
Abstract
Turmeric (Curcuma longa L.) is a rhizomatous species belonging to the Zingiberaceae and known both for its culinary and medicinal uses. Based on an efficient tissue culture and somatic embryogenesis system that we established, we have developed a reliable Agrobacterium-mediated transformation protocol for this species. Calli derived from turmeric inflorescences were used as source tissues for transformation. Factors affecting transformation and regeneration efficiency were evaluated, including callus induction and culture conditions, Agrobacterium strains, co-cultivation conditions, selection agent sensitivity and bacterial elimination, and transformant selection. Optimized transformation conditions were identified, including: use of Agrobacterium strain EHA105 with plasmid pBISN1 for infection; a modified B5 medium system for callus induction, subculture, co-culture and selection; and MS media for transformant regeneration. Transgenic plants and their vegetative (clonal) progeny stably expressed the transgene as indicated by GUS assay, PCR and Southern blot analysis. In addition, a transient gene expression system was developed that involves Agrobacterium infiltration of young turmeric leaves followed by in vitro regeneration of plantlets. This approach established that a MADS-box-GFP fusion protein was localized to the nucleus of turmeric cells. The stable transformation and transient expression systems described herein offer opportunities for assaying gene function in turmeric and for improving turmeric properties.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Turmeric (Curcuma longa L.), a member of the ginger family, the Zingiberaceae, is a perennial rhizomatous sub-tropical and tropical herb species and has a long history of use as a traditional medicine as well as an important spice in southern and southeastern Asia (Gang and Ma 2008). Turmeric rhizomes contain important pharmacologically active metabolites such as curcumin, which is well-known to have chemopreventive activities against Alzheimer’s disease (Balasubramanian 2006) and therapeutic effects related to reducing inflammation in many chronic diseases (Aggarwal and Sung 2009), and has even been shown to help correct cystic fibrosis defects (Egan et al. 2004).
Cultivated turmeric is a sterile polyploid species that is propagated only clonally via its underground rhizomes. This vegetative propagation is susceptible to accumulation and transmittance of pathogens and soil-born diseases, and amplification of particularly useful stocks is a slow process. It is essential to develop new strategies that combine tissue culture and genetic engineering techniques to complement breeding programs, and effective transformation approaches for identification of gene function and improvement of physiological traits for this species. So far, tissue culture techniques associated with this species have been restricted to in vitro micropropagation and plant regeneration from callus culture (Hashemy et al. 2009; Ma and Gang 2006; Mohanty et al. 2008; Salvi et al. 2002; Shirgurkar et al. 2001; Singh et al. 2011).
The use of transgenic techniques has become a common and convenient method for improvement of plants' traits and the functional analysis of genes involved in different physiological, biochemical, and molecular mechanisms of metabolic pathways. Particle bombardment and Agrobacterium-mediated transformation are two main approaches that have been utilized to obtain transgenic plants. Agrobacterium is perceived to have advantages over biolistics because it can introduce larger segments of DNA with minimal rearrangement and with fewer copies of inserted transgenes at higher efficiencies and at lower cost (He et al. 2010a, b; Hiei et al. 1997; Shewry et al. 2008; Shibata and Liu 2000). Although a particle bombardment-based approach has been reported (Shirgurkar et al. 2006), no reliable method describing an Agrobacterium-mediated approach has been published to date for this species. In this report, in addition to establishment of an efficient somatic embryogenesis and regeneration method, we describe the development of a leaf-based transient expression system and callus-based stable transformation system mediated by Agrobacterium tumefaciens in turmeric.
Materials and methods
Plant material, Agrobacterium strains, and plasmid
Plants of turmeric (C. longa) line T3C were grown in the greenhouse of the Institute of Biological Chemistry, Washington State University, in 10 L pots with coconut fiber and with supplemental lighting (sodium vapor lamps) to achieve 16-h photoperiods and were kept well watered as previously described (He et al. 2012; Ma and Gang 2006). Three A. tumefaciens strains, LBA4404 (Ooms et al. 1982), EHA105 (Hood et al. 1993) and AGL1 (Lazo et al. 1991), containing the binary vector pBISN1 (Narasimhulu et al. 1996), were used in this research. The binary vector pBISN1 contains the neomycin phosphotransferase (NPT II) gene under the control of Pnos (nopaline synthase promoter) and the β-glucuronidase (GUS)-intron construct that allows expression in plants but not in bacteria of a functional reporter gene, which is linked to the cauliflower mosaic virus (CaMV) 35S promoter.
Callus induction, subculture, and precultivation
Callus was induced from turmeric rhizome buds, shoot tips, leaf bases, and inflorescence tissue using the method described previously (Ma and Gang 2006), and subcultured and precultured on B5I medium (Table 1).
Selection agent sensitivity experimentation
Non-transformed calli were tested for regeneration on selective regeneration media MSR (Table 1) containing increasing concentrations of kanamycin (Kan: 0, 100, 200, 300, 400, 500 and 600 mg L−1). Approximately 60 explants/calli (four plates/replicates, 15 calli/plate) were used for each treatment. Cultures were grown at room temperature under a 16 h light, 8 h dark photoperiod. Calli were subcultured every 2 weeks. Eight weeks later, the number of calli generating shoots and surviving (excluding necrotic calli) were recorded.
