
Abstract
A Zn(II)-based coordination polymer [Zn3(L)2(dpp)2]n (1) has been solvothermally constructed from a combination of a multifunctional Schiff base 3,5-dibromosalicylaldehyde salicylhydrazone (H2L) and 1,3-di(4-pyridyl)propane (dpp). The photoluminescence properties of the complex have been exploited to use 1 as a dual detection probe for the selective sensing of Cu2+ and 2,4,6-trinitrotoluene (TNT) in the aqueous phase from among a variety of cations and a pool of aromatic nitro compounds, respectively. Competitive fluorometric experiments involving mixtures of cations or nitro compounds established 1 as an efficient and selective sensor for both Cu2+ and TNT in aqueous solutions. The limits of detection for Cu2+ and TNT in aqueous solutions were found to be 1.05 and 49.9 μM, respectively. Additionally, the activity of complex 1 as a photocatalyst for degradation of rhodamine B has been investigated.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
With the development of extensive industrialization, water contamination and associated environmental problems have attracted serious concern [1,2,3]. Efficient detection of hazardous metal ions and nitroaromatic compounds (NACs) has been extensively studied over the past few decades [4,5,6]. Detection of Cu2+ ion is important because short- or long-term exposure to high levels of Cu2+ can lead to Wilson’s disease, Alzheimer’s disease, gastrointestinal disorders and liver/kidney damage [7]. The U.S. Environmental Protection Agency has set the safe limit of Cu2+ ion in drinking water at 1.3 ppm (~ 20 μM) [8]. On the other hand, rapid and selective detection of NACs such as 2,4,6-trinitrotoluene (TNT) has also become a challenging issue in recent times. TNT is one of the most commonly used nitroaromatic explosives, besides being a major source of water pollution resulting from the production of landmines [9, 10].
Coordination polymers (CPs) are a class of materials fabricated through the assembly of metal nodes/clusters and organic ligands to generate multidimensional frameworks [11]. CPs have recently undergone rapid development, not only for their aesthetic structures and topological networks but also for diverse applications in the areas of heterogeneous catalysis, gas storage, gas separation, drug delivery, and molecular sensing [12,13,14,15,16]. Currently, some sophisticated instrumental techniques such as gas chromatography and spectrophotometry can be used for quantitative detection of Cu2+ and NACs, but with several drawbacks such as high cost, technical procedures, and sometimes inefficiency [17,18,19]. Comparatively, fluorescent sensors offer an alternative method for rapid and efficient detection of pollutants. Recently, based on the electron-deficient characteristics of NACs and the photoinduced electron transfer (PET) quenching mechanism, lanthanide and d10 metal node-based luminescent coordination polymers (LCPs) have been designed as efficient fluorescent chemosensors for the detection of NACs in the vapor/liquid phase with good sensitivity and ease of operation [20,21,22,23,24,25,26,27]. Luminescence-based methods have been widely used for detection of TNT [28,29,30,31] as well as Cu2+ [32,33,34]. However, aqueous-phase detection of Cu2+/TNT by LCPs is relatively limited in the literature, perhaps because of the chemical instability of LCPs in the respective analyte solutions [26, 35,36,37,38]. In our continuing quest to devise new LCP sensor materials [39, 40], we here report a hydrazone-based Zn(II) coordination polymer as a dual detection fluorosensor for selective and sensitive detection of Cu2+ and TNT. Thus, a one-dimensional coordination polymer [Zn3(L)2(dpp)2]n (1) with good aqueous chemical stability has been synthesized and characterized. The intrinsic luminescence properties of this d10-based LCP have been utilized for competitive sensing of Cu2+ and TNT from among a variety of cations and NACs, respectively. Furthermore, the potential application of 1 as a photocatalyst for the degradation of rhodamine B under UV irradiation has also been investigated.
