
Abstract
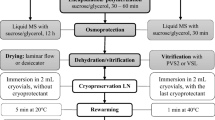
Embryogenic calli of Dioscorea bulbifera L. were successfully cryopreserved using an encapsulation-vitrification method. Embryogenic calli were cooled at 6°C for 5 days on solid MS medium (Murashige and Skoog 1962) containing 2 mg L−1 Kinetin (Kn), 0.5 mg L−1 α-naphthalene acetic acid (NAA) and 0.5 mg L−1 2,4-dichlorophenoxy-acetic acid (2,4-D). These were prior precultured on liquid basal MS medium enriched with 0.75 M sucrose at 25 ± 1°C for 7 days. Embryogenic calli were osmoprotected with a mixture of 2 M glycerol and 1 M sucrose for 80 min at 25°C and dropped in a 0.1 M CaCl2 solution containing 0.4 M sucrose at 25 ± 1°C. After 15 min of polymerization, Ca-alginate beads (about 4 mm in diameter) were dehydrated for 150 min at 0°C in a PVS2 solution [30% glycerol, 15% ethylene glycol, and 15% dimethyl sulfoxide (w/v)] containing 0.5 M sucrose. The encapsulated embryogenic calli were then plunged directly into LN (liquid nitrogen) for 1 h. After rapid thawing in a water bath (37°C; 2 min), the beads were washed 3 times at 10-min intervals in liquid basal MS medium containing 1.2 M sucrose. Following thawing, the embryogenic calli were transferred to fresh solid basal MS media supplemented with Kn 2 mg L−1, 0.09 M sucrose and 0.75% (w/v) agar (embryoid induction medium) and cultured under light conditions of 12-h photoperiod with a light intensity of 36 μmol m−2 s−1 provided by white cool fluorescent tubes after a 2-day dark period at 25 ± 1°C. After 30 days, the embryoids developed from embryogenic calli were transferred to fresh solid basal MS media supplemented with Kn 2 mg L−1, NAA 0.5 mg L−1, 3% (w/v) sucrose and 0.75% (w/v) agar (regeneration medium). After 60 days, the embryogenic calli developed normal shoots and roots. No morphological abnormalities were observed after plating on the regeneration medium. The survival rate of encapsulated vitrified embryogenic callus reached over 70%. This encapsulation-vitrification method appears promising as a routine and simple method for the cryopreservation of Dioscorea bulbifera embryogenic callus.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Yams of the monocotyledonous genus Dioscorea containing 600 species are important tuber-bearing plants that provide a staple food for millions of people in the Caribbean, Central and South America, West Africa, Asia, and the Pacific (Hong et al. 2009). Yams play a unique role in the nutrition economy as well as in social and religious festivities in tropical and subtropical regions of the world (Inoue and Akiko 2009). Dioscorea bulbifera L., commonly called aerial yam, is a herbaceous climber of the family Dioscoreaceae (Alka et al. 2005). The plant has been used as a herbal remedy for many years, for example as an antispasmodic, analgesic, aphrodisiac, diuretic, and a rejuvenative tonic (Tang et al. 2006). Though only traces of diosgenin have been detected, the plant is still used in the indigenous system of medicine (Alka et al. 2007). Diosgenin, a steroidal sapogenin, is a main precursor for the synthesis of cortisones, corticosteroids, sex hormones (progesterone), and oral contraceptives (Alka et al. 2005). Therefore, dried and powdered tubers find application in the treatment of ulcers, hemorrhoids, dysentery, syphilis, carbuncles, lung abscesses, and breast lumps. Recently, Dioscorea bulbifera L., which has been found to exhibit anti-tumor and anti-bacterial activity, and anti-hypoglycaemic and anti-diabetic effects, is often used as clinical medicine to treat thyroid glands and tumors of different types (Marc et al. 2007).
However, as a result of industrial development in the synthesis of steroid hormones, the requirement for Dioscorea bulbifera has increased and the wild resources are rapidly being depleted (Wheeler et al. 2007), so it is important to have techniques to ensure its preservation. Traditionally, Dioscorea bulbifera genetic resources are preserved as whole plants either in field genebanks or in greenhouses. In these cases, preserved plants are subject to risks of losses caused by biological and climatic hazards as well as through human error; in addition, the routine maintenance of plant materials is costly (Dixit et al. 2003). There is a growing need for stable long-term in vitro storage of the germplasm collections of Dioscorea bulbifera as well as those of other vegetatively propagated tropical crops.
