
Abstract
Grapevine (Vitis vinifera L.) was genetically modified with a construct containing a cDNA insert encoding the stilbene synthase gene (Vst1) from grapevine, under the control of the cauliflower mosaic virus 35S promoter in order to test the potential of over-production of resveratrol to protect plants from fungal attack. Southern blot hybridization and quantitative real-time PCR analysis demonstrated the presence and integration of one copy of exogenous DNA sequences in two grapevine-modified lines. Relative expression of the Vst1 gene in different modified lines was confirmed by using gene-specific quantitative real-time PCR. Compared to the control, the concentration of trans-resveratrol quantified by HPLC was up to 7.5 fold higher in the modified plants. The necrotic lesion size of leaves of intact modified plants inoculated by Botrytis cinerea B05.10 strain was consistently smaller and significantly different (p ≤ 0.05) than in control plants, showing that modified grapevine plants were more resistant to the pathogen than the control plants.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Fungal diseases are one of the most serious problems in grapevine (Vitis vinifera L.) causing major economic losses worldwide because they determine a lower yield and quality of berry and wine. These fungi include powdery mildew (Uncinula necator), downy mildew (Plasmopara viticola) or grey mould caused by Botrytis cinerea. Stilbenes are phenolics acting like antifungal compounds and are synthesized by several species, including V. vinifera L., enabling the plant to overcome pathogen attack. Grapevine stilbenes include many compounds such as trans- and cis-resveratrol, their glycosides (piceid or polydatin), viniferins, pterostilbene, astringin, piceatannol, and other resveratrol trimers and tetramers. These substances are present in soft tissues as induced compounds (phytoalexins), and in woody tissues as constitutive ones (Bavaresco and Fregoni 2001). Production of resveratrol and other stilbenes is controlled by a key enzyme, stilbene synthase (STS), which condenses three malonyl-CoA molecules with one molecule of Coumaril-CoA to produce resveratrol, which can be metabolized, producing ε-viniferina (dimerization), pterostilbene (methylation) and piceid (resveratrol glucoside). In grapevine, STS belongs to a multigenic family (Jaillon et al. 2007; Velasco et al. 2007; Wiese et al. 1994) and some of these genes have been identified, isolated and cloned (Melchior and Kindl 1990; Sparvoli et al. 1994; Wiese et al. 1994).
Although defence mechanism in grapevine is not well understood, a positive correlation between the synthesis of resveratrol in leaves and the resistance to B. cinerea infection in different Vitaceae has been found by Langcake and McCarthy (1979). Recently, the effect of resveratrol in Botytis cinerea has been explained in terms of damages in conidia structure (Adrian and Jeandet 2012) similar to damages produced by other phytoalexins (Smith 1982). In addition, it has been shown that stilbenes are mainly produced at the level of the cell wall (Fornara et al. 2008) and that accumulates in high concentrations in contact with pathogen structures (Adrian and Jeandet 2012). Alternatively, the role of resveratrol as inducer of the expression of other phytoalexins has also been proposed (Delanouis et al. 2009). On the other hand, exogen regulation of resveratrol content in cell culture of Vitis amurensis has been achieved by the adition of a DNA demethylation agent (Kiselev et al. 2011). Raising resveratrol levels in grapevine by genetic engineering is an attractive option because of the potential of this substance to protect plants from fungal attack (Jeandet et al. 2002), without any alterations of the essential characters and identity of the cultivar. The process via genetic modification of embryogenic calli using Agrobacterium tumefaciens is available for different grapevine varieties (Coutos-Thévenot et al. 2001; Fan et al. 2008; Dabauza and Velasco 2012; Nookaraju and Agrawal 2012), enabling the transfer and expression of some STS genes and resveratrol accumulation in transformed plants or, for example, exogenous chitinase expression for downie mildew control in grapevine (Nookaraju and Agrawal 2012). Coutos-Thevenot et al. (Coutos-Thévenot et al. 2001) achieved transgenic plants of rootstock 41B with the grapevine stilbene synthase gene (Vst1) under the control of the pathogen-inducible PR10 promoter, and leaves of in vitro plants showed a large reduction of symptoms in response to infection by B. cinerea. Fan et al. (2008) obtained transgenic plants of grapevine Thompson Seedless expressing the STS gene from Vitis pseudoreticulata but infection by Botrytis was not tested. Vitis STS genes have also been transferred to other heterologous plant systems to produce stilbenes (see review by Delaunois et al. 2009). In tobacco and barley the transfer of STS genes led to resveratrol accumulation and increased resistance of transgenic plants to B. cinerea (Hain et al. 1993; Leckband and Lörz 1998).
In the present work genetically-modified plants of grapevine Sugraone over-expressing the Vst1 gene were recovered and the concentration of resveratrol found was up to 7.5 fold higher than in control plants. Infection of leaves of intact plants by B. cinerea showed that modified grapevine plants were more resistant to the pathogen than control plants.
Materials and methods
Bacterial strains and plasmids
Agrobacterium tumefaciens EHA105 strain (Hood et al. 1993), a disarmed derivative of A281, was used for in vitro transformation of grapevine embryogenic calli.
