
Abstract
Sulfamic acid pyridinium chloride-functionalized Fe3O4 nanoparticles as a novel organic-inorganic hybrid heterogeneous catalyst was manufactured and characterized by FT-IR, XRD, TGA, SEM, TEM and VSM techniques. The catalytic activity of the nanomagnetic catalyst was investigated in the multicomponent reactions of arylaldehydes, 7-amino-4-methylcoumarin and dialkyl acetylenedicarboxylate that afford the novel N-coumarin-2-furanones. This green nanocatalytic procedure has good reversibility and provides clean production in a short reaction time.
Graphical Abstract
Preparation and characterization of sulfamic acid pyridinium chloride-functionalized Fe3O4 nanoparticles as a novel magnetic catalyst for synthesis of novel N-coumarin-2-furanone.

Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Nanomaterials are of great interest in organic synthesis due to their extremely small size and large surface-to-volume ratio, which lead to both chemical and physical differences in their properties compared to bulk of the same chemical composition, such as mechanical, biological and satirical properties, higher catalytic activity, thermal and electrical conductivity, optical absorption and melting point [1,2,3]. Surface-functionalized iron oxide magnetic nanoparticles (MNPs) have been widely used in biotechnology and catalysis [4,5,6,7,8]. MNPs can easily be separated and recycled from the products by their response to an external magnetic field. Good biocompatibility and biodegradability as well as basic magnetic characteristics could be designated for functional organic materials grafted to an MNP [9,10,11,12].
Multicomponent reactions (MCRs) belong to the most challenging areas of modern chemistry for several reasons such as reduction of the number of steps, time savings, material savings, higher yields than in comparable multistep reactions and fewer by-products [13,14,15]. Synthesis of medicine and complex molecules should be easy and efficient with minimal workup in this methodology [16, 17].
2-furanone derivatives are applicable synthetic modules in organic synthesis and are the main structural elements present in many pharmaceutical active products [18, 19]. Additionally, the coumarins are present in a variety of naturally occurring compounds that have antiallergic, antidiabetic, analgesic, anticoagulant, anticancer, antiancaphylactia, antibacterial and fungicidal activities [20,21,22,23,24]. These results encouraged us to synthesize novel N-coumarin-2-furanones using MCRs. In continuation of our research on synthesis of heterocyclic compounds in the presence of nanomagnetic catalyst [25, 26], herein we wish to describe the preparation of novel N-coumarin-2-furanone derivatives 4 from reactions of various arylaldehydes 1, 7-amino-4-methylcoumarin 2 and dialkyl acetylenedicarboxylate 3 in the presence of sulfamic acid pyridinium chloride-functionalized Fe3O4 nanoparticles (SA-PYCA-Fe3O4) as a novel green catalyst (Scheme 1).

Synthesis of novel N-coumarin-2-furanones 4 derivatives in the presences of SA-PYCA-Fe3O4 nanocatalyst
Experimental
Chemicals and materials
Melting points were measured on an Electrothermal 9100 apparatus. The X-ray powder diffraction (XRD) of the catalyst was carried out on a Philips PW 1830 X-ray diffractometer with CuKα source (λ = 1.5418 Å) in a range of Bragg’s angle (10–80°) at room temperature. Scanning electron microscopy (SEM) analyses were conducted using a VEGA//TESCAN KYKY-EM 3200 microscope (acceleration voltage 26 kV). Transmission electron microscopy (TEM) experiments were conducted on a Philips EM 208 electron microscope. Thermogravimetric analysis (TGA) was recorded on a Stanton Red craft STA-780 (London, UK). Nuclear magnetic resonance (NMR) spectra were recorded with a Bruker DRX-400 AVANCE instrument (400.1 MHz for 1H, 100.6 MHz for 13C). The spectra were measured in DMSO-d6 as solvent. Mass spectra were recorded on a Finnigan-Matt 8430 mass spectrometer operating at an ionization potential of 70 eV. Elemental analyses were performed using a Heracus CHN-O Rapid analyzer. IR spectra were recorded on an FT-IR Bruker vector 22 spectrophotometer. Magnetic measurements were performed using vibration sample magnetometer (VSM, MDK, and Model 7400) analysis.
Preparation of pyridine-4-carboxylic acid-functionalized Fe3O4 nanoparticles (Fe3O4–PYCA)
FeCl3·6H2O and FeCl2·4H2O (molar ratio 1:2) were added to 80 mL od deionized water and sonicated until the salts dissolved completely. Then, a predetermined amount of pyridine-4-carboxylic acid and ammonium hydroxide solution were added to the above mixture under N2 atmosphere at room temperature until the pH was raised to 11. The color of the reaction solution turned black immediately, indicating the spontaneous formation of nanoparticles. This suspension was then refluxed at 100 °C for a designed period of time and was then cooled to ambient room temperatures. After the complete reaction, the Fe3O4–PYCA nanoparticles were separated by a magnetic field, washed with distilled water five times and then dried in an oven for 12 h (Scheme 2) [25].