Ex situ conservation techniques, including the use of slow growth storage and cryopreservation for in vitro cloning, have also been developed (Leunufna and Keller 2003). For example, slow growth storage techniques of in vitro plants are used in various genebanks as a back-up to Dioscorea bulbifera field collections (Alka et al. 2007). Problems with these in vitro techniques include high costs for maintaining a large number of stocks, space requirements, and risks of contamination and somaclonal variation over time. Cryopreservation is an alternative choice for a long-term conservation of Dioscorea bulbifera germplasm. Cryopreservation at −196ºC in liquid nitrogen (LN) has been considered to be an ideal tool which offers long-term storage capability, maximal stability of phenotypic and genotypic behavior of stored germplasm, and minimal storage space and maintenance requirements (Suzuki et al. 2008). However, the availability or development of efficient and reliable cryogenic protocols which yield a high percentage of recovery is a basic requirement. Recent cryopreservation studies on yams have used simple, economical methods, including the original vitrification and encapsulation-dehydration methods, with various recovery rates attributable to the method and to genetic differences among the species used (Suzuki et al. 2008; Hong and Yin 2009). However, there are few reports (Dixit et al. 2003; Leunufna and Keller 2003) on the cryopreservation of Dioscorea bulbifera. In the preliminary study, embryogenic tissues of Dioscorea bulbifera were cryopreserved using the encapsulation-dehydration technique (Dixit et al. 2003). However, the result indicates low average survival. Even using a protocol of the modified droplet cryopreservation method, recovery of Dioscorea bulbifera still remained low (low to zero survival and regrowth rates) (Leunufna and Keller 2003). Recently, a protocol has been developed for successful cryopreservation of shoot tips of Dioscorea bulbifera by encapsulation-dehydration (Mandal et al. 1996). However, as shown by Kazumasa et al. (2002) and Adriana et al. (2004), encapsulation-dehydration produced relatively lower levels of recovery growth than encapsulation-vitrification, and it was also a longer process that is complicated and time consuming. The development of a simple and efficient method for cryopreservation would facilitate the much wider use for germplasm storage.
Encapsulation-vitrification techniques offer various advantages over encapsulation-dehydration in terms of greater recovery, a simplistic protocol, and high levels of recovery growth (Wang et al. 2004). Vitrification techniques have also been applied to a wide range of plant materials (Engelmann 2000), which enable the cryopreservation of the shoot tips, meristems, and embryogenic tissues (Kazumasa et al. 2002). Proliferating embryogenic callus clumps are known to regenerate easily and to have the potential for genetic transformation. In addition, it has been reported that plants recovered from Dioscorea bulbifera somatic embryos were morphologically similar to non-embryogenic-derived plants (Dixit et al. 2003; Hong et al. 2009), thus, embryogenic calli are fit for cryopreservation of Dioscorea bulbifera germplasm. But to our knowledge, there are few or no reports on the cryopreservation of Dioscorea bulbifera embryogenic calli by encapsulation-vitrification. The present study described for the first time a simple and efficient cryopreservation of Dioscorea bulbifera embryogenic calli by encapsulation-vitrification.
Materials and methods
Plant material, standard callus induction, subculture and photoperiod conditions
Leaves and nodal segments of Dioscorea bulbifera were obtained from greenhouse of Han Hai Ecological Development, Songping Village, Chating Town, Shangrao City, Jiangxi Province, China. Leaf explants about 10 × 10 mm were thoroughly washed under running tap water for 30 min. and then surface-sterilized sequentially with 0.1% mercuric chloride for 10 min and 70% (v/v) alcohol for 30 s. The explants were then thoroughly washed with sterile distilled water before culturing. MS-based (Murashige and Skoog 1962) medium containing 2 mg L−1 Kn, 0.5 mg L−1 NAA, and 0.5 mg L−1 2,4-D was used in all experiments (Wolfgang and Hyeon-Yong 2004). The pH of the medium was adjusted to 5.8–6.0 prior to autoclaving at 121°C for 20 min. Cultures were maintained at 25 ± 1°C with 75 ± 5% relative humidity for 24 h in the dark. In such conditions, two types of calli are produced. A soft, sticky, yellowy, nonnodular and nonembryogenic callus, and a highly nodular, hard, white, embryogenic callus. When subcultured on fresh solid MS-based medium containing 2 mgL−1 Kn, 0.5 mgL−1 NAA, and 0.5 mgL−1 2,4-D three times (cultured for 20 days per time), the friability of the embryogenic callus increases. Loose and white calli, that is friable embryogenic calli, were selected for cryopreservation of Dioscorea bulbifera by encapsulation-vitrification.
Cold-acclimated
Embryogenic calli subcultured for three times were cold-acclimated at 2, 6, and 10°C on solid MS medium containing 2 mg L−1 Kn, 0.5 mg L−1 NAA, and 0.5 mg L−1 2,4-D in the dark for 5 days. Then, the embryogenic calli were precultured in liquid basal MS medium enriched with 0.75 M sucrose at 25 ± 1°C for 7 days. Embryogenic calli were osmoprotected with a mixture of 2 M glycerol and 1 M sucrose for 80 min at 25°C and dropped in a 0.1 M CaCl2 solution containing 0.4 M sucrose at 25 ± 1°C. After 15 min of polymerization, Ca-alginate beads (about 4 mm in diameter) were dehydrated for 150 min at 0°C in a PVS2 solution [30% glycerol, 15% ethylene glycol, and 15% dimethyl sulfoxide (w/v)] containing 0.5 M sucrose. The encapsulated embryogenic calli were then plunged directly into LN (liquid nitrogen) for 1 h. After rapid thawing in a water bath (37°C; 2 min), the beads were washed 3 times at 10-min intervals in liquid basal MS medium containing 1.2 M sucrose. Following thawing, the embryogenic calli were transferred to fresh solid basal MS media supplemented with Kn 2 mg L−1, 0.09 M sucrose, and 0.75% (w/v) agar (embryoid induction medium) as recommended by Wolfgang and Hyeon-Yong (2004). Cultures were maintained for 2 days in the dark and then transferred to light conditions of a 12-h photoperiod with a light intensity of 36 μmol m−2 s−1 provided by white cool fluorescent tubes at 25 ± 1°C. Survival rate was estimated as the percentage of the total number of embryogenic calli that developed an embryoid after 30 days culture.