The binary plasmid pKSTS706 was constructed in our laboratory using the Gateway cloning system (Invitrogen). RNA from Red Globe berries was extracted and cDNA from the stilbene synthase gene was amplified by standard RT-PCR conditions using primers attB1fullVst1U1 (5′ GGGACAAGTTTGTACAAAAAAGCAGGCTCTTCCTCAACTTAATCTTAGGCCT) and attB2polyT (5′ GGGGACCACTTTGTACAAGAAAGCTGGGT). Standard RT-PCR conditions: initial reverse transcription step for 50 min at 37 °C using Mu-MLV and PCR cycles consisting in 2 min at 95 °C followed by 40 × (95 ºc 30 s, 55 °C 30 s, 72 °C 1 min) and a final extension step of 5 min at 72 °C. Next, cloning of cDNA by Gateway BP recombination with plasmid pDONR221 (Invitrogen) was performed to obtain plasmid pEDSTS3105 that was used to transform E. coli DH5α cells. Finally, after Gateway LR recombination between pEDSTS3105 and pK7WG2D (Karimi et al. 2005) plasmid pKSTS706 was obtained and transferred by transformation into E. coli DH5α cells. Restriction and sequencing analyses were carried out to check that the construction was correct. Electroporation of Agrobacterium tumefaciens EHA105 with this plasmid was carried out to obtain the strain EHA105-pKSTS706. The binary plasmid pKSTS706 contains the complete coding region for the Vst1 cDNA (Vvst1 gene) under the control of CaMV 35S promoter and terminator sequences and includes a kanamycin resistance gene under the control of the nopaline synthetase gene promoter and terminator, as well as a GFP expression module provided by the EgfpER gene (Fig. 1a).

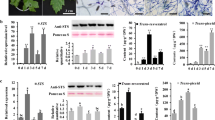
Southern blot hybridisation for the nptII gene and CaMV35S promoter. a Map of the T-DNA of pKSTS706 that includes the Vst1 cDNA with the restriction site for EcoRI used to digest plant genomic DNA and the position of the nptII and p35S probes. b Digested genomic DNA of E38 and E39 genetically-modified plants hybridised with the nptII and p35S probes
Genetic transformation and plant regeneration
Embryogenic calli from Sugraone maintained in 4/1.3 medium [MS (Murashige and Skoog 1962) with 100 mg l−1 casein hydrolysate, 100 mg l−1 calcium pantotenate, 10 mg l−1 biotin, 30 g l−1 sucrose, 4 µM 2,4-D and 1.3 µM BAP] was used for transformation experiments. Agrobacterium tumefaciens EHA105-pKSTS706 at OD600 = 0.2 were co-cultured with embryogenic calli for 2 days following the plant regeneration protocol described by López-Pérez et al. (2008) and Dabauza and Velasco (2012): calli were washed with MS + 900 mg l−1 cefotaxime for 20 min and cultured on ½ MSAC [modified MS (half strength of macroelements ½ MS), 100 mg l−1 myo-inositol, 30 g l−1 sucrose and 2.5 g l−1 of activated charcoal (AC)] (López-Pérez et al. 2005, 2006) with 300 mg l−1 cefotaxime for embryo differentiation. After 10 days, calli were transferred to the same medium with 50 mg l−1 kanamycin to select modified somatic embryos (½MSAC selection medium). The cultures were maintained in darkness at 26 ± 1 °C and sub-cultured to fresh medium every 30 days. GFP-positive somatic embryos were transferred onto germination medium (½ MS medium with 100 mg l−1 casein hydrolysate, 100 mg l−1 calcium pantotenate, 10 mg l−1 biotin, 20 g l−1 sucrose, 2.5 g l−1 AC, 10 µM IAA and 1 µM GA3) with cefotaxime (300 mg l−1) and maintained at 25 °C and 16:8 h photoperiod with a photosynthetic photon flux density of 45 μmol m−2 s−1 provided by Grolux fluorescent tubes (Sylvania). Germinated somatic embryos were cultured into test tubes on plant regeneration medium (½ MS, 100 mg l−1 myo-inositol, 20 g l−1 sucrose) with cefotaxime (300 mg l−1) for development. Plants were micropropagated on the same medium in 500 ml glass pots without cefotaxime. Non-transformed regenerated plants were used as control.
All media were solidified with 8 g l−1 Noble-agar (Difco), pH adjusted to 5.8 and autoclaved at 121 °C and 101 kPa for 15 min. Eight calli (0.08 g fresh weight per callus) were used and the experiment was repeated twice.
Calli were examined periodically under a Leica MZ16F fluorescence stereomicroscope with a GFP-Plant filter that does not block the red auto-fluorescence of chlorophyll. An HBO 100 W high-pressure mercury lamp provided the light source. Photographs were taken using a Canon Power Shot S50 digital camera. The number of genetically-modified somatic embryos was counted.
Acclimatisation of plants was carried out following the protocol described by López-Pérez et al. (2005). Well-rooted and elongated plants were transplanted into 10 cm pots with a mixture of 50 % peat and 50 % perlite, covered with a plastic bag and incubated in a growth chamber under constant conditions, 27 ± 2 °C, and 16:8 h photoperiod at a photosynthetic photon flux density of 70.74 μmol m−2 s−1 provided by white fluorescent tubes (Philips), for 2 weeks. Then, the plastic bag was gradually raised for 2 weeks before being completely removed. After two more weeks in the growth chamber, plants were transferred to greenhouse conditions for resveratrol analysis.