Preparation of Fe3O4-PYCA nanoparticles
Preparation of novel sulfamic acid pyridinium chloride-functionalized Fe3O4 nanoparticles (SA-PYCA-Fe3O4)
The Fe3O4–PYCA (0.5 g) was dispersed in dry CH2Cl2 (10 mL) by ultrasonic bath for 30 min. Eventually, chlorosulfuric acid (0.7 mL) was added dropwise over a period of 25 min at room temperature. Hydrogen gas expelled from the reaction. Then, the prepared functionalized MNPs were separated by magnetic field and washed with dry CH2Cl2 four times to remove the unattached substrates (Scheme 3).

Preparation of SA-PYCA-Fe3O4 nanoparticles
General procedure for the synthesis of novel N-coumarin-2-furanone derivatives by SA-PYCA-Fe3O4
To a mixture of arylaldehydes (1 mmol), 7-amino-4-methylcoumarin (1 mmol) and dialkyl acetylenedicarboxylate (1 mmol) in ethanol (5 mL) was added SA-PYCA-Fe3O4 (15 mg) as a nanocatalyst. The mixture was stirred at 60 °C for 2–3 h. When the reaction was completed (as monitored by thin layer chromatography (TLC)], the solvent was removed under reduced pressure. Then, the mixture was diluted with CH2Cl2 and the SA-PYCA-Fe3O4 nanoparticles were separated by a magnet field. The solution containing the product was evaporated to give the solid. The crude product was purified by washing with hot ethanol to give the desired product as a yellow powder.
Analytical data for all products:
Methyl 4-(4-methyl-2-oxo-2H-chromen-7-ylamino)-2,5-dihydro-5-oxo-2-phenylfuran-3-carboxylate ( 4a ):
Light yellow powder, melting point (m.p.): 220 °C; IR (KBr): νmax = 3340, 3067, 2955, 1717, 1657, 1615, 1434 cm−1; 1H NMR (400.13 MHz, DMSO): δ = 2.34 (s, 3H, CH3), 3.60 (s, 3H, OCH3), 6.21 (s, 1H, CHfuran), 6.28 (s, 1H, CHcoumarin), 7.17–7.26 (m, 3H, 3CHaromatic), 7.33 (d, 2H, J = 7.2 Hz, 2CHaromatic), 7.62–7.72 (m, 3H, 3CHaromatic), 11.77 (s, 1H, N–H) ppm; 13C NMR (100.6 MHz, DMSO): δ = 18.4, 51.6, 60.7, 109.3, 112.9, 113.8, 116.9, 118.0, 126.2, 128.1, 128.5, 128.8, 136.7, 139.7, 152.7, 153.3, 153.5, 160.1, 162.8, 165 ppm. MS (EI, 70 eV) (%) 391 (M+, 100), 359 (25), 332 (16), 263 (29), 202 (50), 189 (46), 158 (13), 130(44); anal. calcd. for C22 H17 NO6 (391.11): C, 67.51; H, 4.38; N, 3.58%; found: C, 67.78; H, 4.40; N, 3.55%.
Ethyl 4-(4-methyl-2-oxo-2H-chromen-7-ylamino)-2,5-dihydro-5-oxo-2-phenylfuran-3-carboxylate ( 4b ):
Light yellow powder, m.p.: 250–251 °C; IR (KBr): νmax = 3369, 3067, 2955, 2925, 1715, 1657, 1615, 1459 cm−1; 1H NMR (400.13 MHz, DMSO): δ = 1.59 (t, 3H, J = 8.4 Hz, CH3), 2.35 (d, 3H, J = 0.8 Hz, CH3), 3.99–4.10 (m, 2H, OCH2), 6.21 (s, 1H, CHfuran), 6.28 (d, 1H, J = 1.2 Hz, CHcoumarin), 7.17 (t, 1H, J = 7.6 Hz, 1CHaromatic), 7.24 (t, 1H, J = 7.6 Hz, 1CHaromatic), 7.34 (d, 1H, J = 7.6 Hz, 1CHaromatic), 7.62–7.68 (m, 2H, 2CHaromatic), 7.72 (d, 1H, J = 2 Hz, 1CHaromatic), 11.71 (s, 1H, N–H) ppm; 13C NMR (100.6 MHz, DMSO): δ = 14.4, 18.4, 60.3, 60.8, 109.3, 113.3, 113.8, 117.0, 118.0, 126.2, 128.2, 128.6, 128.8, 136.7, 139.8, 152.5, 153.4, 153.5, 160.2, 162.3, 165.0 ppm. MS (EI, 70 eV) (%) 405 (M+, 100), 359 (20), 332 (14), 203 (30), 158 (7), 130 (19); anal. calcd. for C23 H19 NO6 (405.12): C, 68.14; H, 4.72; N, 3.46%; found: C, 68.11; H, 4.70; N, 3.41%.
Methyl 4-(4-methyl-2-oxo-2H-chromen-7-ylamino)-2-(4-cyanophenyl)-2,5-dihydro-5-oxofuran-3-carboxylate ( 4c ):
Light yellow powder, m.p.: 200–202 °C; IR (KBr): νmax = 3429, 3067, 2955, 2229, 1726, 1658, 1613, 1428 cm−1; 1H NMR (400.13 MHz, DMSO): δ = 2.35 (d, 3H, J = 0.8 Hz, CH3), 3.60 (s, 3H, OCH3), 6.30 (d, 1H, J = 0.8 Hz, CHcoumarin), 6.35 (s, 1H, CHfuran), 7.60–7.63 (m, 3H, 3CHaromatic), 7.67–7.73 (m, 4H, 4CHaromatic), 12.19 (s, 1H, N–H) ppm; 13C NMR (100.6 MHz, DMSO): δ = 18.4, 51.8, 109.4, 11.4, 112.1, 114.0, 117.2, 117.9, 118.9, 126.4, 129.4, 132.8, 139.4, 142.7, 153.1, 153.3, 153.6, 160.1, 162.7, 164.8 ppm. MS (EI, 70 eV) (%) 416 (M+, 35), 384 (35), 358 (8), 288 (6), 214 (8), 202 (0.30), 155 (10); anal. calcd. for C23 H16 N2O6 (391.11): C, 66.34; H, 3.87; N, 6.73%; found: C, 66.31; H, 3.89; N, 6.74%.