At the same time, the effect of cold-acclimation time on survival of cryopreserved embryogenic calli was also studied: embryogenic calli were cooled at 6°C for in the dark for 0–10 days on solid MS medium containing 2 mg L−1 Kn, 0.5 mg L−1 NAA, and 0.5 mg L−1 2,4-D. Then, the embryogenic calli were precultured in liquid basal MS medium enriched with 0.75 M sucrose at 25 ± 1°C for 7 days. Embryogenic calli were osmoprotected with a mixture of 2 M glycerol and 1 M sucrose for 80 min at 25°C and dropped in a 0.1 M CaCl2 solution containing 0.4 M sucrose at 25 ± 1°C. After 15 min of polymerization, Ca-alginate beads were dehydrated for 150 min at 0°C in a PVS2 solution containing 0.5 M sucrose. The encapsulated embryogenic calli were then plunged directly into LN for 1 h. After rapid thawing in a water bath (37°C; 2 min), the beads were washed 3 times at 10-min intervals in liquid basal MS medium containing 1.2 M sucrose. Following thawing, the embryogenic calli were transferred to fresh solid embryoid induction medium (as described above). Cultures were maintained for 2 days in the dark and then transferred to light conditions of a 12-h photoperiod at 25 ± 1°C. Survival rate was estimated as the percentage of the total number of embryogenic calli that developed an embryoid after 30 days culture.
Finally, the effect of ABA on survival of cryopreserved embryogenic calli was also studied: embryogenic calli subcultured for three times were cold-acclimated on proliferation medium, i.e. basal MS media supplemented with 0.0 or 76 μM ABA at 6°C in the dark for 5 days. Then, the embryogenic calli were precultured in liquid basal MS medium enriched with 0.75 M sucrose at 25 ± 1°C for 7 days. Embryogenic callus were osmoprotected with a mixture of 2 M glycerol and 1 M sucrose for 80 min at 25°C and dropped in a 0.1 M CaCl2 solution containing 0.4 M sucrose at 25 ± 1°C. After 15 min of polymerization, Ca-alginate beads were dehydrated for 150 min at 0°C in a PVS2 solution containing 0.5 M sucrose. The encapsulated embryogenic calli were then plunged directly into LN for 1 h. After rapid thawing in a water bath (37°C; 2 min), the beads were washed 3 times at 10-min intervals in liquid basal MS medium containing 1.2 M sucrose. Following thawing, the embryogenic calli were transferred to fresh solid embryoid induction medium (as described above). Cultures were maintained for 2 days in the dark and then transferred to light conditions of a 12-h photoperiod at 25 ± 1°C. Survival rate was estimated as the percentage of the total number of embryogenic calli that developed an embryoid after 30 days culture.
Preculture
Following cold treatment at 6°C in the dark for 5 days, 0.2 g loose and white callus cells were transferred into 2-ml cryotubes (screw-cap polypropylene ampoules; Shanghai MajorBio Technologies) containing 1.8 ml of liquid basal MS medium supplemented with 0, 0.25, 0.5, 0.75, or 1.0 M sucrose (preculture medium) at 25 ± 1°C for 7 days. Embryogenic callus were osmoprotected with a mixture of 2 M glycerol and 1 M sucrose for 80 min at 25°C and dropped in a 0.1 M CaCl2 solution containing 0.4 M sucrose at 25 ± 1°C. After 15 min of polymerization, Ca-alginate beads were dehydrated for 150 min at 0°C in a PVS2 solution containing 0.5 M sucrose. The encapsulated embryogenic calli were then plunged directly into LN for 1 h. After rapid thawing in a water bath (37°C; 2 min), the beads were washed 3 times at 10-min intervals in liquid basal MS medium containing 1.2 M sucrose. Following thawing, the embryogenic calli were transferred to fresh solid embryoid induction medium (as described above). Cultures were maintained for 2 days in the dark and then transferred to light conditions of a 12-h photoperiod at 25 ± 1°C. Survival rate was estimated as the percentage of the total number of embryogenic calli that developed an embryoid after 30 days culture.
At the same time, the effect of preculture time on survival of cryopreserved embryogenic calli was also studied: following cold treatment at 6°C in the dark for 5 days, 0.2 g loose and white callus cells were transferred into liquid basal MS medium supplemented with 0.75 sucrose at 25 ± 1°C for 0–10 days. Embryogenic callus were osmoprotected with a mixture of 2 M glycerol and 1 M sucrose for 80 min at 25°C and dropped in a 0.1 M CaCl2 solution containing 0.4 M sucrose at 25 ± 1°C. After 15 min of polymerization, Ca-alginate beads were dehydrated for 150 min at 0°C in a PVS2 solution containing 0.5 M sucrose. The encapsulated embryogenic calli were then plunged directly into LN for 1 h. After rapid thawing in a water bath (37°C; 2 min), the beads were washed 3 times at 10-min intervals in liquid basal MS medium containing 1.2 M sucrose. Following thawing, the embryogenic calli were transferred to fresh solid embryoid induction medium (as described above). Cultures were maintained for 2 days in the dark and then transferred to light conditions of a 12-h photoperiod at 25 ± 1°C. Survival rate was estimated as the percentage of the total number of embryogenic calli that developed an embryoid after 30 days culture.