Agrobacterium preparation, infection, and cocultivation
Agrobacterium strains LBA4404 (pBISN1) and EHA105 (pBISN1) and AGL1 (pBISN1) were grown on solid LB medium supplemented with 100 mg L−1 Kan at 28 °C for 3 days. Bacterial cells were resuspended to an OD600 = 0.5 in liquid medium B5A (Table 1), followed by shaking (150 rpm) at 28 °C for 1 h. Freshly precultured (for a week) calli were immersed in bacterial suspension for 20 min with gentle shaking (80 rpm). The excess bacteria were removed by decanting the liquid. The calli were transferred to sterile filter papers for blot-drying and then placed on cocultivation medium B5C (Table 1) to cocultivate in the dark at 24 °C for 3 days.
Selection and regeneration
After coculture, the infected calli were washed (thoroughly and gently) in sterile water five times until the rinse water became clear. They were then washed twice (20 min each time) with sterile water containing 400 mg L−1 of timentin with gentle shaking (80 rpm), blotted dry on filter paper, and placed on resting medium B5R (Table 1) in the dark at 26 ± 2 °C for 2 weeks. The calli were next transferred to selection medium B5S (Table 1) and subcultured every 2 weeks. The cultures were kept in the dark at 26 ± 2 °C for 8–10 weeks until resistant calli proliferated. The Kan-resistant calli, selected on media containing 400 mg L−1 Kan, were moved to preregeneration medium MSP (Table 1) for 2 weeks. Afterwards, the growing calli were cultured on regeneration medium MSR (Table 1) for 6–8 weeks. When the shoots developed into ~2 cm plantlets, they were transferred to magenta boxes containing root-growing medium MSG (Table 1) and were grown under the same conditions for 2–3 weeks. Well-rooted plants were then transferred to soil in pots and grown in a greenhouse.
GUS assay
Calli selected for regeneration and regenerated plantlets were stained for GUS activity by incubation at 37 °C overnight in a buffer containing 1 mM X-gluc (Sigma) dissolved in dimethylsulfoxide (DMSO), 100 mM sodium phosphate buffer pH 7.0, 0.5 mM potassium ferricyanide, 0.5 mM potassium ferrocyanide and 0.1 % Triton X-100. Chlorophyll was removed by soaking the tissues for 2 h in 70 % ethanol after staining.
Transgene detection via PCR and Southern blot analysis
For PCR analysis, DNA was isolated from 100 mg fresh leaves of the control and the Kan-resistant turmeric plantlets using the DNeasy Plant Mini Kit (Qiagen, Valencia, CA, USA). PCR was performed using two sets of primers specific to the gus sequence (5′-TCGCGAAAACTGTGGAATTGATC-3′, 5′-AGCCGACAGCAGCAGTTTCAT-3′), and the nptII sequence (5′-TCGGCTATGACTGGGCACAACAGA-3′, 5′-AAGAAGGCGATAGAAGGCGATGCG-3′), respectively. The PCR reactions were run using the following reaction conditions with a final volume of 25 μL: (1) 10 mM primers (each primer in the pair); (2) 20 ng genomic DNA or 200 ng cDNA; (3) 10× buffer; (4) 0.5 U Taq. PCR was carried out with the following program: 94 °C for 5 min for one cycle; 94 °C for 50 s, 55 °C for 50 s, and 72 °C for 80 s for 34 cycles; 72 °C for 10 min for one cycle. PCR products were analyzed by gel electrophoresis on 1 % agarose gels.
For Southern blot analysis, genomic DNA was extracted from young leaves of the control and T0 transgenic turmeric plants using the hexadecyltrimethylammonium bromide (CTAB, Sigma) method (Sambrook and Russell 2001). 20 μg of genomic DNA was digested overnight with restriction enzymes BamHI and EcoRI, fractionated on 0.8 % agarose gel in 1× TAE buffer for 14 h at 40 V. After depurination, denaturation and neutralization treatments, DNA was transferred onto a Hybond-N+ membrane (Amersham, Inc.). A digoxigenin (DIG)-dUTP labeled probe was prepared using the nptII primers described above and plasmid DNA as template with a PCR DIG probe synthesis kit (Roche). The blot was prehybridized at 65 °C with standard hybridization buffer (5× SSC, 0.1 % N-lauroylsarcosine, 0.02 % SDS and 1 % Blocking Solution (Roche)) for 3 h and hybridized with DIG labeled 722-bp PCR amplified product of the nptII gene for 16 h. Following hybridization, the membrane was washed twice in low stringency buffer (2× SSC containing 0.1 % SDS) for 20 min at room temperature and then twice in high stringency buffer (0.5× SSC containing 0.1 % SDS) for 30 min at 65 °C. Signals and bands on the membrane were visualized using anti-DIG-AP and the chemiluminescent substrate CSPD (Roche), and X-ray film (Kodak) according to the manufacturer’s instructions.