Experimental
Materials and methods
All the chemicals were of commercial origin and used without further purification. Elemental analyses were obtained with an Elementar vario EL cube analyzer. Powder X-ray diffraction (PXRD) data were collected on a Bruker Advance X-ray diffractometer with Cu-Kα radiation (λ = 1.5418 Å) at 50 kV, 20 mA with a scanning rate of 5°/min and a step size of 0.02°. FTIR spectra were measured on KBr pellets using a Nicolet Impact 750 FTIR spectrometer in the range of 400–4000 cm−1. Thermogravimetric analysis was performed with an STA 409PC Luxx Netzsch instrument under an N2 atmosphere from room temperature to 750 °C at a heating rate of 10 °C/min. Solid-state UV–Vis spectra were recorded using a Shimadzu UV-3101PC spectrometer with BaSO4 as a reference. Photoluminescence (PL) spectra were recorded at room temperature utilizing an RF-5301PC spectrofluorophotometer. The photocatalytic activity studies were carried out with a Shimadzu UV–Vis 2501PC recording spectrophotometer.
X-ray crystallography
The single-crystal X-ray diffraction data were collected on a Bruker SMART APEX diffractometer equipped with graphite monochromated MoΚα radiation (λ = 0.71073 Å), using an ω-scan technique. The intensities were corrected for absorption effects using SADABS. The structure was solved with SHELXS-97 and refined with SHELXL-97 [41]. All the hydrogen atoms were generated geometrically and refined isotropically using a riding model. All non-hydrogen atoms were refined with anisotropic displacement parameters. Crystallographic details are listed in Table 1, while selected bond dimensions are listed in Table S1. CCDC 1497314 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/structures.
Synthesis of [Zn3(L)2(dpp)2]n
A mixture of Zn(NO3)2·6H2O (0.029 g, 0.1 mmol), 3,5-dibromosalicylaldehyde salicylhydrazone (0.042 g, 0.1 mmol), 1,3-di(4-pyridyl)propane (0.016 g, 0.1 mmol), DMF (3 mL) and methanol (3 mL) was stirred for 30 min in air. The resulting solution was placed in a Teflon-lined stainless steel vessel (25 mL) and heated at 140 °C for 72 h, and then slowly cooled to room temperature. Yellow block crystals of complex 1 were obtained (yield 58% based on Zn). Anal. Calcd (%) for C54H42Br4N8O6Zn3, C, 45.82; H, 2.99; N, 7.92. Found C, 45.34; H, 3.02; N, 7.96. IR (cm−1): 3058 (m); 2936 (m); 1606 (vs); 1523 (vs); 1450 (v); 1371 (vs); 1222 (m); 1153 (m); 816 (v); 752 (v); 700 (m).
Stability experiments
The aqueous chemical stability of complex 1 was investigated by soaking the material in aqueous solutions with pH values ranging from 4 to 10 (adjusted using HCl and NaOH). Approximately 50 mg of LCP 1 was dispersed into a 25-mL scintillation vial containing the respective aqueous solution to make a suspension. After soaking for 12 h, LCP 1 was removed by centrifugation, washed thoroughly with distilled water and diethyl ether, and dried under vacuum. PXRD data for the recovered solid samples were recorded to confirm the chemical stability.
Photocatalytic method
The photocatalytic experiments were performed as follows. A sample of LCP 1 (50 mg) was dispersed in an aqueous solution of rhodamine B (50 mL, 10 mg/L) under stirring in the dark for 30 min to ensure the establishment of an adsorption–desorption equilibrium. The suspension was then exposed to UV irradiation from an Hg lamp (250 W) and kept under continuous stirring during irradiation for 100 min. Samples of 5 mL volume were removed every 10 min and separated by centrifugation for analysis by UV–Vis spectrometer. A control experiment was also performed under the same conditions, but without adding any catalyst.
Sensing experiments
For the detection of Cu2+ and TNT, fluorimetric experiments were performed with LCP 1 dispersed in aqueous solutions of metal salts (1 × 10−2 M) or NACs (2 × 10−3 M). For detection and sensing studies, mixtures of Al3+, Zn2+, Ca2+, Mg2+, Ni2+, Mn2+, Cd2+, Pb2+, K+, Na+, Li+, Ag+, and Cu2+ and different NACs, namely 4-nitrotoluene (4-NT), 2-nitrotoluene (2-NT), 2,4-dinitrotoluene (2,4-DNT), 2,6-dinitrotoluene (2,6-DNT), 1,3-dinitrobenzene (1,3-DNB) and TNT, were chosen. Generally, a finely ground sample of LCP 1 (3 mg) was added to a screwed vial containing an aqueous solution of cations/nitroaromatics (3 mL). The suspension was then ultrasonically agitated for 30 min before detection. The emission spectrum of each sample was recorded in the range of 400–700 nm upon excitation at 360 nm, and the fluorescence intensity at 495 nm was monitored. All of the titrations were carried out in triplicate to establish the consistency of the results. The quenching efficiency was calculated as [(I0–I)/I0] × 100%, where I0 and I are the fluorescence intensities before and after interaction with the analytes.