Encapsulation-vitrification
Following cold treatment at 6°C in the dark for 5 days and preculture in liquid basal MS medium enriched with 0.75 M sucrose at 25 ± 1°C for 7 days, embryogenic calli were suspended in MS medium containing 2.5% sodium alginate and 0.4 M sucrose. The mixture was dripped with a sterile pipette into a 0.1 M CaCl2 solution containing 0.4 M sucrose (Bachiri et al. 1995). After 15 min polymerization at 25 ± 1°C, Ca-alginate beads (about 4 mm in diameter) containing one embryogenic callus clump each (0.2 g loose and white cells from callus) were rapidly surface-dried on cellulose tissue and transferred to the loading solution. The loading solution containing 2 M glycerol and 1 M sucrose was dispensed in 100-ml Erlenmeyer flasks at 25°C. Various loading durations were tested, ranging from 0 to 120 min. The encapsulated calli were then dehydrated in PVS2 for various durations. PVS2 (Sakai et al. 1990) treatment was either performed at 24 or 0°C (Hirai and Sakai 1999). The PVS2 solution contained 30% glycerol, 15% ethylene glycol, and 15% DMSO (dimethyl sulfoxide) and 0.4 M sucrose (pH 5.8). The ratio of the number of beads to volume of PVS2 was 1:1. After dehydration, the beads were rapidly surface-dried by blotting on cellulose tissue and then 10 beads were transferred into a 2-ml cryotube and immersed directly in LN for 1 h. Thawing was performed quickly in a water bath at 37°C for 2 min. The beads were then washed 3 times at 10-min intervals with liquid basal MS medium containing 1.2 M sucrose (pH 5.8). Following thawing, the embryogenic calli were placed in 9-cm plastic Petri dishes containing two sterile filter papers to absorb the excess PVS2 prior to transfer to fresh solid embryoid induction medium (as described above). Cultures were maintained for 2 days in the dark and then transferred to light conditions of a 12-h photoperiod at 25 ± 1°C. Survival rate was estimated as the percentage of the total number of embryogenic calli that developed an embryoid after 30 days culture.
Effect of culture condition on survival
Embryogenic callus were cooled at 6°C in the dark for 5 days on solid MS medium containing 2 mg L−1 Kn, 0.5 mg L−1 NAA, and 0.5 mg L−1 2,4-D. Then, the embryogenic calli were precultured in liquid basal MS medium enriched with 0.75 M sucrose at 25 ± 1°C for 7 days. Embryogenic callus were osmoprotected with a mixture of 2 M glycerol and 1 M sucrose for 80 min at 25°C, and dropped in a 0.1 M CaCl2 solution containing 0.4 M sucrose at 25 ± 1°C. After 15 min of polymerization, Ca-alginate beads were dehydrated for 150 min at 0°C in a PVS2 solution containing 0.5 M sucrose. The encapsulated embryogenic calli were then plunged directly into LN for 1 h. After rapid thawing in a water bath (37°C; 2 min), the beads were washed 3 times at 10-min intervals in liquid basal MS medium containing 1.2 M sucrose. Following thawing, the embryogenic calli were transferred to fresh solid embryoid induction medium (as described above). In order to study the effect of culture condition on the survival of embryogenic calli after cryopreservation, the cryopreserved embryogenic calli were divided into two groups. One group was incubated in the dark for 2 days, and then under a 12-h photoperiod at 25 ± 1°C, while the other group were directly incubated under a 12-h photoperiod at 25 ± 1°C. Survival rate was estimated as the percentage of the total number of embryogenic calli that developed an embryoid after 30 days culture.
Effect of cryostorage duration on survival
Embryogenic calli were cooled at 6°C in the dark for 5 days on solid MS medium containing 2 mg L−1 Kn, 0.5 mg L−1 NAA, and 0.5 mg L−1 2,4-D. Then, the embryogenic calli were precultured in liquid basal MS medium enriched with 0.75 M sucrose at 25 ± 1°C for 7 days. Embryogenic calli were osmoprotected with a mixture of 2 M glycerol and 1 M sucrose for 80 min at 25°C and dropped in a 0.1 M CaCl2 solution containing 0.4 M sucrose at 25 ± 1°C. After 15 min of polymerization, Ca-alginate beads were dehydrated for 150 min at 0°C in a PVS2 solution containing 0.5 M sucrose. The encapsulated embryogenic calli were then plunged directly into LN for 1–360 days. After rapid thawing in a water bath (37°C; 2 min), the beads were washed 3 times at 10-min intervals in liquid basal MS medium containing 1.2 M sucrose. Following thawing, the embryogenic calli were transferred to fresh solid embryoid induction medium (as described above). Cultures were maintained for 2 days in the dark and then transferred to light conditions of a 12-h photoperiod at 25 ± 1°C. Survival rate was estimated as the percentage of the total number of embryogenic calli that developed an embryoid after 30 days culture.
