
Abstract
A series of zinc coordination polymers, {Zn2X2(BDC)(L)}n (X = Cl (1 and 3) or Br (2 and 4), BDC = 1,4-benzenedicarboxylate, L = 4,4′-bipyridine (1 and 2) or 1,4-diazabicyclo[2.2.2]octane (3 and 4)), have been synthesized by ionothermal reactions and characterized by X-ray crystallography and powder X-ray diffraction (PXRD). Compounds 1, 2 and 4 were also characterized by infrared spectroscopy (IR), thermogravimetric analysis (TGA) and their chemical stability towards various organic solvents were investigated. Compound 1 displays strong luminescence in the solid state and high luminescent selectivity and sensitivity for Fe3+ ions and nitroaromatics (NACs) in ethanol solutions, rendering it a potential fluorescence probe for these substrates. In addition, investigation on organic dye adsorptions shows that, among these coordination polymers, only complex 4 can selectively adsorb Congo red dye from other dye molecules. This demonstrates that the function of these coordination polymers can be regulated through subtle modification of their components.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
In the last two decades, research on metal–organic frameworks (MOFs) or coordination polymers (CPs) have developed rapidly, due in part to their diverse structures given by abundant metal secondary building units (SBUs) and organic linkers [1,2,3]. Moreover, increasing attentions have been drawn to the great potential of these materials in gas storage [4] and separation [5], catalysis [6], magnetism [7], sensing [8], biomedicine [9] and molecular recognition [10]. In recent years, there has been an increasing tendency in employing MOFs or CPs as fluorescent sensors to detect guest species, such as metal ions [11], various anions [12], small organic molecules [13, 14] and nitroaromatics (NACs) [15, 16], because of the easy manipulation and high sensitivity of fluorometric methods.
It is well known that iron is one of the most earth-abundant metals, only second to aluminum, and the Fe3+ ion plays a vital role in various biological processes. For instance, iron deficiency or accumulation will lead to function disorder symptoms such as sleep loss and decreased immunity [17], or severe illness like Parkinson’s or Alzheimer’s diseases [18]. On the other hand, although NACs are widely used in textile, fireworks and pharmaceutical industries, they are considered as toxic pollutants and difficult to be removed or degraded. More importantly, due to their close relation to explosives, these compounds have raised considerable security concerns globally in recent years. Hence, it is urgent to develop reliable and robust materials for the selective and efficient detection of Fe3+ ions and NACs. Nowadays, another serious environmental threat is the water pollution caused by organic dyes. For instance, Congo red is a synthetic industrial colorant broadly used in paper industry, as well as a pH indicator. Because of its low biodegradability, it is highly desirable to develop new materials for efficient dye adsorption from industrial effluents, in order to protect human health and provide environmental sustainability.
Zinc CPs are one of the prototypical MOF materials, whose development witnesses the rapid maturing and evolution of this rapidly expanding research area [4, 19,20,21]. These compounds are usually constructed with various multicarboxylate ligands through solvothermal synthesis. In contrast, using ionic liquids as the reaction media (i.e., ionothermal synthesis) for the preparation of coordination polymers represents a promising alternative route but is far less developed [22, 23]. An evident advantage of ionic liquids is the lack of detectable vapor pressure, which leads to the elimination of safety concerns associated with traditional solvothermal methods. In addition, it has been demonstrated that the ionic liquid acts as not only a solvent but also a template provider, for the assembly of inorganic/organic materials [24,25,26,27]. Xu and Kwon initiated the systematic investigation of ionothermal synthesis of zinc coordination polymers supported by various multicarboxylate ligands, such as BTC (1,3,5-benzenetricarboxylate) [28,29,30], BDC (1,4-benzenedicarboxylate) [31] and BTeC (1,2,4,5-benzenetetracarboxylate) [32]. Other zinc(II) CPs with BDC [33] or NDC (1,4-naphthalenedicarboxylate) [34] ligands were also prepared under ionothermal conditions. Herein, we report ionothermal synthesis of a series of 2D zinc CPs assembled with both BDC and linear bidentate N-donor ligands, i.e., {Zn2X2(BDC)(L)}n (X = Cl (1 and 3) or Br (2 and 4), BDC = 1,4-benzenedicarboxylate, L = 4,4′-bipyridine (4,4′-bpy, 1 and 2) or 1,4-diazabicyclo[2.2.2]octane (dabco, 3 and 4)). X-ray crystallography revealed neutral two-dimensional (2D) layered structures. Furthermore, 1 demonstrates selective and sensitive luminescence sensing for Fe3+ ions and NACs. On the other hand, 4 displays selective adsorption towards Congo red dye.