Results and discussion
Crystal structure of LCP 1
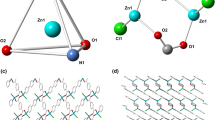
Single-crystal X-ray diffraction analysis reveals that LCP 1 crystallizes in the orthorhombic crystal system with the Ibca space group and features a one-dimensional chain structure. The formula unit contains three independent Zn(II) atoms (2 × Zn1, Zn2), two fully deprotonated L3− ligands, and two 1,3-di(4-pyridyl)propane ligands. LCP 1 is a trinuclear Zn(II) complex with two bridging dpp ligands. As illustrated in Fig. 1a, the two types of Zn(II) center have a five-coordinate tetragonal pyramidal geometry and a four-coordinate distorted tetrahedral geometry, respectively. Zn1 is coordinated by one phenol oxygen atom, one salicyloyl oxygen atom and one hydrazine nitrogen atom from one L ligand, plus two nitrogen atoms from two chelating 1,3-di(4-pyridyl) propane ligands, forming a ZnN3O2 configuration. It is noteworthy that the salicyloyl-O atom of the L ligand deprotonates and enolizes during complexation to bond through carbonylate-O. The square base for Zn1 is formed by O(1), O(2), and N(1) from the NOO donor (L) ligand; the fourth position is occupied by N(3) of the dpp ligand. The apical position is occupied by N(4A) of another dpp ligand. Zn2 is coordinated by two phenol oxygen atoms and two hydrazine nitrogen atoms from two different L ligands. The bond lengths [Zn–O = 1.891 (3)–2.050 (3) Å]; [Zn–N = 2.006 (4)–2.074 (4) Å] and angles [X–Zn–X (X=N, O) = 76.27 (13)°–160.02 (14)°] are in the normal ranges [42].

a The coordination environment of Zn(II) in 1; b view of the 1D chain along the a-axis; c view of the 2D layer constructed by π–π interactions from adjacent chains (pink dotted line represents the π–π interactions; d the 3D framework of 1. (Color figure online)
In LCP 1, two nitrogen atoms from two 1,3-di(4-pyridyl)propane ligands are coordinated with Zn1 in a bridging mode to produce an infinite extending 1D zigzag chain (Fig. 1b). These 1D chains are connected by π–π interactions (the distance between the centroids of two adjacent 3,5-dibromobenzene rings is 3.497 Å) to produce a two-dimensional layer structure (Fig. 1c). Adjacent 2D layers are further connected by π–π interactions to give rise to a three-dimensional supramolecular framework (Fig. 1d).
FTIR and TGA analysis of 1
Comparison of the FTIR spectra (Fig. S1) revealed that the υ(N–H) band observed at 3239 cm−1 for the free Schiff base was absent from the spectrum of complex 1, indicating deprotonation of the NH group of the Schiff base upon complexation. The υ(C=O) band observed at 1651 cm−1 for the free Schiff base was also absent for LCP 1, indicating enolization of the C=O group with concomitant coordination to the zinc centre. The observation of a new υ(C–O)− band for complex 1 in the range of 1370–1360 cm−1 confirms bonding of the ligand to zinc through phenolate O. The υ(C=N) band observed at 1610 cm−1 in the spectrum of the free Schiff base has undergone a redshift in LCP 1, suggesting coordination of the azomethine N. New bands at 760–700 and 570–510 cm−1 in the spectrum of LCP 1 can be tentatively assigned to υ(Zn–O) and υ(Zn–N), respectively.
TGA was carried out to ascertain the thermal stability of LCP 1 (Fig. S2). The TGA curve shows no obvious weight loss until 315 °C. The loss of two 1,3-di(4-pyridyl) propane ligands is then observed in the range of 315–390 °C (observed, 29.5%; calculated, 28.0%). Subsequent decomposition of the Schiff base ligand results in the complete structural collapse of 1.
