs and CRISPR/Cas in zebrafish. Nucleic Acids Res 41:e141
Yang H, Wang H, Shivalila CS, Cheng AW, Shi L, Jaenisch R (2013a) One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell 154:1370–1379
Yang L, Guell M, Byrne S, Yang JL, De Los Angeles A, Mali P, Aach J, Kim-Kiselak C, Briggs AW, Rios X, Huang PY, Daley G, Church G (2013b) Optimization of scarless human stem cell genome editing. Nucleic Acids Res 41:9049–9061
Zhou J, Shen B, Zhang W, Wang J, Yang J, Chen L, Zhang N, Zhu K, Xu J, Hu B, Leng Q, Huang X (2014) One-step generation of different immunodeficient mice with multiple gene modifications by CRISPR/Cas9 mediated genome engineering. Int J Biochem Cell Biol 46:49–55
Acknowledgments
We thank C. Mark for editorial assistance. D.S. is a PhD fellow of the La Caixa Foundation International Fellowship Programme (La Caixa/CNB). This work is supported by MINECO Project BIO2012-39980 and SALAAM BMBS COST Action BM1308 to L.M.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Seruggia, D., Montoliu, L. The new CRISPR–Cas system: RNA-guided genome engineering to efficiently produce any desired genetic alteration in animals. Transgenic Res 23, 707–716 (2014). https://doi.org/10.1007/s11248-014-9823-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11248-014-9823-y