Agrobacterium infiltration of turmeric leaves, GFP transient expression and the fused gene localization
cDNA of a ginger MADS box gene (GT_01880) (without stop codons) was cloned into the pCR8/GW/TOPO vector (Invitrogen) with a BP clonase reaction to generate entry vectors (pCR8/GW/TOPO-gMADS1). Positive clones were then used in an LR clonase reaction for recombination into the pEarleyGate103 destination vector, which contains the 35S promoter and a C-terminal fusion to the enhanced green fluorescent protein (GFP; Earley et al. 2006), by the Gateway system to generate the GFP C-terminal fusion construct pEarleyGate-gMADS1. The construct pEarleyGate-gMADS1 was introduced into turmeric leaves of the regenerated plantlets from in vitro culture to express translational fusion proteins by following the method described previously about A. tumefaciens-mediated transient expression in tobacco plants (Sparkes et al. 2006) with some modifications. The construct pEarleyGate-gMADS1 was electroporated into A. tumefaciens strain EHA105. A single colony was picked to grow in 2 ml liquid YEP medium (10 g L−1 Bacto Peptone, 10 g L−1 yeast extract, 5 g L−1 NaCl, pH 7.0) supplemented with 50 mg L−1 kanamycin at 28 °C for 2 days. The Agrobacterium culture was centrifuged at 2,000×g for 5 min at room temperature (25 °C) to pellet the Agrobacterium cells. Before infiltration, the bacterial pellet was gently resuspended in infiltration medium (27 mM glucose, 10 mM MgCl2, 50 mM 2-(N-morpholino) ethanesulfonic acid (MES), and 0.15 mM acetosyringone). Agrobacteria were infiltrated into turmeric young leaves from the micropropagated plantlets at an optical density OD600 = 0.5. After infiltration, the infiltrated plantlets were kept in a magenta box under the same growth conditions for 3 days. Leaf epidermal cells were screened for GFP. Fluorescent images of GFP were obtained with a confocal microscope, Leica TCS SP5. The GFP fluorescence was excited by a 488-nm argon laser and detected at 500–600 nm. Images were obtained at a scan rate of 1,000 Hz and a resolution of 1,024 × 1,024 pixels. Three images were averaged to improve the signal-to-noise ratio.
Results and discussion
Callus induction, somatic embryogenesis and plant regeneration
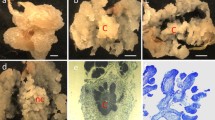
Successful Agrobacterium-mediated transformation depends mainly on an efficient tissue culture system, including callus induction, somatic embryogenesis and plant regeneration. Callus induction and regeneration are affected by many factors, such as genotype, explant type, culture medium, plant growth regulators, and culture conditions. In rhizomatous plants, contamination is a major problem during tissue culture, especially when tissues were initiated from rhizome parts, including rhizome buds and shoots. Establishment of the in vitro cultures in Curcuma species posed considerable problems with contamination in primary cultures, which reappeared even after repeated subculturing (Das et al. 2010). We tested different tissues with media formulations and found that young inflorescence was the best target tissue for callus induction on B5I medium (Table 1). This combination showed much lower contamination rates and higher frequencies in somatic embryo induction and embryo conversion than other tissues/explants, which was consistent with the growing body of evidence showing that immature inflorescence has been recognized as an important source of totipotent cultures in many plants including Curcuma species (Kou et al. 2013; Salvi et al. 2000). After 5–6 weeks of culture, calli initiated from inflorescences (Fig. 1a) were dissected out and subcultured on the same B5I medium. After another 3 weeks, the light yellow, compact, and relatively dry calli (Fig. 1b) were subcultured on B5I medium and maintained for regeneration and transformation experiments. These actively growing, healthy looking, embryogenic calli were transferred to regeneration medium MSR (Table 1) and 85 % of the calli developed green shoots after 2–3 months in culture under light with a 16-h photoperiod. All plantlets regenerated from somatic embryos produced well-developed roots and grew vigorously in magenta boxes and were then transferred to pots for growth in the greenhouse. The regeneration medium MSR (Table 1) was also used for developing the Agrobacterium-mediated transformation system and regenerating transgenic plants.