Fluorescence properties
The solid-state luminescence properties of LCP 1 and the free Schiff base were investigated at room temperature (Figure S3). LCP 1 exhibits a strong emission at 507 nm upon excitation at 360 nm, with a slight blueshift relative to the free Schiff base (λem = 513 nm). The emission peaks of both can be attributed to π*–π and/or π*–n transitions. We further examined the luminescence of 1 dispersed in common solvents, as shown in Figure S4. The spectra of suspensions of 1 in different solvents exhibited broad peaks with emission maxima ranging from 483 to 498 nm, with the highest luminescence intensity observed in DMF dispersion and the lowest in the case of nitrobenzene; i.e., nitrobenzene showed the highest fluorescence quenching. Upon excitation, energy transfer from the organic ligand to the nitrobenzene might take place, resulting in the observed fluorescence quenching [43]. Interestingly, LCP 1 showed strong luminescence when dispersed in water, which inspired us to check its aqueous chemical stability. As shown in Fig. S5, after soaking in a broad range of pH media for 12 h, no noticeable changes appeared in the PXRD patterns, indicating excellent aqueous stability. This in turn suggests the use of LCP 1 as a promising candidate for the aqueous detection of analytes.
Fluorescence studies for Cu2+ detection
In light of the excellent photoluminescence and chemical stability of LCP 1, its sensing and detection capability toward different metal ions in aqueous solution was investigated. A suspension of 1 in water was prepared by adding 3 mg of 1 to 3 mL of an aqueous solution containing 1 × 10−2 M of M(NO3)x (M = Al3+, Zn2+, Ca2+, Mg2+, Ni2+, Mn2+, Cd2+, Pb2+, K+, Na+, Li+, Ag+, and Cu2+) and then ultrasonically agitating for 30 min before detection. The luminescence properties were studied by recording fluorescence spectra in the range of 400–700 nm upon excitation at 360 nm.
As depicted in Fig. 2a, among the metal ions chosen for detection in water by 1, Al3+, Zn2+, and Li+ showed slight increases in the emission intensity, while the rest of the metals showed varying degrees of quenching, with Cu2+ almost completely quenching the emission band. The marked difference between Cu2+ and other cations encouraged us to investigate the ability of 1 to act as a selective chemosensor for Cu2+. Thus, competitive experiments were conducted by addition of 1.5 mL of Cu2+ (2 × 10−2 M) to 1.5 mL of aqueous solutions of other metal ions (1 × 10−2 M). As depicted in Fig. 2b, the response of 1 toward Cu2+ was almost unaffected in the presence of other metal ions. These results confirm that LCP 1 could be used for selective detection of Cu2+ in water from among the above-mentioned cationic series. In order to evaluate the sensitivity of LCP 1 for Cu2+, fluorometric titration of varying concentrations of aqueous Cu2+ (5–800 × 10−5 M) against a suspension of 1 in water was carried out. As shown in Fig. 2c, the emission intensity gradually decreased with incremental addition of Cu2+.

a Fluorescence response of 1 to various cations in the water; b the selective detection of Cu2+ by 1 in the presence of other cations in the water; c emission spectra of 1 in different concentrations of Cu2+ ions in the water (excited at 360 nm); d the Stern–Volmer plot of 1 versus Cu2+ concentrations
The quenching efficiency was calculated by fitting the fluorescence intensity ratio versus concentration of Cu2+ to the Stern–Volmer (SV) equation,
where I0 and I are the emission intensities of the suspension of 1 before and after addition of Cu2+, respectively, [Q] is the molar concentration of Cu2+, and KSV is the quenching constant. As demonstrated in Fig. 2d, the SV plot was nearly linear over the added Cu2+ concentration range (0–100 × 10−5 M), and the calculated quenching constant value is Ksv = 9.7 × 103 M−1 with a linear fit coefficient value of 0.993. The limit of detection (LOD) was calculated to be 1.05 μM on the basis of 3 σ/m [44]. It should be pointed out that aqueous-phase detection of Cu2+ by LCPs has rarely been reported. Known examples of detection of aqueous Cu2+ by LCPs are listed in Table S2. Evidently, the KSV of LCP 1 is much higher than those of known MOF-based sensors for aqueous Cu2+ detection and the LOD in the present investigation is also comparable to previous reports.
