
Abstract
In most humans, obesity is associated with a chronic low-grade inflammatory reaction occurring in several organ tissues, including the adipose tissue. Infiltration of bone marrow derived leukocytes (granulocytes, monocytes, lymphocytes) into expanding adipose depots appears to be an integral component of inflammation in obesity. Circulating leukocytes invade organ tissues mainly through post-capillary venules in the microcirculation. The endothelium of the post-capillary venules acts as a gatekeeper to leukocyte adhesion and extravasation by displacing on its luminal surface adhesion molecules that bind the adhesive receptors expressed on circulating leukocytes. Several studies investigating the impact of obesity on the microcirculation have demonstrated the occurrence of microvascular dysfunction in experimental animal model of obesity, as well as in obese humans. To date though, working hypotheses and study designs have favored the view that microvascular alterations are secondary to adipose tissue dysfunction. Indeed, a significant amount of data exists in the scientific literature to support the concept that microvascular dysfunction may precede and cause adipose tissue inflammation in obesity. Through review of key published data, this article prospectively presents the concept that in response to nutrients overload the vascular endothelium of the microcirculation acutely activates inflammatory pathways that initiate infiltration of leukocytes in visceral adipose tissue, well before weight gain and overt obesity. The anatomical and physiological heterogeneity of different microcirculations is also discussed toward the understanding of how obesity induces different inflammatory phenotypes in visceral and subcutaneous fat depots.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Obesity is a serious health challenge facing the US population with an overall medical cost for managing its complications reaching as high as $147 billion in 2008 [1]. Research has now shown that as body weight increases to reach the levels referred to as “overweight” and “obesity”, incidence of insulin resistance, type 2 diabetes, cardiovascular disease, fatty liver disease, atherosclerosis, neurological disorders including dementia, lung dysfunction, and some cancer also increases. World Health Organization’s estimates indicate that approximately one billion adults are overweight, and 300 million are obese [2]. More importantly, these numbers are expected to rise since the prevalence of childhood obesity is increasing at an alarming rate [3].
Consequently, the past decade has witnessed a highly intensified research effort into the mechanisms that govern adipose tissue dysfunction and obesity-associated complications. A unifying concept emerging from several independent lines or research is the existence of a mechanistic link between nutrients overload and derangements in the cellular and molecular mediators of immunity and inflammation (recently reviewed in [4, 5]). Inflammation is an acute vascular response aimed at restoring tissue integrity following exposure to harmful stimuli. Physiologically, the inflammatory response quickly terminates once the offending agents have been neutralized or removed altogether. If the injurious stimulus persists, a chronic state of low-grade inflammation usually occurs, with local and systemic detrimental consequences.
Even after accounting for lack of physical activity and genetic susceptibility, excessive food energy intake probably remains the most common cause of obesity in western countries [6]. Most individuals who develop overweight and obesity must, therefore, consume high-calorie meals for prolonged periods of time. With prolonged nutrients overload, the inflammatory response found in the obese state becomes of a chronic, low-grade type or, as recently defined, metainflammation [5]. Metainflammation occurs in expanding adipose depots, especially visceral ones. Metainflammation encompasses most components of the classical inflammatory response, including cytokine production [7], recruitment and activation of circulating leukocytes [8], and adipose tissue remodeling [9]. This brief review focuses on the role that leukocyte-endothelium interactions play in adipose tissue inflammation. We prospectively propose the concept that microvascular dysfunction induced by western type high-fat diets may precede and trigger adipose tissue inflammation. The pathophysiological reasoning underlying this working concept is discussed along with supporting experimental and clinical evidence.