Ethyl 4-(4-methyl-2-oxo-2H-chromen-7-ylamino)-2-(4-cyanophenyl)-2,5-dihydro-5-oxofuran-3-carboxylate ( 4d ):
Light yellow powder, m.p.: 255–256 °C; IR (KBr): νmax = 3421, 3061, 2925, 2231, 1724, 1686, 1648, 1613, 1425 cm−1; 1H NMR (400.13 MHz, DMSO): δ = 1.25 (t, 3H, J = 7.2 Hz, CH3), 2.38 (d, 3H, J = 1.2 Hz, CH3), 4.26 (q, 2H, J = 7.2 Hz, OCH2), 5.85 (s, 1H, CHfuran), 6.26 (d, 1H, J = 1.2 Hz, CHcoumarin), 7.36 (d, 1H, J = 2.4 Hz, 1CHaromatic), 7.42 (d, 2H, J = 8.4 Hz, 2CHaromatic), 7.54 (d, 1H, J = 8.8 Hz, 1CHaromatic), 7.22 (d, 2H, J = 8.4 Hz, 2CHaromatic), 7.72 (dd, 1H, J = 8.8, 2.4 Hz, 1CHaromatic), 9.14 (s, 1H, N–H) ppm; 13C NMR (100.6 MHz, DMSO): δ = 14.0, 18.5, 60.5, 61.9, 108.6, 112.8, 113.0, 114.6, 117.0, 117.5, 118.0, 125.5, 128.2, 132.8, 138.9, 140.2, 151.8, 153.9, 156.3, 160.4, 162.7, 164.5 ppm. MS (EI, 70 eV) (%) 430 (M+, 84), 384 (34), 357 (8), 228 (20), 202 (100), 155 (50); anal. calcd. for C24 H18 N2O6 (430.12): C, 66.97; H, 4.22; N, 6.51%; found: C, 66.96; H, 4.20; N, 6.53%.
Methyl 4-(4-methyl-2-oxo-2H-chromen-7-ylamino)-2,5-dihydro-2-(3-nitrophenyl)-5-oxofuran-3-carboxylate ( 4e ):
Light yellow powder, m.p.: 285–286 °C; IR (KBr): νmax = 3263, 3092, 2953, 1730, 1659, 1613, 1528, 1435, 1384 cm−1; 1H NMR (400.13 MHz, DMSO): δ = 2.50 (s, 3H, CH3), 3.61 (s, 3H, OCH3), 6.28 (s, 1H, CHcoumarin), 6.45 (s, 1H, CHfuran), 7.54 (t, 1H, J = 8 Hz, 1CHaromatic), 7.63–7.69 (m, 2H, 2CHaromatic), 7.78 (d, 1H, J = 1.6 Hz, 1CHaromatic), 7.22 (d, 1H, J = 8 Hz, 1CHaromatic), 8.08 (dd, 1H, J = 8, 1.6 Hz, 1CHaromatic), 8.35 (s, 1H, 1CHaromatic), 12.18 (s, 1H, N–H) ppm; 13C NMR (100.6 MHz, DMSO): δ = 18.4, 51.8, 59.7, 109.5, 112.0, 114.0, 117.2, 118.0, 123.6, 123.7, 126.4, 130.5, 134.4, 139.3, 139.4, 148.1, 153.1, 153.2, 153.6, 160.1, 162.7, 164.8 ppm. MS (EI, 70 eV) (%) 436 (M+, 55), 404 (6), 348 (4), 308 (100), 261 (9), 201 (17), 175 (11); anal. calcd. for C22 H16 N2O8 (436.09): C, 60.55; H, 3.70; N, 6.42%; found: C, 60.52; H, 3.71; N, 6.40%.
Ethyl 4-(4-methyl-2-oxo-2H-chromen-7-ylamino)-2,5-dihydro-2-(3-nitrophenyl)-5-oxofuran-3-carboxylate ( 4f ):
Light yellow powder, m.p.: 270–271 °C; IR (KBr): νmax = 3434, 3089, 2986, 1728, 1651, 1613, 1528, 1439, 1357 cm−1; 1H NMR (400.13 MHz, DMSO): δ = 1.13 (t, 3H, J = 6.8 Hz, CH3), 2.34(d, 3H, J = 1.2 Hz, CH3), 4.03 (m, 2H, OCH2), 6.28 (d, 1H, J = 1.2 Hz, CHcoumarin), 6.45 (s, 1H, CHfuran), 7.52 (s, 1H, 1CHaromatic), 7.64 (m, 2H, 2CHaromatic), 7.67 (d, 2H, J = 8 Hz, 2CHaromatic), 8.05 (dd, 1H, J = 8, 1.6 Hz, 1CHaromatic), 8.39 (s, 1H, 1CHaromatic), 12.16 (s, 1H, N–H) ppm; 13C NMR (100.6 MHz, DMSO): δ = 14.4, 18.4, 59.8, 60.4, 109.4, 112.2, 113.9, 117.2, 118.0, 123.7, 123.9, 126.4, 130.5, 134.3, 139.3, 139.5, 148.0, 153.3, 153.6, 160.1, 162.1, 164.8 ppm. MS (EI, 70 eV) (%) 450 (M+, 96), 404 (32), 378 (11), 308 (26), 248 (28), 232 (22), 202 (100), 173 (59); anal. calcd. for C23 H18 N2O8 (450.11): C, 61.33; H, 4.03; N, 6.22%; found: C, 61.32; H, 4.01; N, 6.24%.
Methyl 4-(4-methyl-2-oxo-2H-chromen-7-ylamino)-2-(4-bromophenyl)-2,5-dihydro-5-oxofuran-3-carboxylate ( 4g ):
Light yellow powder, m.p.: 220–222 °C; IR (KBr): νmax = 3431, 3060, 2986, 1725, 1648, 1613, 1437 cm−1; 1H NMR (400.13 MHz, DMSO): δ = 2.35 (s, 3H, CH3), 3.61 (s, 3H, OCH3), 6.23 (s, 1H, CHcoumarin), 6.29 (s, 1H, CHfuran), 7.33 (d, 2H, J = 7.6 Hz, 2CHaromatic), 7.43 (d, 2H, J = 7.6 Hz, 2CHaromatic), 7.61 (d, 1H, J = 8.4 Hz, 1CHaromatic), 7.67 (d, 1H, J = 8.4 Hz, 1CHaromatic), 7.72 (s, 1H, 1CHaromatic), 12.04 (s, 1H, N–H) ppm; 13C NMR (100.6 MHz, DMSO): δ = 18.4, 51.7, 60.0, 109.4, 112.5, 113.9, 117.1, 118.0, 121.7, 126.2, 130.5, 131.8, 136.3, 139.5, 152.7, 153.3, 153.6, 160.1, 162.7, 164.8 ppm. MS (EI, 70 eV) (%) 471 (M+, 100), 437 (20), 411 (9), 343 (40), 267 (29), 202 (47), 157 (5); anal. calcd. for C22 H16 N2O8 (471.01): C, 56.19; H, 3.43; N, 2.98%; found: C, 56.17; H, 3.42; N, 2.99%.