TNT detection studies
The strong emission of LCP 1 in water suspension and its thermal and aqueous stability prompted us to investigate the sensing of nitroaromatic compounds in aqueous media, which has relevance to environmental and security issues. The fluorescence behavior of 1 in the presence of NACs was therefore examined. As shown in Fig. 3a, all of the NAC analytes reduced the emission band of 1 to a certain extent, and significant quenching of up to 93.8% was observed for TNT. The quenching percentages were calculated using the formula:
and found to be in the following order: TNT > 1,3-DNB > 2,6-DNT > 2,4-DNT > 2-NT > 4-NT. Since high specificity is crucial for most probes to be applied in real sample detection, we further studied the fluorescence quenching of 1 by TNT in the presence of other nitroaromatic compounds. As desired, no significant changes of fluorescence intensity were observed in the experiments in which other nitroaromatic compounds were added to the TNT-containing system (Fig. 3b).

a Fluorescence response of 1 to various nitroaromatics in the water; b the selective detection of TNT by 1 in the presence of other nitroaromatics in the water; c emission spectra of 1 in different concentrations of TNT in the water (excited at 360 nm); d the Stern–Volmer plot of 1 versus TNT concentrations
Fluorescence quenching titrations with different TNT concentrations were conducted with an excitation wavelength of 360 nm at room temperature. Upon incremental addition of 0.1, 0.2, 0.3, 0.4, 0.6, and 1.2 mM TNT to an aqueous suspension of 1, the observed fluorescence quenching was 29, 56, 69, 77, 86, and 92%, respectively (Fig. 3c). The photoluminescence quenching efficiency can be quantitatively explained using the SV equation. As shown in Fig. 3d, TNT displays a good linear SV relationship (R2 = 0.989) in the concentration range of 0.1–1.2 mM and the calculated quenching constant value is Ksv = 1.154 × 104 M−1. As seen in Table S2, the KSV of 1 is comparable to those of previously reported MOF-based sensors. The LOD was calculated to be 49.9 μM on the basis of 3 σ/m [44]. As is well known, fast and simple regeneration methods are important concerns for recyclable performance. Hence, we immersed LCP 1 in an aqueous solution of 10−3 M TNT for 30 min, after which the solid was separated by centrifugation and washed several times with distilled water and diethyl ether. After three cycles of repeated fluorescence measurements, the luminescence intensity of the recycled sample was well consistent with the original one (Fig. 4a). Furthermore, PXRD patterns (Fig. 4b) confirmed that the sample retained its crystallinity after these experiments. Since the PXRD patterns demonstrate no collapse of the crystalline structure, the luminescence quenching of 1 caused by analytes may result from energy loss caused by the collisions between the framework and analytes [45].

a The luminescence intensity of three recycles for 1 (a: after the first recycle; b: after the second recycle, c: after the third recycle); b powder XRD profiles of 1 after experiments
Photocatalytic activity
The photocatalytic activity of LCP 1 was evaluated by the photodegradation of rhodamine B (RhB) in aqueous solution under irradiation with a 250 W Hg lamp. The degradation ratio of RhB was monitored by observing the intensity of the characteristic absorption band with time. The absorption intensity of RhB decreased gradually over time in the presence of LCP 1 (Fig. 5a), with 87.0% degradation after 100 min of UV irradiation for 100 min. For the sake of comparison, a control experiment (without the use of catalyst 1) was also carried out (Fig. 5b). The degradation of RhB was merely 14.3% after 100 min under similar reaction conditions in the absence of LCP 1. The UV–Vis diffuse reflectance spectra confirm that 1 can be activated by ultraviolet radiation (Fig. S6). The band gap energy was estimated to be 2.41 eV according to the Kubelka–Munk function (Fig. S7). This suggests that UV radiation promotes an electron from the HOMO to the LUMO of LCP 1, which then abstracts an electron from water to give an ·OH radical. Meanwhile, the electron of the LUMO may reduce O2 to O2− which can also give ·OH. The generated ·OH radicals then oxidize RhB [46, 47].