Plant regeneration
In order to observe whether plant regenerated from cryopreserved embryogenic calli had morphological abnormalities, the regeneration of embryoids originated from cryopreserved embryogenic calli was studied. Embryogenic calli subcultured 3 times were cooled at 6°C for 5 days in the dark on solid MS medium containing 2 mg L−1 Kn, 0.5 mg L−1 NAA, and 0.5 mg L−1 2,4-D. Then, the embryogenic calli were precultured in liquid basal MS medium enriched with 0.75 M sucrose at 25 ± 1°C for 7 days. Embryogenic calli were osmoprotected with a mixture of 2 M glycerol and 1 M sucrose for 80 min at 25°C and dropped in a 0.1 M CaCl2 solution containing 0.4 M sucrose at 25 ± 1°C. After 15 min of polymerization, Ca-alginate beads were dehydrated for 150 min at 0°C in a PVS2 solution containing 0.5 M sucrose. The encapsulated embryogenic calli were then plunged directly into LN for 1–360 days. After rapid thawing in a water bath (37°C; 2 min), the beads were washed 3 times at 10-min intervals in liquid basal MS medium containing 1.2 M sucrose. Following thawing, the embryogenic calli were transferred to fresh solid embryoid induction medium. Cultures were maintained for 2 days in the dark and then transferred to light conditions of a 12-h photoperiod at 25 ± 1°C. After 30 days culture, the embryoids developed from embryogenic calli were transferred to fresh solid basal media supplemented with Kn 2 mg L−1, NAA 0.5 mg L−1, 3% (w/v) sucrose, and 0.75% (w/v) agar (regeneration medium) as recommended by Yin and Hong (2009a). After 60 days, various phenotypic characteristics (height of plantlet, internode length, number of leaves and number of roots) of the regenerated plantlets after cryopreservation and the controls were determined. The controls were embryogenic calli without cryopreservation. They were transferred to fresh solid embryoid induction medium and incubated in the dark for 2 days, and then under a 12-h photoperiod at 25 ± 1°C after being subcultured 3 times. After 30 days culture, the embryoids developed from embryogenic calli were also transferred to fresh solid plant regeneration medium (as described above).
Statistical analysis
In all experiments, approximately 25 callus clumps were used in each treatment. The survival percentages shown in the figures were presented as the mean value with their standard error (SE) of three replicates. ANOVA-analysis and Independent-sample t test (SPSS 10.0 software) was used to evaluate the results. The Duncan’s multiple range test was used as post-hoc test and a significance level of P < 0.05 was used.
Results
Effect of cold-acclimation and ABA on survival
Embryogenic calli cold-acclimated at 2 and 10°C for 5 days showed lower survival than those maintained at 6°C for 5 days (Fig. 1a). As a consequence, cold-acclimation at 6°C was applied in all subsequent experiments. Calli survival after cryopreservation was effected by the cold-acclimation time. The survival of samples cold-acclimated at 6°C for 1–10 days was significantly higher than that of samples non-cold-acclimated (Fig. 1b). Embryogenic calli cold-acclimated for 5 days at 6°C showed the highest survival (74.9%) (Fig. 1b). Therefore, 5 days of cold-acclimation at 6°C was applied in all subsequent experiments. The inclusion of 76 μM ABA in the MS medium did not significantly improve the survival of cold-acclimated embryogenic calli (Fig. 1c). Therefore, ABA treatment was not necessary for cryopreservation of Dioscorea bulbifera embryogenic calli by encapsulation-vitrification.

Survival of cryopreserved embryogenic calli. a Cold-acclimated at 2, 6, and 10°C for 5 days, respectively. b Cold-acclimated at 6°C for 0–10 days. c Treated with 0 and 76 μM ABA, respectively, at 6°C for 5 days. Bars SE for three duplications. Different letters are significantly different according to Duncan’s multiple range test (P < 0.05)
Effect of sucrose concentration in preculture medium and duration of preculture with 0.75 M sucrose on survival
The sucrose concentration in preculture medium showed a strong effect on survival of cryopreserved embryogenic calli (Fig. 2a). Survival increased as sucrose concentration increased. Optimal survival (74.6%) was observed when embryonic calli were precultured in medium containing 0.75 M sucrose. The survival of cryopreserved embryogenic calli increased as the preculture duration on 0.75 M sucrose medium increased from 0 to 7 days with an optimal value of 73.6%. Sugar exposure longer than 7 days resulted in a decrease of survival (Fig. 2b). Therefore, 7 days preculture treatment with 0.75 M sucrose was used in all subsequent experiments.

Survival of cryopreserved embryogenic calli. a Precultured in different sucrose concentrations for 7 days. b Precultured in 0.75 M sucrose for 0–10 days. Bars SE for three duplications. Different letters are significantly different according to Duncan’s multiple range test (P < 0.05)
Effect of loading solution on survival
Encapsulated embryogenic calli were treated with 2 M glycerol and 1 M sucrose for various durations (0–120 min) prior to dehydration by PVS2 at 25°C. Loading solution duration significantly influenced survival (Fig. 3): survival increased as loading time increased. The highest survival (76.3%) of cryopreserved embryogenic calli was observed after 80 min exposure time. Therefore, 80 min loading treatment with 2 M glycerol and 1 M sucrose was used in all subsequent experiments.

Survival of cryopreserved embryogenic calli loaded in a solution containing 2 M glycerol and 1 M sucrose prior to dehydration PVS2 at 25°C. Bars SE for three duplications. Different letters are significantly different according to Duncan’s multiple range test (P < 0.05)
Effect of dehydration with PVS2 on survival
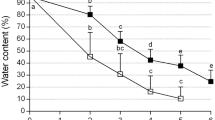
The highest survival (67.6%) were observed after 2 h vitrification at 24°C (Fig. 4a), and while PVS2 has a positive effect right from the beginning of exposure, its effect on survival is noticeable only after 30 min of dehydration. At 0°C, the survival of cryopreserved embryogenic calli also increased as dehydrating time increased, survival reaching a maximum (74.2%) after 150 min and then decreasing (Fig. 4b). Therefore, 150 min dehydration treatment with PVS2 at 0°C was used in all following experiments.