2 Experimental Section
2.1 Materials and Methods
All reagents used were of analytical grade and used without further purification unless otherwise specified. Elemental analyses (C, H and N) were performed with a Perkin-Elmer EA 2400 elemental analyzer. FTIR spectra were obtained from KBr pellets using a Nicolet 6700 infrared spectrometer in the 4000−400 cm−1 region. Powder X-ray diffraction (PXRD) patterns were recorded in the 2θ range of 5°−60° using Cu Kα radiation with a Bruker D8 FOCUS diffractometer. Thermogravimetric analysis (TGA) was carried out under a N2 atmosphere on a Rigaku standard TG-DTA analyzer from 25 to 700 °C with a heating rate of 10 °C min−1. All the fluorescence measurements were recorded on a Hitachi F4500 fluorescence spectrophotometer. The UV–Vis absorption spectra were recorded on a Shimadzu UV-3600 UV-Vis-NIR spectrophotometer.
2.2 Synthesis of Complexes 1–4
1: Zn(NO3)2⋅6H2O (3.0 mmol, 0.893 g), 4,4′-bpy (0.50 mmol, 0.078 g) and H2BDC (0.50 mmol, 0.083 g) were mixed with 1.00 g of EMICl in a 20 mL Teflon-lined stainless-steel autoclave. The mixture was heated at 160 °C for 96 h. After cooling down to room temperature, colorless crystals of 1 were obtained. Yield: 0.163 g. (62% based on H2BDC.) Analysis found (calcd) for C18H12Cl2N2O4Zn2: C, 42.42 (41.33); H, 2.56 (2.30); N, 5.64 (5.35). IR (KBr pellet, cm−1): 3416(m), 1614(m), 1571(s), 1504(w), 1490(w), 1416(s), 1398(s), 1321(w), 1215(w), 1123(w), 1068(m), 831(m), 815(m), 762(m), 725(w), 647(m), 518(w).
2: Zn(NO3)2⋅6H2O (3.0 mmol, 0.893 g), 4,4′-bpy (0.50 mmol, 0.078 g) and H2BDC (0.50 mmol, 0.083 g) were mixed with 1.00 g of EMIBr in a 20 mL Teflon-lined stainless-steel autoclave. The mixture was heated at 160 °C for 96 h. After cooling down to room temperature, colorless crystals of 2 were obtained. Yield: 0.204 g. (67% based on H2BDC.) Analysis found (calcd) for C18H12Br2N2O4Zn2: C, 34.95 (35.37); H, 2.20 (1.97); N, 5.14 (4.60). IR (KBr pellet, cm−1): 3442(m), 1613(m), 1574(s), 1503(w), 1415(s), 1395(s), 1321(w), 1298(w), 1214(w), 1107(w), 1068(m), 828(m), 815(m), 756(m), 725(w), 647(m), 528(w).
3: Zn(NO3)2·6H2O (3.0 mmol, 0.893 g), dabco (1.25 mmol, 0.140 g) and H2BDC (0.50 mmol, 0.083 g) were mixed with 1.00 g of EMICl and 0.1 mL of formic acid in a 20 mL Teflon-lined stainless-steel autoclave. The mixture was heated at 170 °C for 96 h. After cooling down to room temperature, colorless crystals of 3 were obtained. Yield: 0.089 g. (37% based on H2BDC.) Due to the poor reproducibility of this synthetic protocol, further characterization of 3 was not pursued.