Plant tissues at different stages of the Agrobacterium-mediated transformation of turmeric. a The inflorescences used as the starting material to induce calli on induction medium B5I. b Subcultured calli on medium B5I. c Calli on selection medium B5S. d Calli on Preregeneration medium MSP. e The embryogenesis of callus on regeneration medium MSR. f The growth of shoots and roots from the transformed calli after 4 weeks on rooting medium (MSG)
Optimization of selection agent concentration
It is essential to assess selection agents for their utility with specific plant species and to determine their optimal concentration in selection media. Selection agent sensitivity varies by plant species and the different explants used in plant transformation experiments. In strawberry, for example, line LF9 exhibited higher sensitivity to kanamycin (Kan) and hygromycin (Hyg) than other lines. Thus, Kan at 2.5–5 mg/L or Hyg at 1.5 mg/L was used for selection (Folta et al. 2006). On the other hand, for the perennial rhizome grass, Leymus chinensis, Kan at 150 mg/L (Sun and Hong 2012) or Hyg at 100–150 mg/L (Wang et al. 2009) was required for selecting transgenic plants. In this study, turmeric calli grown on regeneration medium without any selection agents present grew healthily, shoots began to initiate at 8–10 weeks and approximately 85 % of the calli developed into vigorous shoots after 2–3 months (Fig. 2). In contrast, the calli showed stress symptoms with slow growth and greatly reduced shoot regeneration when placed on selective regeneration media. None of the calli generated shoots on media containing 400–600 mg/L Kan, and the majority became necrotic after 4 weeks. These results indicated that Kan had a significant impact on callus growth and development in selective media with different concentrations (Fig. 2). The higher the concentration of the selection agent in selective regeneration media, the lower the shoot regeneration frequency was evident and the more necrotic calli were observed (Fig. 2). In this study, 400 mg/L Kan in medium was sufficient to prevent untransformed turmeric calli from producing shoots and was therefore selected as the optimal concentration for turmeric transformation.

Sensitivity to selection agents. Kan was assessed across a range of concentrations in MSR media. The turmeric tissues were treated as described in the text and plated on media under different selection regimens. Error bars indicate standard error of the mean
Effect of different A. tumefaciens strains
We investigated three commonly used A. tumefaciens strains (AGL1, EHA105 and LBA4404) to determine which would be best to use with turmeric. When assayed for GUS activity, a 46.7 % transformation rate was observed when strain EHA105 was used, followed by LBA4404 (35.0 %) and AGL1 (26.7 %; Fig. 3a). Since these three strains harbored the same binary vector pBISN1, this difference in GUS activity might be due to different chromosomal backgrounds and the different virulence (vir) regions of the strains (Hellens et al. 2000). The LBA4404 is an octopine strain containing a disarmed pAL4404 plasmid in the TiAch5 chromosomal background. The EHA105 is a succinamopine strain containing a disarmed pEHA105 plasmid in the C58 chromosomal background. Interestingly, both AGL1 and EHA105 were succinamopine strains with same C58 chromosomal backgrounds, but the GUS-based positive transformation rate of the EHA105 strain was almost 2-fold higher than that of AGL1 (Fig. 3a). These results indicated that there were different transformation efficiencies for different strains and EHA105 was a more suitable strain for turmeric transformation.

Effects of Agrobacterium strains (a) and acetosyringone (b) on transient GUS expression in turmeric calli. a Data were recorded after 3 days of co-cultivation. Both the inoculation and co-cultivation media were supplemented with 150 μM AS. b Calli were inoculated with EHA105 (pBISN1), an infection time of 20 min and co-cultivated for 3 days. Values represent the mean of four independent experiments. The data represent the mean values and SE of four independent experiments
Co-cultivation and Agrobacterium elimination
Co-cultivation was found to be a critical stage in Agrobacterium-mediated transformation of turmeric. The addition of acetosyringone (AS) during co-cultivation is often found to enhance or be essential for plant transformation, because the transfer of T-DNA is mediated by virulence (vir) genes and expression of vir genes were greatly induced and enhanced by phenols such as AS (Pitzschke and Hirt 2010). For example, AS at 100 μM was key to successful transformation of rice (Hiei et al. 1994), while an increased AS concentration (up to 400 μM) led to significantly higher GUS expression and T-DNA delivery efficiency in wheat (He et al. 2010b). In contrast, the results from reed (Phragmites communis) transformation indicated that the highest frequency of GUS-expressing calli was obtained when 200 μM AS was used, and higher concentrations of AS resulted in the reduction in GUS expression (Kim et al. 2013). To determine which concentration of AS would be most effective in turmeric transformation, we evaluated different concentrations (0, 50, 100, 150, 200, 250 and 300 μM) applied within 20 min of infection and maintained in the media for 3 days of co-cultivation. The results (see Fig. 3b) of these experiments showed that the supplementation of AS to both inoculation and co-cultivation media greatly increased GUS expression. The highest percentage (45 %) of GUS positive calli was obtained from the media containing 150 μM AS (Fig. 3b). Higher concentrations of AS resulted in lower GUS expression, as was found for reed, indicating a possible negative effect on T-DNA transfer. Only 8 % of the calli showed GUS expression in the absence of AS, indicating that AS is very important for turmeric transformation via Agrobacterium.
Other factors, such as an acidic pH and the co-cultivation duration, were optimized to develop an efficient transformation procedure for turmeric. It was observed that 2–4 days of co-cultivation with the bacterial density used for infection resulted in highest GUS activity. Longer co-cultivation periods (>4 days) resulted in an abundant proliferation of bacteria and consequently decreased the regeneration frequency of the selected calli. When calli were completely colonized by the Agrobacterium, elimination of bacteria in the subsequent stages became difficult. Therefore, control of Agrobacterium overgrowth after co-cultivation is crucial. In this experiment, the antibiotic timentin (Costa et al. 2000) was more effective in suppressing Agrobacterium proliferation in turmeric than cefotaxime or carbenicillin.