2 The adipose tissue microcirculation
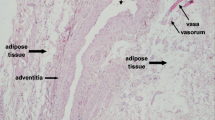
The 1945 work of Gersh and Still was the first to demonstrate the presence of a rich vasculature in adipose tissue [10]. This work changed the original histology based view of adipose tissue as a tissue having only a sparse and irregular vascularity. Later studies demonstrated that the vascularity of adipose tissue is of the same magnitude of that found in organs with intense metabolic activity, such as the skeletal muscle [11]. The use of intravital microscopy to observe directly the intact microcirculation has provided additional evidence that the adipose tissue receives a rich capillary blood supply [12]. Anatomically, the microcirculation can be divided into three segments, each with distinct physiological functions: arterioles, capillaries plus post-capillary venules, and venules. Each segment controls selected microvascular functions, which correspond to specific structures and roles. Arterioles regulate blood flow allocation and they branch into smaller diameter microvessels in which the number of muscular layers progressively decreases until single endothelial cells lying on a basement membrane form the true capillaries. A single terminal arteriole controls blood-perfusion in groups of capillaries, within the same microvascular bed [13]. Capillaries are the sites of true exchange between the blood and tissue compartments. In adipose tissue, capillaries abundantly express on the endothelial luminal surface the lipoprotein lipase (LPL); an enzyme synthesized and secreted in catalytically active form by neighboring adipocytes. LPL is mobilized from adipocytes to the luminal surface of endothelial cells via the action of glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein 1 GPIHBP1 [14]. LPL’s major physiological role is to hydrolyze the triglycerides of chylomicrons and VLDL particles on the luminal side of the capillary endothelium, which results in the release of free fatty acids (FFA). In organ such as the liver, where large fenestrae in the capillaries allow the passage of molecules as large as lipoproteins, circulating FFA have direct access to the parenchymal cells for storage. In adipose tissue, instead, the capillary endothelial cells are bound by tight-junctions, a condition that prevents direct uptake of FFA [15]. Since in adipose tissue, FFA must cross the capillary endothelial cells in order to be available to adipocytes, it is likely that following consumption of high-fat meals excessive FFA formation triggers acute inflammatory events in the adipose tissue microcirculation, even in the absence of primary adipocyte dysfunction. Indeed, elevated plasma FFA correlate with markers of endothelial activation in humans [16]. From a physiologic standpoint, it might appear counterintuitive that the visceral microcirculation initiates inflammatory reactions in response to nutrient absorption. Indeed, such predisposition could be part of an adaptive immunosurveillance function that defends the organism from the many bacteria and toxins that potentially can enter the blood stream during nutrient absorption. In support of this view, recent data have indicated that high-fat food intake is associated with transient endotoxemia in humans [20]
The microcirculation also regulates vascular permeability, an important function in the blood/tissue exchange of fluids, solutes and macromolecules. The main location of both physiological and pathophysiological vascular permeability is the venous, capillary endothelium, although arterioles also exhibit some permeability function [17]. For more detailed information, the reader is referred to a large review on permeability [18]. Abnormally elevated capillary permeability is typically seen in inflamed microvascular networks where it causes leakage of macromolecules into the surrounding organ tissue (e.g., albumin) [19]. In theory then, disruption of the endothelial cell barrier caused by excessive FFA levels following consumption of high-fat meals could result in absorption of bacterial byproducts. In line with this view, emerging research emphasize a role for the gut microbiota in obesity and insulin resistance (recently reviewed in [4]).
3 Microvascular heterogeneity and adipose tissue inflammation
Adipose depots occur in multiple visceral and subcutaneous locations of the body. Clinical evidence indicates that adipose distribution is important since central adiposity, especially visceral obesity, is typically associated with metabolic and vascular complications [21]. Conversely, peripheral and lower body fat accumulation appears to be protective. The physiologic mechanisms underlying these differences remain though unexplained. Perhaps, anatomical and functional differences between the visceral fat and subcutaneous fat microcirculations could help us understand these discrepancies. As a general principle, it should be noted that heterogeneity is a hallmark of the vasculature. Differences are found not only between large and small vessels but also within microcirculations of different organs, within microvascular sections of the same organ and even along the same vessel. To name a few relevant examples, expression of matrix metalloproteinases in microvessels differs from that of macrovascular endothelial cells [22], as do endothelial microdomain signaling complexes [23]. More interestingly, in the mesenteric fat pads, most of the fat cells seem to be uniquely clustered around the venular segment [24]. In the visceral fat, therefore, adipocytes are peculiarly arranged in proximity of the site of the microcirculation where leukocytes adhere and extravasate. In the overweight individual, LPL activity is elevated, particularly at an intermediate waist circumference [25]. In vivo studies of meal FFA uptake provide definitive evidence for heterogeneity in the metabolism of visceral adipose tissue (VAT) and subcutaneous adipose tissue (SAT). Expressed relative to the same mass of adipose tissue, meal FFA uptake is greater in intra-abdominal than abdominal subcutaneous fat in both sexes [26, 27]. The direct uptake of plasma FFA is also greater in omental compared to abdominal subcutaneous fat of women [28]. While high-LPL activity and higher FFA uptake may favor the preferential fat deposition in visceral depots, they may also expose the endothelium of the visceral fat microcirculation to excess FFA levels that are able to initiate the microvascular inflammatory responses discussed above. Overall, this information suggests that a unique anatomical structure, along with a higher LPL activity and FFA uptake are likely to make the visceral fat microcirculation prone to experience inflammation with nutrients overload.