4: Zn(NO3)2⋅6H2O (3.0 mmol, 0.893 g), dabco (1.25 mmol, 0.140 g) and H2BDC (0.50 mmol, 0.083 g) were mixed with 1.00 g of EMIBr and 0.1 mL of formic acid in a 20 mL Teflon-lined stainless-steel autoclave. The mixture was heated at 160 °C for 96 h. After cooling down to room temperature, colorless crystals of 4 were obtained. Yield: 0.119 g. (42% based on H2BDC.) Analysis found (calcd) for C14H16Br2N2O4Zn2: C, 29.33 (29.65); H, 3.07 (2.82); N, 4.86 (4.94). IR (KBr pellet, cm−1): 3433(m), 2960(w), 1586(s), 1502(m), 1462(w), 1439(m), 1396(s), 1322(w), 1106(m), 1054(m), 1011(m), 923(m), 829(m), 807(m), 751(m), 580(w), 541(m).
2.3 X-ray Crystallography
Single crystal X-ray diffraction data of 1–4 were collected on a Rigaku XtaLAB PRO diffractometer with Cu Kα radiation (λ = 1.54178 Å) at 293 K. The empirical absorption corrections were applied using spherical harmonics, implemented in the SCALE3 ABSPACK scaling algorithm [35]. All the structures were solved using direct methods, which yielded the positions of all non-hydrogen atoms. Hydrogen atoms were placed in calculated positions in the final structure refinement. Structure determination and refinement were carried out using SHELXS-2014 and SHELXL-2014 programs, respectively [36]. Detailed crystallographic data and collecting parameters are summarized in Table 1. Selected bond lengths and angles are given in Tables S1–S4. CCDC-1894146 (1), CCDC-1894144 (2), CCDC-1894143 (3) and CCDC-1894145 (4) contain the supplementary crystallographic data for this paper.
2.4 Recyclability Test for the Fluorescence Detection of Fe3+
Finely ground sample of 1 (3 mg) was dispersed in 3 mL ethanol solutions (1 mM) of Fe(NO3)3·9H2O. A fluorescence experiment was performed after 30 min of sonication, and then the suspension was centrifuged to isolate the solid of 1. After that, 3 mL of ethanol was added to the solid, and the sample was sonicated for 10 min, before it was centrifuged again to obtain a solid. The solid was further washed with ethanol multiple times until no color is observed for the supernatant, and dried in the air to obtain a sample of 1 without residual Fe3+. The above procedures were repeated for four more times and luminescence was recorded for each suspension. As we can see from Fig. S16, the luminescence emission is restored to more than 92% of its original value after each cycle of removal of Fe3+, while the quenching efficiency remains at a high level for all five cycles (> 96%).
3 Results and Discussion
3.1 Synthesis of CPs 1–4
Compounds 1–4 were prepared by reacting Zn(NO3)2∙6H2O, H2BDC, N donor ligand and ionic liquid EMIX (EMI = 1-ethyl-3-methylimidazolium, X = Cl or Br) at 160 °C for 96 h. It is noteworthy that although various synthetic conditions have been tested, the preparation of 3 was found to be much less reproducible in comparison with that of 1, 2 or 4. Also, PXRD measurement indicates lower phase purity for the as-synthesized sample of 3 (vide infra). Hence, full characterization of 3 and further study on its luminescence sensing and dye adsorption properties were not further pursued.