Molecular characterization of genetically-modified plants
Genomic DNA was isolated from young leaves according to Dellaporta et al. (1983). A standard PCR technique was used to determine the presence of the EgfpER and nptII genes, using specific primers that amplify the encoding sequences. The primer pairs used were 5′-ATGGTGAGCAAGGGCGAGGA-3′ and 5′-GGACCATGTGATC GCGCTTC-3′, that amplified a 600-bp fragment of the EgfpER gene and 5′-GACGAGGCAGCGCGGCTAT-3′ and 5′-AAGAAGGCGATAGAAGGCGA-3′, that amplified a 660 bp fragment of the nptII gene (Ghorbel et al. 1999). PCR reactions were performed in 25 μl containing 4 ng DNA, 0.4 mM dNTPs, 1.25 mM MgCl2, 0.4 μM of each primer, 1× buffer and 0.5 U of GoTaq DNA polymerase (Promega). Reactions were subjected to 40 cycles of 30 s at 95 °C, 30 s at 55 °C and 1 min at 72 °C and a final extension time of 5 min at 72 °C. In order to detect the introduced foreign Vst1 gene and to discriminate from the endogenous Vst1 gene, a region of 195 bp from the CaMV35S promoter was amplified using the primers 5′-GCTCCTACAAATGCCATCA-3′ and 5′-GATAGTGGGATTGTGCGTCA-3′ (Lipp et al. 1999) and PCR was subjected to 40 cycles in the same conditions described above.
For southern blot hybridization, genomic DNA of the samples (20 μg) was digested with EcoRI, separated on 1 % (w/v) agarose gels and blotted onto positively-charged nylon membranes (Hybond®, Roche, Basel, Switzerland) using a Bio-Rad 785 vacuum blotter system. Following the transfer, the DNA was UV cross-linked (0.120 Joules cm−2, 254 nm) to the membrane prior to overnight hybridisation with DNA DIG-labelled probes (20 ng ml−1) derived from the p35S region and the coding region of the nptII gene, obtained by PCR with the above-described primers (Fig. 1a). Detection of the hybridisation signals was carried out according to the manufacturer′s instructions (Roche).
Additionally, quantitative real-time PCR (RT-qPCR) analysis was performed for foreign Vst1 gene copy number determination and relative expression, by using the Bio-Rad iQ5 Thermal cycler and KAPA SYBR Green FAST qPCR Kit (KAPA Biosystems, Cape Town, South Africa). Individual samples were replicated three times in two independent experiments. All reactions were carried out in a 20 μl final volume containing 1 μl sample DNA (a total of 10 ng), 10 μl of SYBR Green qPCR mix, 2 μl of each primer from the 35S promoter (0.5 μM) and 5 μl sterile water. External controls representing 1–5 gene copies were prepared by diluting EcoRI-linearised plasmid pK7WG2D to a given concentration and adding the DNA to reaction mixtures that contained 10 ng of C2 genomic DNA (control plant). The amount of plasmid DNA corresponding to a single copy of the gene per reaction was calculated as 10 ng (genomic DNA amount) × 1.28 × 104 bp (size of pK7WG2D)/4.75 × 108 bp (1C genome size of grape) = 0.269 pg. The parameter used for foreign Vst1 gene copy number estimation was the crossing point (Cp) in qPCR analysis. The qPCR conditions were the following: 95 °C for 3 min followed by 40 thermal cycles of 95 °C for 15 s and 60 °C for 45 s. The level of SYBR-specific fluorescence (483–533 nm) at the end of each cycle was measured and recorded via a CCD camera connected to the Bio-Rad iQ5 Thermal cycler, and was further analysed using the software Bio-Rad iQ5 System Software v. 2.1. Following the thermal cycling, melting-curve analysis was performed to verify amplicon sequence specificity. The foreign Vst1 gene indicator used for copy number estimation was the 35S promoter.
The relative expression of the Vst1 gene was compared as the means of five biological replicates and three technical replicates from each of the three lines studied (E38, E39 and C2). RNA was extracted from the middle leaf of each plant using the Spectrum Plant Total RNA Kit (Sigma-Aldrich Chemie, GmbH) and treated with RNase-free DNase I (Promega, Madison, WI) to eliminate DNA. The RNA purity and yield were determined using a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE). RNA integrity was checked in 2 % agarose TBE gels. The synthesis of cDNA by RT was performed in a 20-µl reaction with 200 ng of RNA, random nonamers (Takara Bio), and Mu-MLV Reverse Transcriptase, following the manufacturer’s instructions (Eurogentec). The primer pair STS2-For (5′-ATCGAAGATCACCCACCTTG-3′) and STS2-Rev (5′-CTTAGCGGTTCGAAGGACAG-3′) used for the specific amplification of the Vst1 gene were designed with the Primer3 online tool (Rozen and Skaletsky 1999) (http://biotools.umassmed.edu/bioapps/primer3_www.cgi). For the internal reference genes Actin and NAD5, we used the primer pairs described by Gutha et al. (2010).
The qPCR was performed in white 96-well PCR plates (Bioplastics RV, The Netherlands) with 100 ng of cDNA template, 10 µl of SYBR Green qPCR mix and 500 nM of each primer in a 20 µl reaction volume. Samples were subjected to the following conditions: 95 °C for 3 min and 40 cycles of 95 °C for 15 s, 60 °C for 45 s. Product specificity for the Vst1 gene was determined with the software package Bio-Rad Optical System Software v.2.1, by melting-curve analysis of 60 s at 95 °C, 60 s at 55 °C, followed by fluorescence reading at 0.5 °C increments from 55 to 95 °C (Bustin et al. 2009).