4 Leukocyte-endothelium interaction
Infiltration of bone marrow derived inflammatory cells is a key feature of the adipose metainflammation seen in overweight/obese humans as well as in animal models of obesity. It is widely accepted that this inflammatory cell invasion is almost invariably associated with adipose tissue dysfunction and metabolic complications, such as insulin resistance (reviewed in [29, 30]). Of note, recent clinical studies support the concept that adipose tissue inflammation is causative of insulin resistance in complicated obesity. In fact, a subset of obese individuals with less inflammation in their visceral fat remains insulin-sensitive [31, 32]. These studies indicate that the visceral fat of some obese humans is better equipped to deal with calorie overload through mechanisms that remain presently unidentified. Conversely, other experimental and clinical research emphasizes a primary role for non-inflammatory based mechanisms in the adipose tissue dysfunction and insulin resistance of obesity. Thus, primary dysfunction of the fuel-sensing enzyme AMPK in adipocytes has been linked to insulin resistance in obesity [33]. Studies have extensively demonstrated macrophage infiltration of adipose tissue in obese mice and humans [8, 34], and based on experimental results, it has been suggested that expanding adipocytes or neighboring pre-adipocytes might begin to produce chemotactic signals leading to macrophage recruitment [35]. Overall, careful analysis of the literature reveals that mainstream working hypotheses have and continue to consider adipocytes as primary instigators of inflammatory cell recruitment in obesity. Accordingly, only marginal attention has been dedicated to the microcirculation in these processes, despite the unquestionable mandatory role that the microcirculation plays in the regulation of leukocyte trafficking. As result, microvascular alterations are currently considered secondary to adipocyte dysfunction, and, therefore, studied almost exclusively in the context of the cardiovascular complications of obesity [36]. Only recently, adipose tissue angiogenesis studies have emphasized a primary role for the microcirculation in obesity with insulin resistance [37]. Specifically, activation of the transcription factor HIF (hypoxia-inducible factor)-1α due to imbalance between adipose tissue expansion and microvasculature supply has been correlated to adipose tissue inflammation [38]. Indeed, fundamental physiology knowledge supports a primary causative role for the microcirculation in the inflammation of obesity. First, all nutrients absorbed post-prandial must traverse the microcirculation to be stored in adipocytes (see also paragraph above). Accordingly, studies have extensively shown that overload of nutrients such as glucose and FFA acutely induces microvascular dysfunction in non-obese laboratory rodents [39, 40]. Similarly, release of FFA from adipose storage during fasting is acutely associated with accumulation of macrophages in visceral fat depots [41]. Clinical data demonstrate upregulation of endothelial cell adhesion molecules in healthy volunteers following intake of high-fat meals [42]. Second, the microcirculation is the site of the vascular tree where leukocytes infiltrate inflamed organ tissues, including the adipose tissue. Physiologically, leukocytes perform most of their functions in the extravascular compartment. To reach this compartment, they must traverse the vascular endothelium (Fig. 1). Therefore, interaction of circulating leukocytes with the vascular endothelium is a mandatory step in the inflammatory response and, this event occurs on the venular side of the microcirculation [43]. Leukocyte-endothelium interactions occur in three steps, each mediated by a specific set of cell adhesion molecules (eCAMs) expressed on the endothelial cell surface: 1) leukocyte rolling is regulated by the selectin family of adhesion molecules (P-selectin, E-selectin); 2) leukocyte adherence by the immunoglobulin family of adhesion molecules (ICAM-1 and VCAM-1); and 3) leukocyte extravasation is regulated by ICAM-1 and PECAM-1. Although endothelial cells of all vascular segments (arteries, capillaries, and veins) can express eCAMs, post-capillary venules are the preferential site of leukocyte trafficking, probably because of their greatest density in eCAMs expression [44–48] and because of favorable hemodynamic factors [43, 49]. Interestingly, recent studies demonstrate that infiltration of specific leukocyte populations occurs very early with consumption of a high-fat diet; obviously well before excessive calorie storage can affect adipocyte function. Specifically, Dr. Olesfksy’ laboratory has recently shown, that in the mouse, neutrophil accumulation occurs within 48 h from administration of high-fat feeding, with near to peak values at 3 days post-feeding [50]. In the same study, neutrophils-derived elastase was also found to contribute to insulin resistance with prolonged feeding.