3.2 Crystal Structures of 1–4
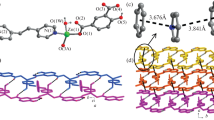
Single crystal X-ray diffraction analysis reveals that 1 and 2 crystallize in the triclinic space group Pī, while 3 and 4 crystallize in the orthorhombic space group P212121 (Table 1). The asymmetric unit of 1 contains one Zn(II) ion, one chlorine atom, half of the 4,4′-bpy ligand and half deprotonated BDC2− ligand (Fig. S1). As shown in Fig. 1a, the central Zn(II) atom displays a tetrahedral coordination geometry, and is four-coordinated by one pyridine N atom, one terminal chlorine atom, and two oxygen atoms from two different BDC2− anions. The Zn–O (1.9673 (15) and 1.9735 (16) Å), Zn–N (2.0326 (17) Å) and Zn–Cl (2.1985 (7) Å) bond distances are in agreement with those observed in other reported Zn(II) complexes [28,29,30,31,32,33,34, 37]. Each carboxylate group of the BDC2− ligand adopts a µ2-η1:η1 bridging mode to coordinate two zinc centers, forming the dimeric {Zn2Cl2(µ-COO)2} secondary building units (SBUs) (Fig. 1b) with a Zn⋯Zn separation of 3.731 Å. Such building units have been observed in a twofold interpenetrated Zn-organic polymer [Zn2(BDC)(BPP)Cl2]n (BPP = 1,3-bis(4-pyridyl)propane) [38]. As shown in Fig. 1c, the assembly of these dimetal SBUs with carboxylate ligands results in 1D chains, which are further connected by the bipyridine linkers to afford a 2D wavy sheet structure. These layers are further assembled into lamellar structures with eclipsed AA stacking mode and an interlayer distance of ca. 2.799 Å (Fig. 1d). Compound 2 is isostructural with 1, only replacing chlorine atoms with bromine atoms, forming Zn−Br bonds of 2.3364 (6) Å (Fig. S2).

a Coordination environment of Zn(II) in 1. b The dimetal SBUs in 1. c The 2D sheet structure of 1. d The stacking of 2D layers in 1
Similarly, 3 and 4 are isostructural with each other, and complex 4 is employed as an example for the following discussion. The asymmetric unit of 4 contains two Zn(II) centers, two bromine atoms, one BDC2− ligand and one dabco ligand (Fig. S4). Both zinc atoms feature similar tetrahedral geometry, each coordinated by one bromine atom, one nitrogen atom from dabco, and two oxygen atoms from different bridging BDC2− linkers (Fig. 2a). These Zn(II) centers form dimetal SBUs {Zn2X2(µ-COO)2} (X = Cl or Br) which are comparable to those in 1 and 2 (Fig. 2b). As depicted in Fig. 2c, the coordination-driven 1D zinc carboxylate chains in 4 are linked by dabco bridges to yield 2D zigzag layers. However, due to the substitution of aromatic 4,4′-bpy ligands with aliphatic dabco ligands in 4, there is no possible π–π interaction to facilitate the layer stacking. As a result, the 2D layers in 4 are stacked in a distinct staggered ABAB packing mode, with halogen atoms pointing towards the dabco ligands of adjacent layers, as illustrated in Fig. 2d.

a Coordination environment of Zn(II) in 4. b The dimetal SBUs in 4. c The 2D sheet structure of 4. d The stacking of 2D layers in 4
3.3 PXRD, Thermal Stability and IR Spectroscopy
The thermal stability of compounds 1, 2 and 4 was studied using thermogravimetric analysis (TGA) under N2 atmosphere (Figs. S5–S7). All three TGA curves show gradual slight weight loss in low temperature ranges (ca. 310 to 450 °C for 1, 280 to 430 °C for 2, 220 to 430 °C for 4), possibly due to loss of absorbed moisture as well as partial N donor pillar molecules (i.e., 4,4′-bpy or dabco). At higher temperatures (ca. 450 to 620 °C for 1, 430 to 570 °C for 2, 430 to 580 °C for 4), sharp weight losses occur for all complexes, indicating the decomposition of organic ligands and collapse of the coordination networks. The PXRD patterns of 1, 2 and 4 are shown in Fig. 3a, b, d, respectively, which show good accordance with the corresponding simulated patterns from single crystal data. In contrast, PXRD study indicates relatively poor phase purity for the synthesized sample of 3 (Fig. 3c). In addition, the experimental and simulated PXRD patterns for 2 show some difference in peak intensity, possibly due to preferred orientation of the samples. The IR spectra of 1, 2 and 4 (Figs. S8–S10) show the strong characteristic bands at 1571−1586 cm−1 for the carboxylate asymmetric stretching, and at 1395−1398 cm−1 for the carboxylate symmetric stretching. The large discrepancy between these two bands (173−190 cm−1) confirms the bridging coordination mode of BDC ligands revealed by X-ray crystallography.