a UV–Vis absorption spectra of the RhB solution during the decomposition reaction under 250 W Hg lamp irradiation in the presence of 1; b photodegradation of RhB solution with the change in A/A0 of 1, and the control experiment without any catalyst; c the Langmuir–Hinshelwood plot of 1 against irradiation time
In order to evaluate the recycling potential of CP 1 as a photocatalyst, the repeated photocatalytic degradation of a constant RhB concentration was investigated. The degradation percentages of RhB showed no significant reduction when the photocatalyst 1 was used three times in the same procedures, being 87.2, 86.5, and 85.1% for the three times. The kinetics of RhB photocatalytic degradation by 1 can be described by the Langmuir–Hinshelwood model as expressed by ln(C/C0) = −kt (k = apparent reaction rate constant). C0 is the initial concentration of RhB, t is the reaction time, and C is the concentration of RhB at time t. A plot of ln(C0/C) versus irradiation time (t) is approximately linear and approximates a first-order kinetic process (Fig. 5c), with a calculated apparent rate constant k value of 1.91 × 10−2 min−1.
Conclusions
In summary, using 3,5-dibromosalicylaldehyde salicylhydrazone and 1,3-di(4-pyridyl)propane, we have prepared a new functional CP 1, which can be used for the selective and sensitive detection of both Cu2+ and TNT in aqueous solution through fluorescence quenching. LCP 1 exhibited exceptional thermal and chemical stability in aqueous solutions with a wide range of pH values (4–10). Notably, the Ksv and LOD values of 1 toward Cu2+ and TNT are comparable to those reported for MOF materials to date. The high stability and recyclability of 1 for the degradation of RhB make it an outstanding candidate in the field of photocatalytic degradation of organic pollutants.
References
Rachuri Y, Parmar B, Bisht KK, Suresh E (2017) Cryst Growth Des 17:1363–1372
Zhang C, Yan Y, Sun LB, Liang ZQ, Li JY (2016) Cryst Eng Commun 18:4102–4108
Liu C, Yan B (2016) Sensor Actuat B Chem 235:541–546
Gong YN, Huang YL, Jiang L, Lu TB (2014) Inorg Chem 53:9457–9459
Wu L, Zhang XF, Li ZQ, Wu F (2016) Inorg Chem Commun 74:22–25
Vishnoi P, Walawalkar MG, Sen S, Datta A, Patwari GN, Murugavel R (2014) Phys Chem Chem Phys 16:10651–10658
Su YT, Lan GY, Chen WY, Chang HT (2010) Anal Chem 82:8566–8572
Lan GY, Huang CC, Chang HT (2010) Chem Commun 46:1257–1259
Germain MN, Arechederra RL, Minteer SD (2008) J Am Chem Soc 130:15272–15273
Dasary SSR, Senapati D, Singh AK, Anjaneyulu Y, Yu HT, Paresh CR (2010) ACS Appl Mater Interfaces 2:3455–3460
Zhou HC, Long JR, Yaghi OM (2012) Chem Rev 112:673–674
Chughtai AH, Ahmad N, Younus HA, Laypkov A, Verpoort F (2015) Chem Soc Rev 44:6804–6849
Croitor L, Coropceanu EB, Masunov AE, Rivera-Jacquez HJ, Siminel AV, Zelentsov VI, Datsko TY, Fonari MS (2014) Cryst Growth Des 14:3935–3948
Kang ZX, Fan LL, Sun DF (2017) J Mater Chem A 5:10073–10091
Huxford RC, Della Rocca J, Lin W (2010) Curr Opin Chem Biol 14:262–268
Zhao D, Cui Y, Yang Y, Qian G (2016) Cryst Eng Commun 18:3746–3759
Hakansson K, Coorey RV, Zubarev RA, Talrose VL, Hakansson P (2000) J Mass Spectrom 35:337–346
Sylvia JM, Janni JA, Klein JD, Spencer KM (2000) Anal Chem 72:5834–5840
Kandpal M, Bandela AK, Hinge VK, Rao VR, Rao CP (2013) ACS Appl Mater Interfaces 5:3448–13456