Effect of dehydration with PVS2 on survival of cryopreserved embryogenic calli. a Dehydrated with PVS2 at 24°C. b Dehydrated with PVS2 at 0°C. Bars SE for three duplications. Different letters are significantly different according to Duncan’s multiple range test (P < 0.05)
Effect of photoperiod on survival
Following cryopreservation, embryogenic calli were cultured under continuous light (12-h photoperiod) with or without a prior period of 2 days darkness. The culture conditions had a significant influence on survival. The highest survival (74.5%) of cryopreserved embryogenic calli was observed following 2 days in the dark (Fig. 5). Consequently, it is recommended that, after cryopreservation by encapsulation-vitrification, the embryogenic calli should first be cultured under continuous darkness for 2 days, prior to transfer to 12-h photoperiod conditions.

Survival of cryopreserved embryogenic calli cultured under continuous light with or without a prior period of 2 days darkness. Bars SE for three duplications. Different letters are significantly different according to Duncan’s multiple range test (P < 0.05)
Effect of cryostorage duration on survival
Figure 6 shows that whatever the cryostorage duration, the survival of embryogenic calli remained constant.

Survival of embryogenic calli cryopreserved for 1–360 days. Bars SE for three duplications. The same letter indicates no significant difference according to Duncan’s multiple range test (P>0.05)
Morphological comparison of cryopreserved-derived or non cryopreserved-derived tissues
Morphological indexes between plantlets regenerated from embryogenic calli treated with and without cryopreservation were compared (Fig. 7). Through Independent-sample t test, morphologies of recovered plantlets were similar to those of the control seedlings (P > 0.05). These data imply that it is appropriate to preserve Dioscorea bulbifera using cryopreservation by encapsulation-vitrification as genetic stability of Dioscorea bulbifera germplasm is maintained.

Comparison of morphological indexes between plantlets regenerated from D. bulbifera embryogenic calli with or without prior cryopreservation via encapsulation-vitrification. Bars SE for three duplications. The same letter indicates no significant difference according to Duncan’s multiple range test (P > 0.05)
Discussion
The cryopreservation of Dioscorea bulbifera embryogenic calli by vitrification was reported by us earlier (Hong et al. 2009). This is the first report on a successful protocol for the cryopreservation of D. bulbifera embryogenic calli by encapsulation-vitrification. The procedure described in the protocol does not require sophisticated or expensive equipment and leads to the regeneration of embryogenic calli.
Many factors can influence post-thaw recovery of cryopreserved embryogenic calli (Škrlep et al. 2008). The main ones are cold acclimation, ABA treatment, and preculture. Cold acclimation has been reported to be important for successful cryopreservation of most in vitro-grown plants. Cold acclimation is likely to enhance the accumulation of endogenous solutes that increase cell tolerance to osmotic excursion and dehydration as well as the glass-forming tendency within the cytoplasm (Steponkus et al. 1992). Our study indicated that cold-acclimated embryogenic calli of Dioscorea bulbifera showed a higher freezing tolerance than controls, and that ABA did not affect the freezing tolerance responses. However, in the shoot-tip cryopreservation of Dioscorea bulbifera explants using vitrification and modified droplet, cold acclimation at 10°C had no beneficial effects on survival and regrowth in comparison to culture at 25°C (Leunufna and Keller 2003). ABA treatments were previously shown to confer tolerance to cryopreservation in cell cultures of Alfalfa (Reaney and Gusta 1987); in the present study, ABA could not substitute for the effects of cold acclimation.
Preculturing in media containing sugars seems to be a critical step in the successful cryostorage of tissues using the vitrification procedure (Yin and Hong 2009b). High sugar concentration and its treatment time in the cytoplasm helps to establish a vitrified state during cryopreservation and enables cells to tolerate dehydration that can cause freezing damage (Yin and Hong 2009b). Precultured in two different sucrose concentration (10%, 20%) for 5 days, the survival rate of cryopreserved shoots of Dioscorea bulbifera using vitrification methods is up to 17% (Leunufna and Keller 2003). While Dioscorea deltoidea Wall shoot tips were precultured with 0.75 M sucrose, the highest survival of cryopreserved shoot tips was obtained after 3 days preculture (Mandal and Sonali 2007). The positive effect of extending duration of the preculture with high sucrose concentration has been reported for Dioscorea floribunda (Mandal and Sangeeta 2007), Dioscorea spp. (Mandal et al. 1996), and Dioscorea deltoidea (Sonali et al. 2005). The present study obtained a similar result: culturing embryogenic calli on medium supplemented with 0.75 M sucrose for 7 days prior to LN treatments was effective in improving the survival of cryopreserved embryogenic calli of Dioscorea bulbifera by encapsulation-vitrification.
It is indispensable for the precultured embryogenic calli to be osmoprotected with a loading solution during the encapsulation. A previous study showed that the highest survival of cryopreserved Dioscorea deltoidea shoot tips was obtained after 20 min loading with 2 M glycerol plus 0.4 M sucrose at 25°C (Mandal and Sonali 2007). A similar effect was observed in the present study. Although the precise mechanism by which the loading treatment induces osmotolerance to PVS2 is as yet unknown, the loading solution may minimize the osmotic stress caused by severe dehydration and thus protect the cell membrane from injury during dehydration and subsequent freezing (Yin and Hong 2009b).