Simulated and as-synthesized PXRD patterns of 1 (a), 2 (b), 3 (c) and 4 (d)
3.4 Luminescent Sensing of Fe3+
It is well known that CPs constructed from d10 metals and electron rich π-conjugated organic ligands are promising candidates for luminescent sensing because of their excellent photoluminescence properties. Therefore, the luminescence properties of 1 and free ligands (H2BDC and 4,4′-bpy) were investigated in the solid state (Fig. S11). A strong emission of compound 1 and H2BDC ligand was observed at 450 and 383 nm upon excitation at 353 and 330 nm, respectively. In contrast, 4,4′-bpy exhibits much weaker emission at 447 nm (λex = 360 nm). The red shift of the emission band of 1 in comparison with those of both ligands might be due to the packing of ligands in the framework. The relatively intense emission of 1 could be ascribed to the reduction of energy loss through non-radiative pathway upon coordination of the ligands and assembly of the network structure. In contrast, complexes 2 and 4 show much weaker emissions at 438 nm and 421 nm (Figs. S11 and S12), respectively. These results indicate the potential of 1 as a fluorescence sensing material. Hence, its fluorescence properties in different organic solvent dispersions were investigated (Fig. S13). Furthermore, the chemical stability of 1 were tested by comparison the PXRD patterns of 1 after being soaked in various solvents for 24 h (Fig. S14). It is found that 1 demonstrates excellent stability in these organic solvents, but decomposes gradually upon contact with water. Hence, ethanol was chosen as the solvents for the luminescent sensing studies.
Samples of 1 (3 mg) were ground and immersed in ethanol solutions (3 mL) with 1.0 × 10−3 mol L−1 M(NO3)n (M = Mn2+, Co2+, Mg2+, Zn2+, Ni2+, Cd2+, Ag+, Al3+, Cr3+, Cu2+, Fe3+) for 30 min. As shown in Fig. 4a, the observed luminescence intensities are highly dependent on the presence of metal ions, and that with Fe3+ displays the most significant quenching effect. Moreover, the addition of another metal ion into Fe3+ solutions does not interfere with the quenching effect (Fig. 4b), indicating the highly selective sensing for Fe3+ ions. The relationship between the luminescence intensity and the concentration of Fe3+ was explored by gradually increasing the Fe3+ concentration from 1.3 × 10−5 to 8.3 × 10−4 mol L−1. As depicted in Fig. 4c, the luminescence intensity is gradually decreased as the Fe3+ concentration increases. The correlation between luminescence intensity and Fe3+ concentration can be fitted well with the Stern–Volmer equation, I0/I = 1 + KSV[M], where [M] is the concentration of the metal ion, I0 and I are the luminescence intensities before and after metal ion incorporation, respectively (Fig. 4d). The quenching constant KSV was calculated to be 9.2 × 103 M−1. The detection limit was calculated according to the formula reported in the literature (3δ/k) [39, 40] to be 9.8 × 10−7 M (Fig. S15 and Table S5), which is lower than that of some other reported d10 metal based Fe3+ sensors [41]. Furthermore, 1 was demonstrated to be reusable as a fluorescence probe for Fe3+ for at least 5 cycles (Fig. S16). To elucidate the mechanism of luminescence quenching by Fe3+, the PXRD patterns of samples after Fe3+ detection were measured and found to match well with the simulated single-crystal data and experimental patterns for as-synthesized samples of 1 (Fig. S17). This suggests that the luminescence quenching is not caused by structural collapse of the coordination polymer. Moreover, the UV–visible absorption spectrum of Fe3+ shows little overlap with the emission spectrum of 1, but is largely overlapped with the excitation spectrum of 1 (Fig. S18). Therefore, the competition of absorption of the excitation wavelength energy between Fe3+ and 1 should be the reason for the observed luminescence quenching.