Methyl 4-(4-methyl-2-oxo-2H-chromen-7-ylamino)-2-(4-chlorophenyl)-2,5-dihydro-5-oxofuran-3-carboxylate ( 4h ):
Light yellow powder, m.p.: 265–266 °C; IR (KBr): νmax = 3434, 2954, 1724, 1656, 1614, 1427 cm−1; 1H NMR (400.13 MHz, DMSO): δ = 2.35 (d, 3H, J = 0.8 Hz, CH3), 3.60 (s, 3H, OCH3), 6.23 (s, 1H, CHfuran), 6.29 (d, 1H, J = 0.8 Hz, CHcoumarin), 7.29 (d, 2H, J = 8.4 Hz, 2CHaromatic), 7.39 (d, 2H, J = 8.4 Hz, 2CHaromatic), 7.61 (dd, 1H, J = 8.8, 2 Hz, 1CHaromatic), 7.67 (d, 1H, J = 8.8 Hz, 1CHaromatic), 7.72 (d, 1H, J = 2 Hz, 1CHaromatic), 11.82 (s, 1H, N–H) ppm; 13C NMR (100.6 MHz, DMSO): δ = 18.4, 51.6, 60.0, 109.4, 112.0, 113.9, 117.0, 118.0, 126.2, 128.9, 130.1, 133.0, 136.1, 139.6, 153.3, 153.4, 153.6, 160.1, 162.9, 165.1 ppm. MS (EI, 70 eV) (%) 425 (M+, 100), 393 (19), 366 (9), 297 (14), 223 (46), 202 (48), 155 (18); anal. calcd. for C22 H16 ClNO6 (425.07): C, 62.05; H, 3.79; N, 3.29%; found: C, 62.03; H, 3.78; N, 3.26%.
Methyl 4-(4-methyl-2-oxo-2H-chromen-7-ylamino)-2,5-dihydro-2-(4-nitrophenyl)-5-oxofuran-3-carboxylate ( 4i ):
Light yellow powder, m.p.: 227 °C; IR (KBr): νmax = 3437, 3076, 2954, 1728, 1658, 1613, 1522, 1434, 1354 cm−1; 1H NMR (400.13 MHz, DMSO): δ = 2.51 (s, 3H, CH3), 3.60 (s, 3H, OCH3), 6.30 (d, 1H, J = 0.8 Hz, CHcoumarin), 6.42 (s, 1H, CHfuran), 7.63–7.75 (m, 5H, 5CHaromatic), 8.09 (d, 2H, J = 8.4 Hz, 2CHaromatic), 12.26 (s, 1H, N–H) ppm; 13C NMR (100.6 MHz, DMSO): δ = 18.4, 51.7, 59.8, 109.4, 111.6, 114.0, 117.2, 118.0, 124.0, 126.4, 129.7, 139.4, 144.9, 147.4, 147.7, 153.3, 153.6, 160.1, 162.8, 165.0 ppm. MS (EI, 70 eV) (%) 436 (M+, 88), 404 (36), 308 (22), 201 (100), 175 (59), 158 (26), 59 (28); anal. calcd. for C22 H16 N2O8 (436.09): C, 60.55; H, 3.70; N, 6.42%; found: C, 60.53; H, 3.69; N, 6.44%.
Ethyl 4-(4-methyl-2-oxo-2H-chromen-7-ylamino)-2,5-dihydro-2-(4-nitrophenyl)-5-oxofuran-3-carboxylate ( 4j ):
Light yellow powder, m.p.: 201–203 °C; IR (KBr): νmax = 3427, 3113, 2980, 1728, 1660, 1613, 1522, 1434, 1355 cm−1; 1H NMR (400.13 MHz, DMSO): δ = 1.13 (t, 3H, J = 7.2 Hz, CH3), 2.34 (d, 3H, J = 1.2 Hz, CH3), 4.03 (m, 2H, OCH2), 6.29 (d, 1H, J = 1.2 Hz, CHcoumarin), 6.42 (s, 1H, CHfuran), 7.64 (dd, 2H, J = 8.8, 1.6 Hz, 2CHaromatic), 7.70 (d, 2H, J = 8.8 Hz, 2CHaromatic), 7.75 (d, 1H, J = 1.6 Hz, 1CHaromatic), 8.09 (d, 2H, J = 8.8 Hz, 2CHaromatic), 12.15 (s, 1H, N–H) ppm; 13C NMR (100.6 MHz, DMSO): δ = 14.5, 18.4, 59.9, 60.4, 109.3, 112.0, 114.0, 117.2, 117.9, 123.9, 126.4, 129.8, 139.4, 144.9, 147.7, 153.3, 153.5, 153.6, 160.1, 162.2, 164.9 ppm. MS (EI, 70 eV) (%) 450 (M+, 100), 404 (27), 378 (18), 308 (5), 248 (5), 202 (21), 175 (9), 151 (10); anal. calcd. for C23 H18 N2O8 (450.11): C, 61.33; H, 4.03; N, 6.22%; found: C, 61.30; H, 4.00; N, 6.25%.
Results and discussion
Characterization of the prepared Fe3O4–PYCA and SA-PYCA-Fe3O4 nanoparticles
X-ray diffraction (XRD) analysis
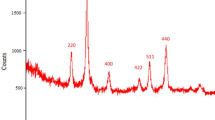
X-ray diffraction using a Cu Kα irradiation was used to characterize the preservation of the crystal structure of the samples after the functionalization step. The result shown in Fig. 1 was fitted for observed six peaks with the following miller indices: (2 2 0), (3 1 1), (4 0 0), (4 2 2), (5 1 1) and (4 4 0) that existing phases were identified as Fe3O4 nanoparticles. The average particle diameter was determined by the Scherrer equation [27, 28]. The calculation led for PYCA-Fe3O4 to particle sizes of about 15 nm and SA-PYCA-Fe3O4 nanoparticles sizes of about 21 nm which is in a good agreement with TEM observations.