In cryopreservation by encapsulation-vitrification, precultured embryogenic calli have to be dehydrated by exposure to a vitrification solution such as PVS2, prior to direct immersion in LN. However, the direct exposure of embryogenic calli to PVS2 without osmoprotection caused harmful effects, because of osmotic stress or chemical toxicity, which has been described as a major factor in determining the success of cryopreservation by encapsulation-vitrification (Hirai and Sakai 1999); therefore, it is essential to induce a high level of osmotolerance to PVS2. Incubation duration and temperature in PVS2 are two important factors affecting survival of cryopreserved materials, since they determine the extent of cell dehydration and the amount of cryoprotectants permeated into the cells (Wang et al. 2004). Mandal and Sangeeta (2007) reported that when treated with PVS2 for 90 min at 0°C using the vitrification method, the survival of Dioscorea floribunda was remarkably improved. Dehydrated with PVS2 for 20 min using vitrification methods, the survival of Dioscorea bulbifera cryopreserved explants also increased to about 58.9% (Leunufna and Keller 2003). Therefore, the optimal incubation time in PVS2 varies with plant species, size of materials, and temperature during incubation. In the present study, the optimal duration and temperature to PVS2 were respectively 150 min and 0°C. Many reports have shown that dehydration at 0°C can reduce the toxicity of vitrification solutions, compared with that at 24°C, usually yielding higher survival (Yin and Hong 2009b). Also, the incubation time at 0°C was greatly extended compared with that at 24°C, thus allowing much more flexibility for handling a large number of samples at the same time (Yin and Hong 2009b). When Dioscorea deltoidea Wall shoot tips were dehydrated with PVS2 for 90 min at 0°C using encapsulation-dehydration, the highest regeneration frequency was 76% (Mandal and Sonali 2007). The results reported here are consistent with those of Dixit et al. (2003). However, Leunufna and Keller (2003) found that dehydration at 0°C or at room temperature has no significant effects on the survival and regrowth of cryopreserved yam explants using vitrification methods.
Cryostorage duration has no obvious influence on plant regeneration. Our results showed that the survival of cryopreserved embryogenic calli of Dioscorea bulbifera, maintained in LN for up to 1 year, remain above 68.1%. Similarly, when cryopreserved for 360 days by encapsulation-vitrification, the Dioscorea deltoidea Wall shoot tips maintained their viability and an unaltered level of regeneration capability (Mandal and Sonali 2007). Furthermore, the morphological indexes between the regenerated plantlets treated with or without cryopreservation showed no significant differences. These research results are in agreement with some previous reports on cryopreservation of Dioscorea bulbifera embryogenic tissues (Dixit et al. 2003) confirming that long-term conservation is feasible. The culture conditions applied during recovery had an effect on the survival of cryopreserved embryogenic calli. In particular, culture in darkness for a short time after post-thawing allowed a significant improvement in survival. This result is in accordance with the observations of cryopreservation of Dendrobium candidum Wall. ex Lindl. protocorm-like bodies (Yin and Hong 2009b).
In the present study, we have developed a simple and efficient method for cryopreservation of Dioscorea bulbifera embryogenic calli. With the optimized parameters reported here, a high survival (about 70%) of cryopreserved embryogenic calli was achieved. The present study has shown that the cryopreservation by encapsulation-vitrification is easy to apply, and that it appears to be a promising technology for the conservation of Dioscorea bulbifera embryogenic calli and other plant germplasm.
Abbreviations
- MS:
-
Murashige and Skoog
- NAA:
-
α-naphthalene acetic acid
- Kn:
-
Kinetin
- LN:
-
Liquid nitrogen
- 2,4-D:
-
2,4-dichlorophenoxy-acetic acid
References
Adriana S, Mirta F, Ricardo M, Sof′ıa O, Luis M (2004) Plant recovery of cryopreserved apical meristem-tips of Melia azedarach L. using encapsulation/dehydration and assessment of their genetic stability. Euphytica 135:29–38
Alka N, Sanjeev K, Srivastava PS (2005) Abiotic metal stress enhances diosgenin yield in Dioscorea bulbifera L. cultures. Plant Cell Rep 24:250–254
Alka N, Sanjeev K, Srivastava PS (2007) Genetic fidelity of in vitro regenerants, encapsulation of shoot tips and high diosgenin content in Dioscorea bulbifera L., a potential alternative source of diosgenin. Biotechnol Lett 29:623–629
Bachiri Y, Gazeau C, Hansz J, Morisset C, Dereuddre J (1995) Successful crypreservation of suspension cells by encapsulation-dehydration. Plant Cell Tiss Organ Cult 43:241–248
Dixit S, Mandal BB, Ahuja S, Srivastava PS (2003) Genetic stability assessment of plants regenerated from cryopreserved embryogenic tissues of Dioscorea bulbifera l. using RAPD, biochemical and morphological analysis. Cryoletters 24:77–84