a Luminescence intensity of 1 upon the addition of different metal ions in ethanol solutions (1.0 × 10−3 mol L−1). b The influence of interfering ions on the luminescence intensity of 1 with Fe3+. c Comparison of luminescence spectra of 1 upon incremental addition of Fe3+ ions (10 mM). d Stern–Volmer plot for the fluorescence intensity of 1 upon addition of Fe3+ ions
3.5 Luminescent Sensing of NACs
The possibility of sensing NACs with 1 was also examined. Compound 1 (3 mg) was dispersed into 3 mL of 10 mM ethanol solutions containing nitromethane (NM), nitrobenzene (NB), m-dinitrobenzene (m-DNB), p-nitrotoluene (p-NT), o-nitrotoluene (o-NT), p-nitrophenol (p-NP) and o-nitrophenol (o-NP), and the emission spectra of the suspensions were measured (λex = 310 nm). As shown in Fig. 5, the luminescence intensity for all NAC-containing solutions are significantly quenched, while that for the solution with NM only slightly decreased. This indicates that 1 can selectively detects NACs over other nitro compounds. The fluorescence quenching efficiency is further investigated by fluorescence titration experiment and Stern–Volmer equation analysis (Fig. 6 and Fig. S19-S23). The order of calculated KSV values and quenching efficiency at low concentrations was p-NP > o-NT > o-NP > p-NT > NB > m-DNB, with p-NP showing the highest KSV value of 1.4 × 104 M−1 (Fig. 6), which is only slightly lower than that of a previously reported NAC sensor based on a Zn(II) coordination polymer (4.39 × 104 M−1) [42]. This demonstrates that 1 has great potential for the detection of trace amount of p-NP in ethanol solutions.

Luminescence intensity of 1 upon the addition of different nitro compounds in the ethanol solution (1.0 × 10−2 mol L−1) at room temperature, λex = 310 nm

a Comparison of luminescence spectra of 1 upon incremental addition of p-nitrophenol (p-NP) at room temperature, λex = 310 nm. b Stern–Volmer plot for the fluorescence intensity of 1 upon addition of p-nitrophenol (p-NP)
3.6 Dye Adsorption Property
The dye adsorption properties of 1, 2 and 4 were further explored. Samples of these CPs (20 mg) were soaked in 3 mL of 5 × 10−5 mol L−1 ethanol solutions of each organic dyes (Congo red (CR), malachite green (MAG), methylene blue (MEB), methyl orange (MO) and rhodamine B (RB)) at ambient temperature and the adsorption process was monitored by UV–Vis spectroscopy. Interestingly, it was found that 4 could selectively adsorb CR (Fig. 7a), but has no effects on the other four types of dyes (Fig. S24). In contrast, neither 1 nor 2 can adsorb any of the tested dyes from their ethanol solutions (Figs. S25 and S26). Considering the fact that both MO and CR are anionic dyes but only CR can be absorbed by 4 from its ethanol solution, it is reasonable to assume that intermolecular host–guest π–π interactions or hydrogen bonding might be responsible for the selective dye adsorption, rather than electrostatic interactions. The disparity between the dye adsorption properties of structurally similar compounds 2 and 4 also suggests that alteration of the bridging ligands (4,4′-bpy vs. dabco) could regulate the dye adsorption properties of CPs. Further selectivity test demonstrated that CR can be selectively adsorbed by 4 from a mixture dye solution containing both CR and MAG (Fig. 7b). In addition, PXRD data show that the coordination network in 4 remains intact after the dye adsorption experiment (Fig. S27).