The analysis of the relative expression of the Vst1 gene was based on the standard curve method described by Pfaffl (2001). Briefly, fold ratio determination was derived from the formula: ratio = Etarget EXP(∆Cq(target)(control-sample))/ErefEXP(∆Cq(ref)(control-sample)) where Etarget refers to the amplification efficiency for the gene (Vst1) and Eref is the amplification efficiency for the reference genes (Actin or NAD5). The ΔCq values of samples were determined and normalised to the mean value of five plants of the control line C2 included in each plate. For the calculus of the efficiency (E = 10EXP[−1/slope]), the Cq of a dilution series of the positive control (plasmid) in three replicates ranging from 100, 10−1, 10−2, 10−3, 10−4 and 10−5 were determined and the intercept and slope were calculated based on the linear regression. Similarly, the amplification efficiencies for the Actin and NAD5 reference genes were obtained. Because the Cq data were not normally distributed, the values were log10-transformed for the calculations.
Resveratrol analyses
For resveratrol analyses, two independent genetically-modified lines (E38 and E39) and one control plant (C2) were chosen. Five clones of each plant were transferred to the greenhouse and individual plant leaves were collected at 0, 15, 30, and 269 days after transfer (DAT). Sampling was performed on 2008/09/19, 2008/10/04, 2008/10/19 and 2009/06/16. Fully expanded third or fourth leaves on plants with 10–12 leaves were detached, rinsed with demineralised water and stored at −20 °C in the freezer until further analysis. Frozen leaves were ground with a mortar and pestle in liquid nitrogen and a known concentration of keracyanin chloride was added as internal standard. Resveratrol was extracted with methanol, followed by centrifugation at 10,000 rpm (14,136×g) for 20 min to separate the insoluble residue. The extracts were evaporated to dryness under vacuum and dissolved in methanol/formic acid (97:3 v/v) before HPLC analysis. All procedures were carried out under reduced light.
Samples of 20 μl of extract were analysed using an HPLC system (Hewlett-Packard, Germany) equipped with photodiode array UV/Vis and fluorescence detectors. Separation was performed on a 250 mm × 4 mm i.d., 5 µm reversed phase Lichrocart C18 column (Merck, Germany) with 6 % acetic acid (A) and 5:30:65 acetic acid: acetonitrile: ultra pure water (B) as the mobile phase at a flow-rate of 0.4 ml min−1. The chromatograms were recorded at 510 and 306 nm by a UV detector and, for fluorimetric detection the maximum excitation wavelength was measured at 270 nm and emission at 372 nm (Roggero and García-Parrilla 1995). Pure resveratrol (trans-3, 4′5-trihydroxystilbene) and keracyanin chloride were purchased from Sigma Chemical Co. (USA) and used as external and internal standards, respectively. The peak assignment of resveratrol was based on the retention time of the standard and calibration curves were plotted versus known concentrations of commercial metabolite. Resveratrol concentration was expressed as μg g−1 fresh weight (FW). Each analysis was carried out in triplicate.
Plant inoculation with Botrytis cinerea
B. cinerea strain B05.10 is a putative haploid strain obtained after treatment with benomyl of a field isolate collected from Vitis (Buttner et al. 1994). It was grown on potato dextrose agar (Difco Laboratories, Detroit, MI) in dark chamber at 22–24 °C. Grapevine plants grown in the greenhouse for 2 years were used. For inoculation experiments (June, 2010), a total of five E38 genetically-modified clones and three C2 clones (control) were transferred to the same growth chamber described for plant acclimatisation, inoculated with B. cinerea and maintained into a methacrylate box (73 × 55 × 50 cm) in order to obtain high humidity conditions (provided by glass bottles filled with water). For inoculations, agar plugs (5 mm in diameter) from the growing edge of non-sporulating 3-day-old PDA B05.10 cultures were placed with the mycelium in contact with the leaf surface on intact plants. For control treatment, plugs of sterile PDA agar were used. Two leaves per plant were inoculated, each with two plugs (20 replicates for E38 and 12 for C2), and the experiment was repeated twice. The position of each infection site was marked for future reference. Daily (1–8 days), for each lesion (replicate) the two perpendicular diameters (length and width) were measured and the mean was calculated. Necrotic lesion diameter was estimated by calculating the average of the lesion size for each genotype. The values were log10-transformed for the calculations.
Statistical analyses
One-way analysis of variance was used to compare the means for resveratrol analyses, Vst1 gene expression and necrotic lesion size. The Student–Newman–Keuls (SNK) multiple range test was used to determine significant differences among groups for Vst1 gene expression and the Least Significant Differences (LSD) test for resveratrol analyses and for B. cinerea necrotic lesion development. Data were subjected to log-transformation. All effects were tested at the 5 % significance level. For the statistical analysis, the SPSS v.15 (Chicago, IL, USA) statistical package was used.