Engelmann F (2000) Importance of crop for conservation of plant genetic resources. In: Engelmann F, Takagi H (eds) Cryopreservation of tropical plant germplasm. JIRCAS, Tsukuba, Japan, pp 8–20
Hirai D, Sakai A (1999) Cryopreservation of in vitro-grown axillary shoot-tip meristems of mint (Mentha spicata L.) by encapsulation vitrification. Plant Cell Rep 19:150–155
Hong SR, Yin MH (2009) High-efficiency vitrification protocols for cryopreservation of in vitro grown shoot tips of rare and endangered plant Emmenopterys henryi Oliv. Plant Cell Tiss Organ Cult 99:217–226
Hong S, Yin M, Shao X, Wang A, Xu W (2009) Cryopreservation of embryogenic callus of Dioscorea bulbifera by vitrification. CryoLetters 30:64–75
Inoue M, Akiko T (2009) Secondary dispersal of Dioscorea japonica (Dioscoreaceae) bulbils by rodents. J For Res 14:95–100
Kazumasa H, Misuzu M, Saeko G, Masumi IK, Kenji Y, Sakai A, Kazuhisa M (2002) Cryopreservation of hairy root cultures of Vinca minor (L.) by encapsulation-dehydration. Biotechnol Lett 24:371–376
Leunufna S, Keller ERJ (2003) Investigating a new cryopreservation protocol for yams (Dioscorea spp.). Plant Cell Rep 21:1159–1166
Mandal BB, Sangeeta AG (2007) Regeneration of Dioscorea floribunda plants from cryopreserved encapsulated shoot tips: effect of plant growth regulators. CryoLetters 28:329–336
Mandal BB, Sonali DS (2007) Cryopreservation of in vitro shoot tips of Dioscorea deltoidea Wall., an endangered medicinal plant: effect of cryogenic procedure and storage duration. CryoLetters 28:461–470
Mandal BB, Dwivedi S, Chandel KPS (1996) Cryopreservation of yam (Dioscorea spp) shoot apices by encapsulation-dehydration. CryoLetters 17:165–174
Marc S, Anne-Claire MO, Marie-Aleth LD (2007) The Dioscorea genus: a review of bioactive steroid saponins. J Nat Med 61:91–101
Murashige T, Skoog F (1962) A revised medium for rapid growth and bio assays with tobacco tissue cultures. Plant Physiol 15:473–497
Reaney MJ, Gusta LV (1987) Factors influencing the induction of freezing tolerance by abscisic acid in cell suspension cultures of Bromus inermis Leyss and Medicago sativa L. Plant Physiol 83:423–427
Sakai A, Kobayashi S, Oiyama I (1990) Cryopreservation of nucellar cells of navel orange (Citrus sinensis Osb. var. brasiliensis Tanaka) by vitrification. Plant Cell Rep 9:30–33
Škrlep K, Bergant M, Winter GMD, Bohanec B, Žel J, Verpoorte R, Van Iren F, Camloh M (2008) Cryopreservation of cell suspension cultures of Taxus × media and Taxus floridana. Biol Plant 52:329–333
Sonali DS, Ahuja GS, Mandal BB, Srivastava PS (2005) Metabolic stability of plants regenerated from cryopreserved shoot tips of Dioscorea deltoidea—an endangered medicinal plant. Sci Hortic 105:513–517
Steponkus PL, Langis LR, Fujikawa S (1992) Cryopreservation of plant tissues by vitrification. In: Steponkus PL (ed) Advances in low-temperature biology, vol 1. JAI Press, London, pp 1–61
Suzuki M, Tandon P, Ishikawa M, Toyomasu T (2008) Development of a new vitrification solution, VSL, and its application to the cryopreservation of gentian axillary buds. Plant Biotechnol Rep 2:123–131
Tang ZX, Zhou Y, Zeng YK, Zang SL, He PG, Fang YZ (2006) Capillary electrophoresis of the active ingredients of Dioscorea bulbifera L. and its medicinal preparations. Chromatographia 63:617–622
Wang QC, Munir M, Nachman S, Li P, Colova-Tsolova V, Ron G, Ilan S, Edna T, Avihai P (2004) Cryopreservation of grapevine (Vitis spp.) embryogenic cell suspensions by encapsulation–vitrification. Plant Cell Tiss Organ Cult 77:267–275
Wheeler GS, Pemberton RW, Raz L (2007) A biological control feasibility study of the invasive weed-air potato, Dioscorea bulbifera L. (Dioscoreaceae): an effort to increase biological control transparency and safety. Nat Areas J 27:269–279
Wolfgang W, Hyeon-Yong P (2004) The apparent loss of tissue culture competence during leaf differentiation in yams (Dioscorea bulbifera L.). Plant Cell Tiss Organ Cult 34:101–105
Yin MH, Hong SR (2009a) Establishment of regeneration system of leaves and stems of Dioscorea bulbifera L. virus-free plantlets. Bulletin of Botanical Res 29:492–499
Yin MH, Hong SR (2009b) Cryopreservation of Dendrobium candidum wall. ex Lindl. protocorm-like bodies by encapsulation-vitrification. Plant Cell Tiss Organ Cult 98:179–185
Acknowledgments
This research was supported by Science and Technology Foundation of the Education Department of Jiangxi province (No. GJJ09374) and key scientific and technological plan of Shangrao Normal University in 2010-2011 (SR1014).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ming-Hua, Y., Sen-Rong, H. A simple cryopreservation protocol of Dioscorea bulbifera L. embryogenic calli by encapsulation-vitrification. Plant Cell Tiss Organ Cult 101, 349–358 (2010). https://doi.org/10.1007/s11240-010-9695-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11240-010-9695-7