Two steps were adopted to overcome the problem of Agrobacterium overgrowth and subsequent contamination. First, the co-cultivated calli were fully washed at the end of the co-cultivation period with a solution containing 400 mg L−1 timentin. Next, a resting step was used in the transformation process. After the calli were cultured on the resting medium (400 mg L−1 timentin added) for 1 week, Agrobacterium growth appeared to be fully restrained. The resting cultivation step might help the calli recover from the stress of Agrobacterium inoculation. If the inoculated calli were cultured on the selection medium immediately following co-cultivation, these calli would suffer from the combined stresses of Agrobacterium inoculation and selection agent (Kan) application, resulting a reduction in transformation efficiency.
Regeneration of transgenic plants
Selection duration was very important to obtain transgenic plants. Based on the selection agent sensitivity information obtained (see above), we used stringent conditions with 400 mg L−1 Kan for 8–10 weeks. Most calli became necrotic and died, but new (resistant) calli were formed on selection medium during this period (Fig. 1c). After 8–10 weeks of selection culture, the frequency of resistant calli was about 15 % (Table 2). The resistant calli were transferred to pre-regeneration medium for two more weeks of selection and pre-regeneration (Fig. 1d), and then to regeneration media without Kan for regeneration of plantlets (Fig. 1e). Calli selected by Kan for several cycles were regenerated on regeneration medium in the absence of Kan, thereby maintaining a balance between stringency of selection and regeneration efficiency. About 37 % of the resistant calli could differentiate into shoots (Table 2). To further avoid false positives, regenerated shoots were transferred to root-growing medium containing 400 mg L−1 Kan for further selection (Fig. 1f). After selection in the root-growing medium, 52 % of the shoots could grow normally and were identified as potential transformants (Table 2). Therefore, the addition of Kan to the root-growing medium (with low sucrose content, 10 g L−1) was a very effective means to obtain true transgenic plants and improving selection efficiency. This two-step selection procedure, an initial selection of transformed calli on selection medium and a second selection of putatively transformed plantlets on root-growing medium, significantly improved the efficiency of transformation with regards to true transformants to false positive ratio.
On average, each resistant callus could produce 3–5 shoots that likely originated from the same transformation event, constituting a transgenic line (transformant). This result was consistent with the report of turmeric transformation via particle bombardment (Shirgurkar et al. 2006). In the present study, the average transformation efficiency was about 2.8 % (Table 2).
GUS assay and molecular analysis of transgenic turmeric plants
The transformed calli growing on selection medium and the regenerated transgenic plantlets were tested for GUS activity. GUS assays showed transgene expression in resistant calli and shoots, while there was no GUS staining in the control calli and the non-transformed shoots (Fig. 4a, b). PCR analysis was further used to confirm the presence and the integration of T-DNA into the turmeric genome. As shown in Fig. 4c and d, the amplification of genomic DNAs from different transgenic lines with gus and nptII specific primers produced the expected 988 bp band for gus and 722 bp band for nptII, while the same band was not detected in amplification with DNA from the control plants.

GUS assay and PCR analysis of turmeric transformants. a GUS expression in the transformed calli (left), control (right). b GUS expression in the transgenic plantlet (left), control (right). c PCR analysis of genomic DNA to detect the presence of the gus-coding regions in transformed plants, showing amplification of the predicted specific gus fragment of 998 bp. Lanes 1–12 are from turmeric plants regenerated from 12 independent, Kan-resistant calli. d PCR analysis of genomic DNA to detect the presence of the nptII-coding regions in transformed plants, showing amplification of the predicted specific nptII sequence 722 bp fragment. Lanes 1–12 are from transformed turmeric plants. Lane M molecular weight markers, lane P positive control pBISN1 plasmid DNA, Lane C untransformed control plant
To confirm the T-DNA integration and copy number into genomic DNA, we performed Southern blot analysis on four independent lines to demonstrate that the transgene was inserted into the turmeric genome (Fig. 5). The number of hybridizing bands indicates the npt gene copy number. Single copy (Fig. 5, Lanes 2–4) and multiple copies (Fig. 5, Lane 1) were shown in the transformed lines, whereas no hybridization signal was detected from the non-transformed control plant (Fig. 5, lane C). The insertion patterns observed in this experiment are consistent with the observation that Agrobacterium-mediated transformation shows the relatively simple T-DNA insertions and tends to produce transgenic lines with a low copy number of transgenes integrated (He et al. 2010b; Hiei et al. 1997).