Results and discussion
Regeneration of modified plants and molecular characterization
After culture on selection medium (½ MSAC with kanamycin), 26 independent modified plants were obtained, micropropagated and acclimatised following the regeneration conditions specified in “Materials and methods” section. In grapevine, the stilbene synthase is encoded by a multigene family and several chalcone synthase genes have 70 % homology with the stilbene synthase (Jeandet et al. 2002), which makes difficult to analyse by PCR or Southern blot the integration of any grapevine STS genes. In our study, the expression of the Vst1 gene was controlled by the constitutive CaMV35S promoter, and the use of specific primers for this promoter allowed us to analyse the integration of the cisgene Vst1 gene by PCR, Southern blot and RT-qPCR analyses. PCR of several putative genetically-modified plants showed the amplification of the three genes transferred in our transformation approach, a 600 bp fragment for EgfpER, a 660 bp fragment for nptII and a 195 bp fragment for the CaMV35S promoter, the latter revealing the integration of the Vst1 gene. Then, Southern Blot analysis for the nptII gene and for the p35S carried out for two genetically-modified plants (E38 and E39) indicated that just one single, stable integration event had occurred in each plant (Fig. 1B). No hybridisation signals were detected in non-modified control C2 for either probe. In addition, the Vst1-gene copy number was estimated by quantitative real-time PCR (as Dutt et al. 2008 and Dhekney et al. 2009; 2011) according to the Cp values of the 35S promoter compared with an equivalent copy number of plasmid dilutions. The results for E38 and E39 indicated that one single copy of the foreign Vst1 gene was found in both lines and none in the non-modified control (Table 1).
Different strategies have been adopted to analyse grapevine plants engineered with genes of the same species. Coutos-Thévenot et al. (2001) modified the 41B rootstock grapevine with the cDNA of the Vst1 gene and performed Southern blot with a probe of the nptII gene, as an indirect approach to test transformation by the chimerical gene. Dhekney et al. (2011) used a specific forward primer at the junction region between the core promoter (duplicated CaMV35S enhancer) and the vvtl-1 cisgene (thaumatin-like protein gene) start codon, and a reverse primer at the junction region between the stop codon of the cisgene and the terminator sequence (35S-T) to characterise engineered grapevines. Fan et al. (2008) transformed V. vinifera cv. Thompson Seedless with an STS gene from V. pseudoreticulata and used specific STS primers and probes to analyse the transgenic plants.
Relative expression of Vst1 gene and resveratrol analyses
Quantification of molecular targets using real-time RT-qPCR requires a linear relationship between the threshold PCR cycle (Ct) defined during the exponential increase of the reporter’s fluorescence and the logarithm of the initial target quantity in the corresponding sample. The expression pattern of the Vst1 gene was examined in the control line and in E38 and E39 modified lines. Using two previously-described reference genes, Actin and NAD5 (Gutha et al. 2010), Cq data from Vst1 real-time PCR experiments were normalised and the relative expression levels are shown in Fig. 2a.

Relative expression of Vst1 gene and resveratrol analyses. a Relative expression level of the Vst1 gene in leaves of the C2 control and E38 and E39 genetically-modified grapevine plants by using gene-specific RT-qPCR. Columns represent mean values from five biological replicates. Fold increase values were normalised to the reference genes Actin (black) or NAD5 (grey). Bars represent the SE of the means. b Concentration of trans-resveratrol (μg g−1 FW) in leaves of grapevine at 0, 15, 30 and 269 days after transfer (DAT) of plants to greenhouse conditions. Significant differences between the control and the genetically-modified plants were determined by one-way ANOVA, using the SPSS software v. 15 (p < 0.05), values are mean ± SE (n = 5)
For the specific amplification of the Vst1 mRNA, the primer pair STS2-for and STS2-rev was designed (see “Materials and methods”). The cDNA obtained was of the expected size (163 bp) and was cloned and sequenced, confirming the identity of the gene. Dissociation curve analysis done at the end of the RT-qPCR also demonstrated that the primers pair amplified a single PCR product (data not shown). Serial dilutions in three replicas of target Vst1 and the reference genes Actin and NAD5 cloned DNAs were performed in order to obtain solutions containing from 100 to 10−5 copies of the target and internal controls sequences, in 1 μl. The standard curves showed a linear relationship between the amount of input DNA and the CT values for the three templates over a range of six log units (not shown). Curves showed high correlation coefficients and were linear within the range of the standard and experimental DNA concentrations examined. Amplification efficiencies were calculated resulting in 2.10 for Vst1, 1.97 for Actin and 1.89 for NAD5.
According to SNK analysis, Vst1 gene expression was significantly (p < 0.05) higher in both E38 and E39 modified lines with respect to the C2-control plant due to the additional copy of this gene engineered introduced. When the reference gene Actin was used, Vst1 expression was, on average, eight-fold higher for the E38 than for the control. For the E39, the fold increase was five times with respect to the control. When the reference gene considered is NAD5, Vst1 expression in E38 was 15-times higher with respect to the control while only 3.5-fold higher regarding E39.
As resveratrol production depends on both biotic and abiotic factors, including the physiological stage of the plant (Jeandet et al. 2002), leaves of E38 and E39 modified plants were analysed for resveratrol concentration by HPLC from the day of transfer to greenhouse conditions (day 0) until several months after. The concentration of trans-resveratrol was higher in modified plants than in the control, regardless of the sampling time (Fig. 2b). At 15 days after transfer to the greenhouse, trans-resveratrol was increased in modified plants, up to 7.5 and 5.9 μg g−1 FW for E38 and E39, respectively, and then was stabilised. Thus, at 269 DAT, the same plants maintained the resveratrol levels.