XRD powder patterns of PYCA-Fe3O4 nanoparticles and SA-PYCA-Fe3O4 nanoparticles
Fourier transform infrared (FT-IR) analysis
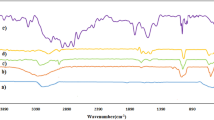
FT-IR measurements were carried out to identify the organic group for capping and efficient stabilization of the synthesized Fe3O4 nanoparticles. The FT-IR spectra of Fe3O4 appear at 632 and 590 cm−1, which can be ascribed to the vibrations of the Fe–O group. The specific absorption peaks of 1624, 2924 and 2854 cm−1 are attributed to the C=O, C–O and C–H stretching of the PYCA unit, respectively. Reaction of PYCA-Fe3O4 with chlorosulfuric acid produces SA-PYCA-Fe3O4 in which the presence of a sulfonyl moiety is asserted with 1204 and 1128 cm−1 bands in the FT-IR spectra. Therefore, the data obtained from FT-IR spectroscopy can be confirmed the existence of the nanomagnetic particle and organic group moiety in the structure of SA–PYCA-Fe3O4 nanoparticles (Fig. 2).

FT-IR spectra for Fe3O4 nanoparticles, PYCA-Fe3O4 and SA-PYCA-Fe3O4 nanoparticles
Thermogravimetric analysis (TGA)
Information about loading of Fe3O4 nanoparticles with an organic group was obtained by TGA. The results revealed that the SA-PYCA-Fe3O4 nanoparticles contain about 13% of organic material (volatile components disappearing until a temperature of about 100 °C were neglected; Fig. 3). The weight loss of PYCA-modified MNPs appears to be about 6.2% at 300 °C which contributes to the thermal decomposition of the pyridine-4-carboxylic groups. For SA-PYCA-Fe3O4, there is a well-defined mass weight loss of 6.8% between 300 and 860 °C related to the breakdown of the SA moieties. On the basis of these results, the good grafting of PYCA and SA groups on the Fe3O4 nanoparticles is confirmed.