The obese organism experiences an abnormal infiltration of leukocytes in the adipose tissue, a process that results in adipose tissue inflammation and dysfunction. An integrated, maladaptive response involving the endothelium of the adipose tissue microcirculation may be the initiating factor in this phenomenon. Following consumption of high-fat meals, excessive levels of FFA acutely upregulate P-selectin (P-sel) in post-capillary venules of the visceral fat microcirculation by reducing AMPK/eNO signaling. P-sel initiates rolling of circulating leukocytes, especially neutrophils. Rolling leukocytes are primed for enhanced secretion and degranulation, thus releasing, among other inflammatory factors, the powerful oxidant myeloperoxidase (MPO). MPO quickly initiates adipocyte damage leading to impaired adiponectin function (early adipocyte dysfunction). With repeated high-fat meals, loss of adiponectin function further exacerbates infiltration of leukocytes via reduced AMPK/eNOS signaling in the vascular endothelium (metainflammation)
The results of this study raise new important questions on the mechanism(s) and consequences of this early infiltration of inflammatory cells associated with consumption of high-food. A possible explanation to this phenomenon can be found in the endothelial regulation of the inflammatory response. In response to inflammatory stimuli, the vascular endothelium quickly displaces the adhesion molecule P-selectin on its luminal surface. P-selectin is constitutively expressed and, therefore, readily upregulated at the cell surface with a time lag of approximately 15–20 min from recognition of inflammatory stimuli [51]. In contrast, a time lag of at least 2 h or more is required for upregulation of other eCAMs (E-selectin, ICAM-1 and VCAM-1) since they require de novo synthesis through transcriptional activity [52]. Thus, it is widely accepted that P-selectin initiates leukocyte recruitment in inflamed microvascular networks. Accordingly, it is reasonable to argue that perturbations of the microcirculation caused by high-fat meals, could rapidly and transiently induce trafficking of leukocytes in the adipose tissue microcirculation via upregulation of P-selectin. In line with this concept, two laboratories have recently demonstrated that deletion of the counter-receptor for P-selectin, P-selectin glycoprotein ligand-1 (PSGL-1), is protective against adipose tissue inflammation in obese mice [53, 54]. PSGL-1 is expressed on all myeloid and lymphoid lineages including dendritic cells. PSGL-1 binds endothelial expressed P- and/or E-selectin with much higher affinity for P-selectin (recently reviewed in [55]). Although PSGL-1 is constitutively expressed on the cell surface of all circulating leukocytes, it cannot initiate or sustain leukocyte trafficking in the absence of endothelial selectin expression. Thus, the function of PSGL-1 is indirectly regulated by the vascular endothelium of the microcirculation where upregulation of selectins restricts its engagement to inflamed vascular networks only. Interestingly, studies in the literature have reported upregulation of P-selectin in humans after consumption of a single high-fat meal [56, 57]. Others have also implicated P-selectin in obesity with insulin resistance [58–61].