The two modified Sugraone plants analysed by Southern blot and qPCR (E38 and E39) showed only one copy of the engineered Vst1 gene introduced by transformation. The insertion of this gene under the control of a CaMV35S promoter enhances the synthesis of the Vst1 specific mRNA. The increased levels of Vst1 transcripts may explain the higher resveratrol production in E38 and E39 plants compared with the non-modified control. Moreover, the increase in the synthesis of specific mRNA is in the same order that the level of resveratrol quantified by HPLC (7.5–8 fold). The grapevine transgenic line tested by Fan et al. (2008) also carried a single copy of the STS gene and it was able to produce fivefold greater resveratrol than the control. Coutos-Thévenot et al. (2001) refer to the expression (by RNA blot analysis) of the Vst1 gene in modified 41B plants, and resveratrol over-production after UV-induction or fungus infection (2–100-fold increase over the control), but do not refer to the copy number of the gene. Single gene insertion in transgenic plants is much more useful for eventual breeding purposes (Fan et al. 2008) although some studies showed no definite relationship between copy number of inserted genes and level of enzyme activity (Flavell 1994; Stam et al. 1997; Vaucheret et al. 1998; Zhu et al. 2004).
It has been described that constitutive expression of STS gene and resveratrol accumulation caused alterations in plant development, such as modification of the morphology and the colour of the flowers, and induced male sterility in transgenic tobacco (Fischer et al. 1997, using a duplicated upstream region of the 35S promoter from CaMV). An increase in the levels of soluble antioxidants of primary metabolism in transgenic tomato fruit (Giovinazzo et al. 2005, using a CaMV 35S promoter) or alterations in the phenylpropanoid metabolism (Hanhineva et al. 2009) has also been described. In our case the use of the constitutive 35S promoter did not alter the normal growth of grapevine-modified plants. Hain et al. (1993) obtained also normal development of transgenic tobacco plants.
Susceptibility of genetically modified plants to Botrytis cinerea
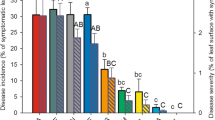
Possible alterations in the resistance of E38-modified plant to B. cinerea in comparison to the C2-control plant were investigated by evaluating the virulence of the B. cinerea B05.10 strain in artificial inoculations carried out on intact plants under controlled conditions. Leaves were inoculated with agar plugs from actively growing fungal culture plates and lesion progression was followed during 8 days. At all the time points considered, both the control and modified plants developed clear lesions (Fig. 3). However, the necrotic lesion size measured in the group of modified plants was consistently smaller and significantly different (p ≤ 0.05) than in the group of control plants (Fig. 4). Moreover, disease progression was much slower in the modified plants; and from day 6 onwards lesion expansion in these plants was almost undetectable (Fig. 3) suggesting a higher level of resistance to B. cinerea than the control plants.

Development of disease and symptom production in C2 control and E38 genetically-modified grapevine plants inoculated with B. cinerea B05.10 strain. a–f C2 plant inoculated at 3, 4, 5, 6, 7 and 8 days post-inoculation (DPI), g–l E38 plant inoculated at 3, 4, 5, 6, 7 and 8 DPI. Bar 10 mm

Necrotic lesion size in C2 control and E38 genetically-modified grapevine plants inoculated with B. cinerea B05.10. The values show the average of the lesion size of 20 inoculations in modified plants (5 E38 plants × 4 inoculations per plant) and 12 inoculations in control plants (3 C2 plants × 4 inoculations per plant) ± SE, at different days post-inoculation. Significant differences between the control and the genetically-modified plants were determined by one-way ANOVA, using the SPSS software v. 15 (p < 0.05), values are mean ± SE
Our results are in agreement with Langcake and McCarthy (1979) and Douillet-Breuil et al. (1999) that found a positive correlation between the resveratrol level in grapevine cultivars and their resistance to B. cinerea. Coutos-Thévenot et al. (2001), reported that in vitro transgenic plants of grapevine with the chimeric Pr10prom-Vst1 construct increased resveratrol production during fungal infection, and were more tolerant, showing a large reduction of symptoms. In some heterologous plant systems, like in tobacco and barley, the transfer and expression of STS genes led to resveratrol accumulation in transgenic plants and to a higher resistance to B. cinerea infection (Hain et al. 1993; Leckband and Lörz 1998). The mode of action of resveratrol as antimicrobial agent either as a direct effect on the pathogen or due to its role as a precursor of other phytoalexins reinforces its present and future utilization in the control of microbial diseases using transgenic approach or exogen compounds as regulators (Delaunois et al. 2009; Kiselev et al. 2011).
In the current work, genetically-modified grapevine plants showed a positive correlation between the expression level of the Vst1 gene (one additional copy), the resveratrol content and the increased resistance to B. cinerea compared to the control, showing the benefits of application of the recombinant DNA technology for enhancing resistance of plants to fungal pathogens.