TGA thermograms of Fe3O4 nanoparticles, PYCA-Fe3O4 and SA-PYCA-Fe3O4 nanoparticles
Scanning electron microscopy (SEM)
The morphological features and size details of synthesized PYCA-Fe3O4 nanoparticles and SA-PYCA-Fe3O4 nanoparticles were studied by SEM (Fig. 4). The SEM image shows that Fe3O4–PYCA nanoparticles have a mean diameter of about 35 nm. Fig. 4a, b shows SA-PYCA-Fe3O4 nanoparticles greater than 43 nm in size. Comparison of experimental results showed the good grafting of PYCA and SA groups on the Fe3O4 nanoparticles is confirmed.

SEM images of a Fe3O4–PYCA and b SA-PYCA-Fe3O4 nanoparticles
Transmission electron microscopy (TEM)
The morphologies of the PYCA-Fe3O4 nanoparticles andSA-PYCA-Fe3O4 nanoparticles were investigated by TEM (Fig. 5). As can be seen from the TEM images, the average particle size distribution increases from 14 nm in PYCA-Fe3O4 nanoparticles to 19 nm in SA-PYCA-Fe3O4 nanoparticles, which is in very good agreement with the crystallite size estimated from XRD. Comparison of experimental results showed the good grafting of PYCA and SA groups on the Fe3O4 nanoparticles is confirmed.

The TEM images of a Fe3O4–PYCA and b SA-PYCA-Fe3O4 nanoparticles
Vibrating sample magnetometer (VSM)
Information about magnetic properties of Fe3O4 nanoparticles with an organic group supported on Fe3O4 nanoparticles was obtained by VSM (Fig. 6). The saturation magnetization values of MNPs, Fe3O4–PYCA nanoparticles and SA-PYCA-Fe3O4 nanoparticles were 91.69 emu/g, , 73.39 emu/g and 71.20 emu/g, respectively. These differences were caused by the different coating layers and their thicknesses on the surface of Fe3O4 nanoparticles. The resulting high values of saturation magnetization of SA-PYCA-Fe3O4 nanoparticles enables them to be easily be separated and recycled from the products by their response to an external magnetic field.

Room temperature magnetization curves of Fe3O4, Fe3O4-PYCA and SA-PYCA-Fe3O4 nanoparticles
Catalytic application of SA-PYCA-Fe3O4 nanoparticles
First, to find optimization conditions, the one-pot reaction of 4-cyanobenzaldehyde, 7-amino-4-methylcoumarin and diethyl acetylenedicarboxylate in the presence of the SA-PYCA-Fe3O4 as catalyst was selected as a model. The reaction was carried out with different amounts of SA-PYCA-Fe3O4 as catalyst (5, 10, 15, 20 mg) in different temperatures (25, 60 °C). The obtained results in Table 1 show that optimal condition was 15 mg of SA-MNPs at 60 °C (Table 1, entry 4).