5 FFA regulation of P-selectin cell surface expression
As discussed above, data in the literature demonstrate that uptake of FFA is elevated in visceral fat depots, which is likely to expose the microvascular endothelium of the visceral fat to higher rates of FFA fluxes than that of other adipose depot. This process can in theory cause fast displacement of P-selectin to the endothelial cell surface via activation of specific signaling pathways. Under resting conditions, P-selectin is stored inactive in Webel-Palade bodies [62]. Inflammatory stimuli quickly (≈15 min) upregulate P-selectin at the endothelial cell surface to initiates tethering of circulating leukocytes [63, 64]. In endothelial cells, endothelial nitric oxide (eNO) is a major inhibitor of Weibel-Palade body exocytosis, a step of the endothelial, inflammatory cascade necessary for surface expression of P-selectin [65]. Accordingly, loss of physiologic levels of eNO quickly causes upregulation of P-selectin [66, 67]. Studies have shown that the 5′ AMP-activated protein kinase (AMPK) regulates eNOS activity at the posttranslational level (recently reviewed in [68]). Interestingly, administration of high-fat diets to laboratory rodents reduces AMPK activity in multiple tissues [69]. Moreover, FFA acutely reduce AMPK activity in endothelial cells [70]. Conversely, stimulation of AMPK activity attenuates activation of pro-inflammatory pathway and increases NO release in human endothelial cells exposed to FFA [71]. Thus, excess FFA formation following ingestion of high-fat meals may cause loss of basal eNO levels and upregulation of P-selectin via downregulation of AMPK activity.
6 Functional implication of P-selectin upregulation
Leukocytes that adhere to endothelia expressing P-selectin are activated and primed for degranulation and enhanced secretion [51]. Among other factors, activated leukocytes release the powerful oxidant, myeloperoxidase (MPO). MPO is a heme protein abundantly expressed in neutrophils, and, to a lesser extent, in monocyte and macrophages [72]. MPO has long been viewed to function primarily as a bactericidal enzyme centrally linked to innate host defense. Indeed, recent work demonstrates that MPO levels are increased in the plasma and visceral organs of obese humans [73]. Others have demonstrated that MPO accumulates in the visceral epididymal fat of C57BL/6 J mice fed a high-fat diet [74].
One of the main toxic byproduct of MPO is hypochlorous acid, a powerful oxidant that is able to initiate modification of signaling proteins, including transcription factor [75], and to cause tissue dysfunction [76, 77] (Fig. 2). Interestingly, studies in the literature implicate the neutrophil/MPO axis in adipocyte function, measured as adiponectin abundance. In particular, acute reduction in plasma and adipose tissue levels of adiponectin mRNA have been described during pulmonary allergic reactions [78], a condition in which neutrophils become highly activated. Adiponectin function is also rapidly impaired by oxidants (recently reviewed in [79]).

Potential signaling targets of MPO chlorination in adipocytes, which can impair adiponectin function in adipose depots experiencing leukocyte infiltration through inflamed microvascular networks
Interestingly, we have reported that loss of adiponectin causes chronic leukocyte-endothelium interactions in the microcirculation via upregulation of cell adhesion molecules, including selectins [80]. We have also demonstrated that physiologic levels of adiponectin prevent eCAMs upregulation in the microcirculation via an eNO-mediated mechanism. Others have reported that adiponectin stimulates nitric oxide production in vascular endothelial cells [81]. Furthermore, mice with reduced levels of adiponectin experience microvascular and adipose tissue inflammation [80, 82, 83], while mice overexpressing adiponectin are protected from infiltration of leukocyte-derived inflammatory cells in the visceral fat [84]. Thus, upregulation of P-selectin in the visceral fat microcirculation could impair acutely adiponectin signaling via promoting MPO release. With repeated insults by high-fat meals, progressive loss of adiponectin function can result in sustained upregulation of endothelial cell adhesion molecules in the microcirculation (Fig. 1), a process that ultimately sustains chronic leukocyte trafficking into expanding visceral fat depots. Accordingly, further studies should be undertaken to understand fully the role that the neutrophil/MPO axes plays in the adipocyte tissue dysfunction of obesity.
7 Conclusion
Obesity is associated with low-grade inflammation mostly in visceral fat depots. In the present article, we have reviewed studies suggesting that unique anatomical and functional features make the visceral fat microcirculation vulnerable to acute inflammatory responses following nutrients overload. We have also discussed evidence indicating that microvascular dysfunction induced by nutrients overload initiates leukocyte trafficking in the visceral adipose tissue acutely and before weight gain and/or insulin resistance. The present discussion may foster new studies on the pathophysiology underlying adipose tissue inflammation and related complications. It may also lead to new therapeutic approaches that specifically target microvascular driven causes of obesity-related disorders.