Abbreviations
- GFP:
-
Green fluorescent protein
- MS:
-
Murashige and Skoog (1962)
- ½ MSAC:
-
Half strength of MS macronutrients salts plus active charcoal
- Vst1 :
-
Vitis stilbene synthase 1 gene
- RT-qPCR:
-
Quantitative real-time PCR
- CaMV35S (p35S):
-
Cauliflower mosaic virus 35S promoter
References
Adrian A, Jeandet P (2012) Effects of resveratrol on the ultrastructure of Botrytis cinerea conidia and biological significance in plant/pathogen interactions. Fitoterapia 83:1345–1350
Bavaresco L, Fregoni C (2001) Physiological role and molecular aspects of grapevine stilbenic compounds. In: Roubelakis-Angelakis KA (ed) Molecular biology and biotechnology of the grapevine. Kluwer Academic Pub, Dordrecht, pp 153–182
Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT (2009) The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem 55(4):611–622
Buttner P, Koch F, Voigt K, Quidde T, Risch S, Blaich R, Bruckner B, Tudzynski P (1994) Variations in ploidy among isolates of Botrytis cinerea: implications for genetic and molecular analyses. Curr Genet 25:445–450
Coutos-Thévenot P, Poinssot B, Bonomelli A, Yean H, Breda C, Buffars D, Esnault R, Hain R, Boulay M (2001) In vitro tolerance to Botrytis cinerea of grapevine 41B rootstock in transgenic plants expressing the stilbene synthase Vst1 gene under the control of a pathogen-inducible PR 10 promoter. J Exp Bot 52:901–910
Dabauza M, Velasco L (2012) Development of highly efficient genetic transformation protocols for table grape Sugraone and Crimson Seedless. In: Dunwell JM, Wetten AC (eds) Transgenic Plants: Methods and Protocols. Methods in Molecular Biology, 2nd edn, vol 847. Springer Science + Business Media, LLC, pp 227–235
Delaunois B, Cordelier S, Conreux A, Clément C, Jeandet P (2009) Molecular engineering of resveratrol in plants. Plant Biotechnol J 7:2–12
Dellaporta SL, Wood J, Hicks JB (1983) A plant DNA minipreparation: version II. Plant Mol Biol Rep 1(14):19–21
Dhekney SA, Li ZT, Zimmerman TW, Gray DJ (2009) Factors influencing genetic transformation and plant regeneration of Vitis. Am J Enol Vitic 60:285–292
Dhekney SA, Li ZT, Gray DJ (2011) Grapevines engineered to express cisgenic Vitis vinifera thaumatin-like protein exhibit fungal disease resistance. In Vitro Cell Dev Biol Plant 47:458–466
Douillet-Breuil AC, Jeandet P, Adrian M, Bessis R (1999) Changes in the phytoalexin content of various Vitis spp. in response to ultraviolet C elicitation. J Agric Food Chem 47:4456–4461
Dutt M, Li ZT, Dhekney SA, Gray DJ (2008) A co-transformation system to produce transgenic grapevines free of marker genes. Plant Sci 175:423–430
Fan C, Pu N, Wang X, Wang Y, Fang L, Xu W, Zhang J (2008) Agrobacterium-mediated genetic transformation of grapevine (Vitis vinifera L.) with a novel stilbene synthase gene from Chinese wild Vitis pseudoreticulata. Plant Cell Tissue Organ Cult 92:197–206
Fischer R, Budde I, Hain R (1997) Stilbene synthase gene expression causes changes in flower colour and male sterility in tobacco. Plant J 11:489–498
Flavell RB (1994) Inactivation of gene expression in plants as a consequence of specific sequence duplication. Proc Natl Acad Sci USA 91:3490–3496
Fornara V, Onelli E, Sparvoli F, Rossoni M, Aina R, Marino G et al (2008) Localization of stilbene synthase in Vitis vinifera L. during berry development. Protoplasma 233:83–93
Ghorbel R, Juárez J, Navarro L, Peña L (1999) Green fluorescent protein as a screenable marker to increase the efficiency of generating woody fruit plants. Theor Appl Genet 99:350–358
Giovinazzo G, d’Amico L, Paradisio A, Bollini R, Sparvoli F, DeGara L (2005) Antioxidant metabolite profiles in tomato fruit constitutively expressing the grapevine stilbene synthase gene. Plant Biotechnol J 3:57–69
Gutha LR, Casassa LF, Harbertson JF, Naidu RA (2010) Modulation of flavonoid biosynthetic pathway genes and anthocyanins due to virus infection in grapevine (Vitis vinifera L.) leaves. BMC Plant Biol 10:187. doi:10.1186/1471-2229-10-187
Hain R, Reif HJ, Krause E, Langebartels R, Kindl H, Vornam B, Wiese W, Schmelzer E, Schreier PH, Stöcker RH, Stenzel K (1993) Disease resistance results from foreign phytoalexin expression in a novel plant. Nature 361:153–156
Hanhineva K, Kokko H, Siljanen H, Rogachev I, Aharoni A, Kärenlampi SO (2009) Stilbene synthase gene transfer caused alterations in the phenylpropanoid metabolism of transgenic strawberry (Fragaria × ananassa). J Exp Bot 60(7):2093–2106
Hood EE, Gelvin SB, Melchers LS, Hoekema A (1993) New Agrobacterium helper plasmids for gene transfer to plants. Transgenic Res 2:208–218
Jaillon O, Aury JM, Noel B et al (2007) The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449:463–467
Jeandet P, Douillet AC, Debord S, Sbaghi M, Bessis R, Adrian M (2002) Phytoalexins from the Vitaceae: biosynthesis, phytoalexin gene expression in transgenic plants, antifungal activity, and metabolism. J Agric Food Chem 50:2731–2741. doi:10.1021/jf011429s