Next, the reactions were performed with various aromatic aldehydes including electron-donating groups and electron-withdrawing substituents in the presence of SA-PYCA-Fe3O4 catalyst. As shown in Table 2, aldehyde-containing electron-withdrawing groups such as NO2, CN, Cl and Br (entries 3–10) led to the corresponding products in high yields, but the aldehyde-containing electron-donating groups such as CH3, OCH3 and OH (entries 11–16) did not afford the desired products.

The structure of 4d was confirmed by FT-IR, 1H NMR, 13C NMR and mass spectra, as well as elemental analysis. The FT-IR spectrum of 4d showed a broad band at 3421 cm−1 for the N–H stretching, a medium signal at 2231 cm−1 for the nitrile group and two peaks at 1724 and 1648 cm−1 for the two C=O groups of the ester and amide moieties, respectively. The 1H NMR spectrum of 4d exhibited a triplet at δ = 1.24 ppm (3J HH = 7.2 Hz) for the methyl group, a doublet at δ = 2.38 ppm (4J HH = 1.2 Hz) for the methyl group of coumarian, a quartet 4.25 ppm (3J HH = 7.2 Hz) for the OCH2, a singlet at δ = 5.85 ppm for the CH group of furan-2(5H)-one moiety, a doublet at δ = 6.23 ppm (4J HH = 8.8 Hz) for the = CH groups of coumarin and two doublets at δ = 7.42 and 7.61 ppm (3J HH = 8.4 Hz) for the four CH groups of the para-substituted benzene ring and two doublets at δ = 7.36 (4J HH = 2.4 Hz) and 7.53 (3J HH = 8.8 Hz) as well as a doublet of a doublet at δ = 7.71 ppm (3J HH = 8.8 Hz, 4J HH = 2 Hz) for the three protons of the phenyl group of coumarin. The NH group appeared at δ = 9.14 ppm as a broad signal. The 13C NMR spectrum of 4a exhibited 22 signals in agreement with the proposed structure. The mass spectrum of this compound displayed a molecular ion peak at m/z 430 (M +) and the other fragments at 384, 357, 201, 155 and 127 in accordance with the structure of 4d.
A proposed mechanism for the reaction was shown in Scheme 2. First, the enamine 5 was formed from the reaction of 7-amino-4-methylcoumarin 1 and dialkyl acetylenedicarboxylate 2. Then, enamine 5 attacked to the activated aromatic aldehyde 6 that protonated with SA-PYCA-Fe3O4 as acidic catalyst. The intermediate 7 is converted to intermediate 8 by loss of a proton. Finally, compound 8 perform a cyclization reaction with loss of ROH to produce compound 4 (Scheme 4).

Suggested mechanism for the synthesis of N-coumarin-2-furanones derivatives in the presences of SA-PYCA-Fe3O4 nanocatalyst
We also investigated the recycling of the SA-PYCA-Fe3O4 as the catalyst using the model reaction of 4-cyanobenzaldehyde, 7-amino-4-methylcoumarin and diethyl acetylenedicarboxylate in ethanol (5 mL; Table 2, entry 4). When the reaction was complete, the reaction mixture was cooled to ambient temperature and solvent was removed on a rotary evaporator. Then, the mixture was diluted with CH2Cl2 and the SA-PYCA-Fe3O4 nanoparticles were separated by magnetic field for separation of the catalyst, and the reusability under similar reaction conditions was checked. The results showed that SA-PYCA-Fe3O4 is a stable catalyst in reaction media and can be reused ten times without significant loss of its catalytic activity (Figs. 7, 8). The SEM, TEM and XRD analysis of recycled SA-PYCA-Fe3O4 was provided and shown in Fig. 9.

Recycling of the SA-PYCA-Fe3O4 as catalyst

Image showing SA-PYCA-Fe3O4 nanoparticles can be separated by an applied magnetic field. A reaction mixture in the absence (right) or presence of a magnetic field (left)