Southern blot analysis of transgenic turmeric plants. Genomic DNA isolated from transgenic turmeric plants (lanes 1–4) and a non-transformed turmeric plant (lane C), and plasmid pBISN1 DNA (lane P) were digested with BamHI and EcoRI. PCR-amplified product of the nptII gene was used as the probe for Southern hybridization
Sub-cellular localization of the MADS box fusion protein by Agroinfiltration
Agroinfiltration allows efficient transfer and transient expression of T-DNA constructs in young leaf tissues and seedlings to provide valuable insight into many biological processes, such as investigating gene function, protein localization and protein–protein interaction. This approach has been widely used not only in dicot species, such as tobacco (Li et al. 2013; Sparkes et al. 2006; Voinnet et al. 2003), Arabidopsis (Tsuda et al. 2012; Wroblewski et al. 2005), tomato (Li et al. 2009; Wroblewski et al. 2005), potato (Bhaskar et al. 2009), sunflower (Manavella and Chan 2009), citrus (Figueiredo et al. 2011) and poplar (Takata and Eriksson 2012), but also in monocot species, including rice (Andrieu et al. 2012; Li et al. 2009) and switchgrass (Chen et al. 2010; Li et al. 2009). However, the use of this technology has not been reported to date in monocot rhizome species such as turmeric.
The level of transient expression in an agroinfiltration system depends on the efficiency of transformation and the transcriptional and translational activity of the monitored marker. In preliminary experiments, we tested three different A. tumefaciens strains (AGL1, EHA105 and LBA4404), and found that EHA105 resulted in consistently higher transformation rates than the other strains.
To determine the subcellular localization of the transcription factors identified in previous work (Koo et al. 2013), a C-terminal translational fusion of GFP to the MADS box gene (GT_01880) ORF (encoding 243 aa) was cloned into the pEarleyGate103 vector, driven by the constitutive 35S CaMV promoter. The construct containing the fusion-GFP reporter carried by Agrobacterium strain EHA105 was infiltrated into turmeric leaves and was found to be highly expressed in transformed leaf cells, with the fusion-GFP protein apparently exclusively localized in the nucleus (Fig. 6c). We did not find any independent GFP-fluorescence signals in any other organelles or in the control leaf tissue (Fig. 6d). Since MADS-box proteins generally present a nuclear localization signal (NLS), which is positioned in the N-terminal binding domain of the protein (Immink et al. 2002), this transcription factor is mobilized into the nucleus to exert its regulatory action. Indeed, the protein used in this fusion construct contained both the MADS-box superfamily binding domain and a putative NLS domain in the N-terminal domain. The localization observed in this transient expression experiment demonstrated that this NLS domain was indeed functional and that the transient expression system could rapidly and easily be used with turmeric to test cellular and subcellular localization of specific proteins in this plant.

Sub-cellular localization of a MADS-box protein in agroinfiltrated turmeric leaves. The GFP-tagged transcription factor (a MADS-box protein) was expressed in epidermal cells and analyzed by laser-scanning confocal microscopy. a Turmeric plant regenerated from callus in vitro culture as used for the transient GFP fusion expression assay. b Leaf epidermal cells and GFP signals were observed in guard cells from the infiltrated leaf. c MADS-box-GFP protein (green) accumulates in the nucleus of leaf epidermal cells of the infiltrated leaf. d The control leaf tissue (no GFP signals were observed). c and d Three-dimensional projections of confocal z-stacks. Chloroplasts appear red due to autofluorescence. Bars represent a scale of 50 μm
In conclusion, this study established an efficient regeneration protocol for turmeric via direct somatic embryogenesis and developed a reliable procedure for Agrobacterium-mediated transformation in turmeric. The efficient regeneration and transformation system opens a new avenue for improving this rhizomatous and medicinal plant, as well as for testing gene function within medicinal rhizomes. Furthermore, the transient gene expression method was demonstrated to be highly effective with young turmeric leaves, and validated the nuclear localization of a MADS-box protein. This study outlines a valuable tool set for investigating gene function and metabolic pathways related to rhizome development and germplasm improvement for this species.