Karimi M, De Meyer B, Hilson P (2005) Modular cloning and expression of tagged fluorescent protein in plant cells. Trends Plant Sci 10(3):103–105
Kiselev KV, Tyunin AP, Manyakhin AY, Zhuravlev YN (2011) Resveratrol content and expression patterns of stilbene synthase genes in Vitis amurensis cells treated with 5-azacytidine. Plant Cell Tisse Organ Cult 105(1):65–72
Langcake P, McCarthy W (1979) The relationship of resveratrol production to infection of grapevine leaves by Botrytis cinerea. Vitis 18:244–253
Leckband G, Lörz H (1998) Transformation and expression of a stilbene synthase gene of Vitis vinifera L. in barley and wheat for increased fungal resistance. Theor Appl Genet 96:1004–1012
Lipp M, Brodmann P, Pietsch K, Pauwels J, Anklam E (1999) IUPAC collaborative trial study of a method to detect the presence of genetically modified organisms in soy beans and maize. J AOAC Int 82:923–928
López-Pérez AJ, Carreño J, Martinez-Cutillas A, Dabauza M (2005) High embryogenic ability and plant regeneration of table grapevine cultivars (Vitis vinifera L.) induced by activated charcoal. Vitis 44:79–85
López-Pérez AJ, Carreño J, Dabauza M (2006) Somatic embryo germination and plant regeneration of three grapevine cvs: effect of IAA, GA3 and embryo morphology. Vitis 45:141–143
López-Pérez AJ, Velasco L, Pazos-Navarro M, Dabauza M (2008) Development of highly efficient genetic transformation protocols for table grape Sugraone and Crimson Seedless at low Agrobacterium density. Plant Cell Tissue Organ Cult 94(2):189–199
Melchior F, Kindl H (1990) Grapevine stilbene synthase cDNA only slightly differing from chalcone synthase cDNA is expressed in Escherichia coli into a catalytically active enzyme. FEBS Lett 268:17–20
Murashige T, Skoog F (1962) A revised medium for rapid growth and bio-assays with tobacco tissue cultures. Physiol Plant 15:473–497
Nookaraju A, Agrawal D (2012) Enhanced tolerance of transgenic grapevines expressing chitinase and β-1,3-glucanase genes to downy mildew. Plant Cell Tissue Organ Cult 111(1):15–28
Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29(9):e45
Roggero JP, García-Parrilla C (1995) Effects of ultraviolet irradiation on resveratrol and changes in resveratrol and various of its derivatives in the skins of ripening grapes. Sci Aliment 15:411–422
Rozen S, Skaletsky H (1999) Primer3 on the WWW for general users and for biologist programmers. In: Misener S, Krawetz SA (eds) Bioinformatics methods and protocols: methods in molecular biology, vol 132. Humana Press, Clifton, pp 365–386
Smith DA (1982) Toxicity of phytoalexins. In: Bailey JA, Mansfield JW (eds) phytoalexins. Blackie & Sons, Ltd., Glasgow, pp 218–252
Sparvoli F, Martin C, Scienza A, Gavazzi G, Tonelli C (1994) Cloning and molecular analysis of structural genes involved in flavonoid and stilbene biosynthesis in grape (Vitis vinifera L.). Plant Mol Biol 24:743–755
Stam M, Mol JNM, Kooter JM (1997) The silence of genes in transgenic plants. Ann Bot Lond 79:3–12
Vaucheret H, Beclin C, Elmayan T, Feuerbach F, Godon C, Morel JB, Mourrain P, Palauqui JC, Vernhettes S (1998) Transgene induced gene silencing in plants. Plant J 16:651–659
Velasco R, Zharkikh A, Troggio M, Cartwright DA, Cestaro A, Pruss D, Pindo M, FitzGerald LM, Vezzulli S et al (2007) High quality draft consensus sequence of the genome of a heterozygous grapevine variety. PLoS ONE 2:e1326
Wiese W, Vornam B, Krause E, Kindl H (1994) Structural organization and differential expression of three stilbene synthase genes located on a 13 kb grapevine DNA fragment. Plant Mol Biol 26:667–677
Zhu YJ, Agbayani R, Jazckson MC, Tang CS, Moore PH (2004) Expression of the grapevine stilbene synthase gene VST1 in papaya provides increased resistance against diseases caused by Phytophthora palmivora. Planta 12:807–812
Acknowledgments
We thank Dr. L. Peña for providing the EHA105 Agrobacterium strain, M. López-Romero, A. Palazón and V. Arnau for their excellent technical assistance and Dr. Walker for his critical review of the manuscript. This research was supported by the Instituto Murciano de Investigación y Desarrollo Agrario y Alimentario (IMIDA, PR06-002 Project) and by a fellowship provided by IMIDA to M. Pazos-Navarro. M. Dabauza, P. Flores and P. Hellín were co-supported by the European Social Fund and IMIDA.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Dabauza, M., Velasco, L., Pazos-Navarro, M. et al. Enhanced resistance to Botrytis cinerea in genetically-modified Vitis vinifera L. plants over-expressing the grapevine stilbene synthase gene. Plant Cell Tiss Organ Cult 120, 229–238 (2015). https://doi.org/10.1007/s11240-014-0598-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11240-014-0598-x