a SEM, b TEM and c XRD analysis of SA-PYCA-Fe3O4 nanoparticles after recycling four times
Conclusions
In this research, we described synthesis, characterization and catalytic applications of novel SA-PYCA-Fe3O4 MNPs for preparation of novel N-coumarin-2-furanones derivatives from arylaldehydes, 7-amino-4-methylcoumarin and dialkyl acetylenedicarboxylate. The attractive features of this procedure are a simple procedure, cleaner reaction and use of a nanocatalyst with good reversibility.
References
A. Tiwari, A.K. Mishra, H. Kobayashi, A.P.F. Turner, Intelligent Nanomaterials (Wiley, New Jersey, 2012)
C.N.R. Rao, A. Müller, A.K. Cheetham, The Chemistry of Nanomaterials: Synthesis, Properties and Applications, vol. 1 (Wiley, Weinheim, 2006)
K.J. Klabunde, R. Mulukutla, Nanoscale Materials in Chemistry (Wiley, NewYork, 2001)
A. Wang, X. Liu, Z. Su, H. Jing, Catal. Sci. Tech. 4, 71–80 (2014)
S. Fan, W. Dong, X. Huang, H. Gao, J. Wang, Z. Jin, J. Tang, G. Wang, ACS Catal. 7, 243–249 (2016)
K. Mishra, T.N. Poudel, N. Basavegowda, Y.R. Lee, J. Catal. 344, 273–285 (2016)
H.N. Dadhania, K.R. Dipak, N.D. Abhishek, Catal. Sci. Tech. 5, 4806–4812 (2015)
X. An, D. Cheng, L. Dai, B. Wang, H.J. Ocampo, J. Nasrallah, X. Jia, J. Zou, Y. Long, Y. Ni, Appl. Catal. B 206, 53–64 (2017)
K. He, Y. Ma, B. Yang, C. Liang, X. Chen, C. Cai, Spectrochim. Acta Mol. Biomol. Spectrosc. 173, 82–86 (2017)
S. Liu, H. Wang, L. Chai, M. Li, J. Colloid Interface Sci. 478, 288–295 (2016)
R. Mirzajani, S. Ahmadi, J. Ind. Eng. Chem. 23, 171–178 (2015)
M.A. Rahman, U. Culsum, A. Kumar, H. Gao, N. Hu, Int. J. Biol. Macromol. 87, 488–497 (2016)
C.M. Volla, I. Atodiresei, M. Rueping, Chem. Rev. 114, 2390–2431 (2013)
A. DeAngelis, M.T. Taylor, J.M. Fox, J. Am. Chem. Soc. 131, 1101–1105 (2009)
J. Sun, E.Y. Xia, Q. Wu, C.G. Yan, Org. Lett. 12, 3678–3681 (2010)
H. Dong, L. Xu, S.S. Li, L. Wang, C.L. Shao, J. Xiao, ACS. Comb. Sci. 18, 604–610 (2016)
S. Mirza, S.A. Naqvi, K.M. Khan, U. Salar, M.I. Choudhary, Bioorg. Chem. 70, 133–143 (2017)
M.R.M. Shafiee, S.S. Mansoor, M. Ghashang, A. Fazlinia, C. R. Chim. 17, 131–134 (2014)
L. Nagarapu, U.N. Kumar, P. Upendra, R. Bantu, Synth. Commun. 42, 2139–2148 (2012)
A. Kathuria, N. Priya, K. Chand, P. Singh, A. Gupta, S. Jalal, S. Gupta, H.G. Raj, S.K. Sharma, Bioorg. Med. Chem. 20, 1624–1638 (2012)
M. Mazzei, E. Nieddu, M. Miele, A. Balbi, M. Ferrone, M. Fermeglia, M.T. Mazzei, S. Pricl, P. La Colla, F. Marongiu, C. Ibba, Bioorg. Med. Chem. 16, 2591–2605 (2008)
A.M. Hamdy, Z. Khaddour, N.A. Al-Masoudi, Q. Rahman, C. Hering-Junghans, A. Villinger, P. Langer, Bioorg. Med. Chem. 24, 5115–5126 (2016)
M. Bozdag, A.M. Alafeefy, A.M. Altamimi, D. Vullo, F. Carta, C.T. Supuran, Bioorg. Med. Chem. 25, 677–683 (2017)
A. Montagut-Romans, M. Boulven, M. Jacolot, S. Moebs-Sanchez, C. Hascoët, A. Hammed, S. Besse, M. Lemaire, E. Benoit, V. Lattard, F. Popowycz, Bioorg. Med. Chem. Lett. 27, 1598–1601 (2017)
S. Asghari, M. Mohammadnia, Res. Chem. Intermed. 42, 1899–1911 (2016)
S. Asghari, M. Mohammadnia, Synth. React. Inorg. Met. Org. Chem. 47, 1004–1011 (2017)
T. Wejrzanowski, R. Pielaszek, A. Opalińska, H. Matysiak, W. Łojkowski, K. Kurzydłowski, J. Appl. Surf. Sci. 253, 204–208 (2006)
R. Pielaszek, J. Appl. Crystallogr. 1, 43–50 (2003)
Acknowledgements
This research was supported by the Research Council of the University of Mazandaran in Iran.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Asghari, S., Mohammadnia, M. Preparation and characterization of sulfamic acid pyridinium chloride-functionalized Fe3O4 nanoparticles as a novel magnetic catalyst for synthesis of novel N-coumarin-2-furanones. Res Chem Intermed 43, 7193–7209 (2017). https://doi.org/10.1007/s11164-017-3068-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11164-017-3068-3