Abbreviations
- AS:
-
Acetosyringone
- CaMV:
-
35S Cauliflower mosaic virus 35S promoter
- GFP:
-
Green fluorescent protein
- GUS:
-
β-Glucuronidase
- Kan:
-
Kanamycin
- MS:
-
Murashige and Skoog
- NAA:
-
α-Naphthalene acetic acid
- NLS:
-
Nuclear localization signal
- npt :
-
Neomycin phosphotransferase gene
References
Aggarwal BB, Sung B (2009) Pharmacological basis for the role of curcumin in chronic diseases: an age-old spice with modern targets. Trends Pharmacol Sci 30:85–94
Andrieu A, Breitler JC, Siré C, Meynard D, Gantet P, Guiderdoni E (2012) An in planta, Agrobacterium-mediated transient gene expression method for inducing gene silencing in rice (Oryza sativa L.) leaves. Rice 5:1–12
Balasubramanian K (2006) Molecular orbital basis for yellow curry spice curcumin’s prevention of Alzheimer’s disease. J Agric Food Chem 54:3512–3520
Bhaskar PB, Venkateshwaran M, Wu L, Ane JM, Jiang J (2009) Agrobacterium-mediated transient gene expression and silencing: a rapid tool for functional gene assay in potato. PLoS ONE 4:e5812
Chen X, Equi R, Baxter H, Berk K, Han J, Agarwal S, Zale J (2010) A high-throughput transient gene expression system for switchgrass (Panicum virgatum L.) seedlings. Biotechnol Biofuels 3:9
Costa MGC, Nogueira FTS, Figueira ML, Otoni WC, Brommonschenkel SH, Cecon PR (2000) Influence of the antibiotic timentin on plant regeneration of tomato (Lycopersicon esculentum Mill.) cultivars. Plant Cell Rep 19:327–332
Das A, Kesari V, Rangan L (2010) Plant regeneration in Curcuma species and assessment of genetic stability of regenerated plants. Biol Plant 54:423–429
Earley KW, Haag JR, Pontes O, Opper K, Juehne T, Song K, Pikaard CS (2006) Gateway-compatible vectors for plant functional genomics and proteomics. Plant J 45:616–629
Egan ME, Pearson M, Weiner SA, Rajendran V, Rubin D, Glockner-Pagel J, Canny S, Du K, Lukacs GL, Caplan MJ (2004) Curcumin, a major constituent of turmeric, corrects cystic fibrosis defects. Science 304:600–602
Figueiredo JF, Romer P, Lahaye T, Graham JH, White FF, Jones JB (2011) Agrobacterium-mediated transient expression in citrus leaves: a rapid tool for gene expression and functional gene assay. Plant Cell Rep 30:1339–1345
Folta KM, Dhingra A, Howard L, Stewart PJ, Chandler CK (2006) Characterization of LF9, an octoploid strawberry genotype selected for rapid regeneration and transformation. Planta 224:1058–1067
Gang DR, Ma XQ (2008) Ginger and turmeric, ancient spices and modern medicines. Springer, Berlin
Hashemy T, Maki H, Yamada Y, Kaneko TS, Syono K (2009) Effects of light and cytokinin on in vitro micropropagation and microrhizome production in turmeric (Curcuma longa L.). Plant Biotech 26:237–242
He RF, Pan J, Zhu LL, He GC (2010a) Agrobacterium-mediated transformation of large DNA fragments using a BIBAC vector system in rice. Plant Mol Biol Rep 28:613–619
He Y, Jones HD, Chen S, Chen XM, Wang DW, Li KX, Wang DS, Xia LQ (2010b) Agrobacterium-mediated transformation of durum wheat (Triticum turgidum L. var. durum cv Stewart) with improved efficiency. J Exp Bot 61:1567–1581
He R, Kim MJ, Nelson W, Balbuena TS, Kim R, Kramer R, Crow JA, May GD, Thelen JJ, Soderlund CA, Gang DR (2012) Next-generation sequencing-based transcriptomic and proteomic analysis of the common reed, Phragmites australis (Poaceae), reveals genes involved in invasiveness and rhizome specificity. Am J Bot 99:232–247
Hellens R, Mullineaux P, Klee H (2000) A guide to Agrobacterium binary Ti vectors. Trends Plant Sci 5:446–451
Hiei Y, Ohta S, Komari T, Kumashiro T (1994) Efficient transformation of rice (Oryza sativa L.) mediated by Agrobacterium and sequence analysis of the boundaries of the T-DNA. Plant J 6:271–282
Hiei Y, Komari T, Kubo T (1997) Transformation of rice mediated by Agrobacterium tumefaciens. Plant Mol Biol 35:205–218
Hood EE, Gelvin SB, Melchers LS, Hoekema A (1993) New agrobacterium helper plasmids for gene-transfer to plants. Transgenic Res 2:208–218
Immink RG, Gadella TW Jr, Ferrario S, Busscher M, Angenent GC (2002) Analysis of MADS box protein–protein interactions in living plant cells. Proc Natl Acad Sci USA 99:2416–2421
Kim YG, Sharmin SA, Alam I, Kim KH, Kwon SY, Sohn JH, Kim SH, Liu GS, Lee BH (2013) Agrobacterium-mediated transformation of reed (Phragmites communis Trinius) using mature seed-derived calli. GCB Bioenergy 5:73–80
Koo HJ, McDowell ET, Ma X, Greer KA, Kapteyn J, Xie Z, Descour A, Kim H, Yu Y, Kudrna D, Wing RA, Soderlund CA, Gang DR (2013) Ginger and turmeric expressed sequence tags identify signature genes for rhizome identity and development and the biosynthesis of curcuminoids, gingerols and terpenoids. BMC Plant Biol 13:27
Kou YP, Ma GH, da Silva JAT, Liu NA (2013) Callus induction and shoot organogenesis from anther cultures of Curcuma attenuata wall. Plant Cell, Tissue Organ Cult 112:1–7