UV–Vis spectra of 4 in ethanol solutions with different dyes (3 mL, 5 × 10−5 mol L−1): a CR; b CR and MAG (1:1 mixture)
4 Conclusion
In summary, a series of two-dimensional coordination polymers (1–4) were constructed by ionothermal synthesis. 1 can maintain its crystal structure in various organic solvents and exhibit highly sensitive and selective fluorescence quenching effect to Fe3+ ions in ethanol solutions. Meanwhile, 1 can also be used as a luminescence probe for the detection of NACs. In contrast, 4 can preferentially adsorb CR over other organic dyes. Altogether, the results reported here indicate that ionothermally prepared CPs could be potential multi-functional materials for luminescence sensing and dye adsorption.
References
H. Furukawa, K.E. Cordova, M. O’Keeffe, O.M. Yaghi, Science 341, 1230444 (2013)
O.M. Yaghi, M. O’Keeffe, N.W. Ockwig, H.K. Chae, M. Eddaoudi, J. Kim, Nature 423, 705 (2003)
S. Kitagawa, R. Kitaura, S.I. Noro, Angew. Chem. Int. Ed. 43, 2334 (2004)
M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O’Keeffe, O.M. Yaghi, Science 295, 469 (2002)
J.R. Li, R.J. Kuppler, H.C. Zhou, Chem. Soc. Rev. 38, 1477 (2009)
J.Y. Lee, O.K. Farha, J. Roberts, K.A. Scheidt, S.T. Nguyen, J.T. Hupp, Chem. Soc. Rev. 38, 1450 (2009)
M. Kurmoo, Chem. Soc. Rev. 38, 1353 (2009)
L.E. Kreno, K. Leong, O.K. Farha, M. Allendorf, R.P. Van Duyne, J.T. Hupp, Chem. Rev. 112, 1105 (2012)
P. Horcajada, R. Gref, T. Baati, P.K. Allan, G. Maurin, P. Couvreur, G. Férey, R.E. Morris, C. Serre, Chem. Rev. 112, 1232 (2012)
B. Chen, S. Xiang, G. Qian, Acc. Chem. Res. 43, 1115 (2010)
B. Chen, L. Wang, Y. Xiao, F.R. Fronczek, M. Xue, Y. Cui, G. Qian, Angew. Chem. Int. Ed. 48, 500 (2009)
K.L. Wong, G.L. Law, Y.Y. Yang, W.T. Wong, Adv. Mater. 18, 1051 (2006)
Z. Hu, B.J. Deibert, J. Li, Chem. Soc. Rev. 43, 5815 (2014)
Z.Z. Lu, R. Zhang, Y.Z. Li, Z.J. Guo, H.G. Zheng, J. Am. Chem. Soc. 133, 4172 (2011)
S.S. Nagarkar, B. Joarder, A.K. Chaudhari, S. Mukherjee, S.K. Ghosh, Angew. Chem. Int. Ed. 52, 2881 (2013)
A. Lan, K. Li, H. Wu, D.H. Olson, T.J. Emge, W. Ki, M. Hong, J. Li, Angew. Chem. Int. Ed. 48, 2334 (2009)
K.P. Carter, A.M. Young, A.E. Palmer, Chem. Rev. 114, 4564 (2014)
L. Zecca, M.B.H. Youdim, P. Riederer, J.R. Connor, R.R. Crichton, Nat. Rev. Neurosci. 5, 863 (2004)
S.S. Kaye, A. Dailly, O.M. Yaghi, J.R. Long, J. Am. Chem. Soc. 129, 14176 (2007)
J. Kim, B. Chen, T.M. Reineke, H. Li, M. Eddaoudi, D.B. Moler, M. O’Keeffe, O.M. Yaghi, J. Am. Chem. Soc. 123, 8239 (2001)