Lan A, Li K, Wu H, Olson DH, Emge TJ, Ki W, Hong M, Li J (2009) Angew Chem Int Ed 48:2334–2338
Lee JH, Jaworski J, Jung JH (2013) Nanoscale 5:8533–8540
Sun X, Brückner C, Nieh MP, Lei Y (2014) J Mater Chem A 2:14613–14621
Roy S, Katiyar AK, Mondal SP, Ray SK, Biradha K (2014) ACS Appl Mater Interfaces 6:11493–11501
Gole B, Bar AK, Mukherjee PS (2011) Chem Commun 47:12137–12139
Yang J, Wang Z, Hu K, Li Y, Feng J, Shi J, Gu J (2015) ACS Appl Mater Interfaces 7:11956–11964
Park IH, Medishetty R, Kim JY, Lee SS, Vittal JJ (2014) Angew Chem Int Ed 53:5591–5595
Rachuri Y, Parmar B, Bisht KK, Suresh E (2015) Inorg Chem Front 2:228–236
Nie HR, Zhao Y, Zhang M, Ma YG, Baumgartenb M, Mullen K (2011) Chem Commun 47:1234–1236
Yang X, Wang JH, Su DY, Xia QD, Chai F, Wang CG, Qu FY (2014) Dalton Trans 43:10057–10063
Nie HR, Lv Y, Yao L, Pan YY, Zhao Y, Li P, Sun GN, Ma YG, Zhang M (2014) J Hazard Mater 264:474–480
Kovalev IS, Taniya OS, Slovesnova NV, Kim GA, Santra S, Zyryanov GV, Kopchuk DS, Majee A, Charushin VN, Chupakhin ON (2016) Chem Asian J 11:775–781
Zhang S, Yu T, Sun MT, Yu H, Zhang ZP, Wang SH, Jiang H (2014) Talanta 126:185–190
Li M, Liu ZJ, Wang SW, Calatayud DG, Zhu WH, James TD, Wang LD, Mao BY, Xiao HN (2018) Chem Commun 54:184–187
Elmizadeh H, Soleimani M, Faridbod F, Bardajee GR (2017) J Fluoresc 27:2323–2333
Liu B, Wu WP, Hou L, Wang YY (2014) Chem Commun 50:8731–8734
Tian D, Li Y, Chen RY, Chang Z, Bu XH (2014) J Mater Chem A 2:1465–1470
Zhang SR, Du DY, Qin JS, Bao SJ, Su ZM (2014) Chem Eur J 20:3589–3594
Zhang M, Feng G, Song Z, Zhou YP, Zhao D (2014) J Am Chem Soc 136:7241–7244
Wu Y, Wu J, Luo ZD, Wang J, Li YL, Han YY, Liu JQ (2017) RSC Adv 7:10415–10423
Wu Y, Wu J, Xie B, Zou LK, Li YL, Han YY, Wu XR (2017) J Lumin 192:775–782
Sheldrick GM (1997) SADABS, program for siemens area detector absorption corrections. University of Göttingen, Germany
Roztocki K, Jędrzejowski D, Hodorowicz M, Senkovska I, Kaskel S, Matoga D (2010) Inorg Chem 55:9663–9670
Huang XL, Liu L, Gao ML, Han ZB (2016) RSC Adv 6:87945–87949
Wu LL, Wang Z, Zhao SN, Meng X, Song XZ, Feng J, Song SY, Zhang H (2016) Chem Eur J 22:477–480
Tang Q, Liu SX, Liu YW, Miao J, Li SJ, Zhang L, Shi Z, Zheng ZP (2013) Inorg Chem 52:2799–2801
Yang H, He XW, Wang F, Zhang J (2012) J Mater Chem 22:21849–21851
Kan WQ, Liu B, Yang J, Liu YY, Ma JF (2012) Cryst Growth Des 12:2288–2298
Acknowledgements
The authors acknowledge financial assistance from National Natural Science Foundation of China (Nos: 21501124 and 41603124), the program of Science and Technology Department of Sichuan Province (Nos. 2016JY0048, 2016GZ0172, 2017JY0194), the Education Committee of Sichuan Province (No. 18ZB0425, 18ZA0337), the Start-up Foundation of Sichuan University of Science and Engineering (No. 2017RCL02), and Student’s Platform for Innovation and Entrepreneurship Training Program in Sichuan Province (201710622066).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Wu, Y., Gu, Z., Luo, W. et al. Crystal structure, luminescent sensing and photocatalytic activity of a multifunctional hydrazone-based zinc(II) coordination polymer. Transit Met Chem 43, 673–681 (2018). https://doi.org/10.1007/s11243-018-0256-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11243-018-0256-y