Lazo GR, Stein PA, Ludwig RA (1991) A DNA transformation-competent Arabidopsis genomic library in Agrobacterium. Nat Biotechnol 9:963–967
Li JF, Park E, von Arnim AG, Nebenfuhr A (2009) The FAST technique: a simplified Agrobacterium-based transformation method for transient gene expression analysis in seedlings of Arabidopsis and other plant species. Plant Methods 5:6
Li W, Yadeta KA, Elmore JM, Coaker G (2013) The Pseudomonas syringae effector HopQ1 promotes bacterial virulence and interacts with tomato 14-3-3 proteins in a phosphorylation-dependent manner. Plant Physiol 161:2062–2074
Ma X, Gang DR (2006) Metabolic profiling of turmeric (Curcuma longa L.) plants derived from in vitro micropropagation and conventional greenhouse cultivation. J Agric Food Chem 54:9573–9583
Manavella PA, Chan RL (2009) Transient transformation of sunflower leaf discs via an Agrobacterium-mediated method: applications for gene expression and silencing studies. Nat Protoc 4:1699–1707
Mohanty S, Panda MK, Subudhi E, Nayak S (2008) Plant regeneration from callus culture of Curcuma aromatica and in vitro detection of somaclonal variation through cytophotometric analysis. Biol Plant 52:783–786
Narasimhulu SB, Deng XB, Sarria R, Gelvin SB (1996) Early transcription of Agrobacterium T-DNA genes in tobacco and maize. Plant Cell 8:873–886
Ooms G, Hooykaas PJJ, Vanveen RJM, Vanbeelen P, Regensburgtuink TJG, Schilperoort RA (1982) Octopine Ti-plasmid deletion mutants of Agrobacterium tumefaciens with emphasis on the right side of the T-region. Plasmid 7:15–29
Pitzschke A, Hirt H (2010) New insights into an old story: Agrobacterium-induced tumour formation in plants by plant transformation. EMBO J 29:1021–1032
Salvi ND, George L, Eapen S (2000) Direct regeneration of shoots from immature inflorescence cultures of turmeric. Plant Cell, Tissue Organ Cult 62:235–238
Salvi ND, George L, Eapen S (2002) Micropropagation and field evaluation of micropropagated plants of turmeric. Plant Cell, Tissue Organ Cult 68:143–151
Sambrook J, Russell DW (2001) Molecular cloning—a laboratory manual, 3rd edn. Cold Spring Harbor Laboratory Press, New York
Shewry PR, Jones HD, Halford NG (2008) Plant biotechnology: transgenic crops. Adv Biochem Eng Biotechnol 111:149–186
Shibata D, Liu YG (2000) Agrobacterium-mediated plant transformation with large DNA fragments. Trends Plant Sci 5:354–357
Shirgurkar MV, John CK, Nadgauda RS (2001) Factors affecting in vitro microrhizome production in turmeric. Plant Cell, Tissue Organ Cult 64:5–11
Shirgurkar MV, Naik VB, von Arnold S, Nadgauda RS, Clapham D (2006) An efficient protocol for genetic transformation and shoot regeneration of turmeric (Curcuma longa L.) via particle bombardment. Plant Cell Rep 25:112–116
Singh S, Kuanar A, Mohanty S, Subudhi E, Nayak S (2011) Evaluation of phytomedicinal yield potential and molecular profiling of micropropagated and conventionally grown turmeric (Curcuma longa L.). Plant Cell, Tissue Organ Cult 104:263–269
Sparkes IA, Runions J, Kearns A, Hawes C (2006) Rapid, transient expression of fluorescent fusion proteins in tobacco plants and generation of stably transformed plants. Nat Protoc 1:2019–2025
Sun YL, Hong SK (2012) Agrobacterium tumefaciens-mediated transformation of the halophyte Leymus chinensis (Trin.). Plant Mol Biol Rep 30:1253–1263
Takata N, Eriksson ME (2012) A simple and efficient transient transformation for hybrid aspen (Populus tremula × P. tremuloides). Plant Methods 8:30
Tsuda K, Qi Y, le Nguyen V, Bethke G, Tsuda Y, Glazebrook J, Katagiri F (2012) An efficient Agrobacterium-mediated transient transformation of Arabidopsis. Plant J 69:713–719
Voinnet O, Rivas S, Mestre P, Baulcombe D (2003) An enhanced transient expression system in plants based on suppression of gene silencing by the p19 protein of tomato bushy stunt virus. Plant J 33:949–956
Wang LJ, Li XF, Chen SY, Liu GS (2009) Enhanced drought tolerance in transgenic Leymus chinensis plants with constitutively expressed wheat TaLEA (3). Biotechnol Lett 31:313–319
Wroblewski T, Tomczak A, Michelmore R (2005) Optimization of Agrobacterium-mediated transient assays of gene expression in lettuce, tomato and Arabidopsis. Plant Biotechnol J 3:259–273
Acknowledgments
We gratefully acknowledge the US National Science Foundation (Grant IOS-1044821) for financial support of this research.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
He, R., Gang, D.R. Somatic embryogenesis and Agrobacterium-mediated transformation of turmeric (Curcuma longa). Plant Cell Tiss Organ Cult 116, 333–342 (2014). https://doi.org/10.1007/s11240-013-0407-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11240-013-0407-y