H. Deng, S. Grunder, K.E. Cordova, C. Valente, H. Furukawa, M. Hmadeh, F. Gándara, A.C. Whalley, Z. Liu, S. Asahina, H. Kazumori, M. O’Keeffe, O. Terasaki, J.F. Stoddart, O.M. Yaghi, Science 336, 1018 (2012)
E.R. Parnham, R.E. Morris, Acc. Chem. Res. 40, 1005 (2007)
R.E. Morris, Chem. Commun. 2990 (2009)
E.R. Parnham, E.A. Drylie, P.S. Wheatley, A.M.Z. Slawin, R.E. Morris, Angew. Chem. Int. Ed. 45, 4962 (2006)
H. Xing, J. Li, W. Yan, P. Chen, Z. Jin, J. Yu, S. Dai, R. Xu, Chem. Mater. 20, 4179 (2008)
L.M. Li, K. Cheng, F. Wang, J. Zhang, Inorg. Chem. 52, 5654 (2013)
L. Xu, B. Liu, S.X. Liu, H. Jiao, B. de Castro, L. Cunha-Silva, CrystEngComm 16, 10649 (2014)
L. Xu, E.Y. Choi, Y.U. Kwon, Inorg. Chem. 46, 10670 (2007)
L. Xu, E.Y. Choi, Y.U. Kwon, Inorg. Chem. Commun. 11, 150 (2008)
L. Xu, E.Y. Choi, Y.U. Kwon, Inorg. Chem. Commun. 11, 1190 (2008)
Z.H. Zhang, B. Liu, L. Xu, H. Jiao, Dalton Trans. 44, 17980 (2015)
M.M. Wang, Z. Wei, L. Xu, B. Liu, H. Jiao, Eur. J. Inorg. Chem. 2018, 932 (2018)
W. Tapala, T.J. Prior, A. Rujiwatra, Polyhedron 68, 241 (2014)
J.J. Wei, Q.Y. Liu, Y.L. Wang, N. Zhang, W.F. Wang, Inorg. Chem. Commun. 15, 61 (2012)
CrysAlis Pro, Version 1.171.38.43 (Rigaku OD, 2015)
G.M. Sheldrick, Acta Crystallogr. C 71, 3 (2015)
W. Xiong, W. Xu, Y. Zhou, D. Huang, J. Wang, M. Hong, C. Xiong, J. Mol. Struct. 1060, 75 (2014)
J. Zhang, Y.B. Chen, Z.J. Li, Y.Y. Qin, Y.G. Yao, Inorg. Chem. Commun. 9, 449 (2006)
W. Xu, Y. Zhou, D. Huang, W. Xiong, M. Su, K. Wang, S. Han, M. Hong, Cryst. Growth Des. 13, 5420 (2013)
X.L. Zhao, D. Tian, Q. Gao, H.W. Sun, J. Xu, X.H. Bu, Dalton Trans. 45, 1040 (2016)
W.Q. Tong, W.N. Liu, J.G. Cheng, P.F. Zhang, G.P. Li, L. Hou, Y.Y. Wang, Dalton Trans. 47, 9466 (2018)
X. Zhang, X. Luo, N. Zhang, J. Wu, Y.Q. Huang, Inorg. Chem. Front. 4, 1888 (2017)
Acknowledgements
This work was supported by Pearl River S&T Nova Program of Guangzhou (No. 201610010118), Natural Science Foundation of Guangdong Province (No. 2018A0303130021), the National Natural Science Foundation of China (No. 21301070), the Fundamental Research Funds for the Central Universities (No. 21615401), and Jinan University.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Mai, Z., Luo, X. & Lei, H. Ionothermal Synthesis of Zn(II) Coordination Polymers with Fluorescent Sensing and Selective Dye Adsorption Properties. J Inorg Organomet Polym 29, 1746–1754 (2019). https://doi.org/10.1007/s10904-019-01136-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10904-019-01136-w