References
Finkelstein EA, Trogdon JG, Cohen JW, Dietz W. Annual medical spending attributable to obesity: payer-and service-specific estimates. Health Aff (Millwood). 2009;28(5):w822–31.
WHO. WHO fact files: ten facts on obesity. http://www.who.int/features/factfiles/obesity/en/index.html (2010).
Ogden CL, Carroll MD, Curtin LR, Lamb MM, Flegal KM. Prevalence of high body mass index in US children and adolescents, 2007–2008. JAMA. 2010;303(3):242–9.
Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annu Rev Immunol. 2011;29:415–45.
Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444(7121):860–7.
Bauer KW, Hearst MO, Earnest AA, French SA, Oakes JM, Harnack LJ. Energy content of U.S. Fast-food restaurant offerings: 14-year trends. Am J Prev Med. 2012;43(5):490–7.
Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259(5091):87–91.
Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante Jr AW. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112(12):1796–808.
Itoh M, Suganami T, Hachiya R, Ogawa Y. Adipose tissue remodeling as homeostatic inflammation. Int J Inflam. 2011;2011:720926.
Gersh I, Still MA. Blood vessels in fat tissue. Relation to problems of gas exchange. J Exp Med. 1945;81(2):219–32.
Hausberger FX, Widelitz MM. Distribution of labeled erythrocytes in adipose tissue and muscle in the rat. Am J Physiol. 1963;204:649–52.
Kroeger S. The influence of intravenously administered fat emulsions on the microcirculation of pancreas and mesenteric adipose tissue. Adv Microcirc. 1970;3:1–66.
Lo A, Fuglevand AJ, Secomb TW. Oxygen delivery to skeletal muscle fibers: effects of microvascular unit structure and control mechanisms. Am J Physiol Heart Circ Physiol. 2003;285(3):H955–63.
Davies BS, Beigneux AP, Barnes 2nd RH, Tu Y, Gin P, Weinstein MM, et al. GPIHBP1 is responsible for the entry of lipoprotein lipase into capillaries. Cell Metab. 2010;12(1):42–52.
Stremmel W, Pohl L, Ring A, Herrmann T. A new concept of cellular uptake and intracellular trafficking of long-chain fatty acids. Lipids. 2001;36(9):981–9.
Mathew M, Tay E, Cusi K. Elevated plasma free fatty acids increase cardiovascular risk by inducing plasma biomarkers of endothelial activation, myeloperoxidase and PAI-1 in healthy subjects. Cardiovasc Diabetol. 2010;9:9.
Sarelius IH, Kuebel JM, Wang J, Huxley VH. Macromolecule permeability of in situ and excised rodent skeletal muscle arterioles and venules. Am J Physiol Heart Circ Physiol. 2006;290(1):H474–80.
Michel CC, Curry FE. Microvascular permeability. Physiol Rev. 1999;79(3):703–61.
Kurose I, Argenbright LW, Anderson DC, Tolley J, Miyasaka M, Harris N, et al. Reperfusion-induced leukocyte adhesion and vascular protein leakage in normal and hypercholesterolemic rats. Am J Physiol. 1997;273(2 Pt 2):H854–60.
Amar J, Burcelin R, Ruidavets JB, Cani PD, Fauvel J, Alessi MC, et al. Energy intake is associated with endotoxemia in apparently healthy men. Am J Clin Nutr. 2008;87(5):1219–23.
Fox CS, Massaro JM, Hoffmann U, Pou KM, Maurovich-Horvat P, Liu CY, et al. Abdominal visceral and subcutaneous adipose tissue compartments: association with metabolic risk factors in the Framingham Heart Study. Circulation. 2007;116(1):39–48.
Jackson CJ, Nguyen M. Human microvascular endothelial cells differ from macrovascular endothelial cells in their expression of matrix metalloproteinases. Int J Biochem Cell Biol. 1997;29(10):1167–77.
Sandow SL, Haddock RE, Hill CE, Chadha PS, Kerr PM, Welsh DG, et al. What’s where and why at a vascular myoendothelial microdomain signalling complex. Clin Exp Pharmacol Physiol. 2009;36(1):67–76.
Ballard K, Malmfors T, Rosell S. Adrenergic innervation and vascular patterns in canine adipose tissue. Microvasc Res. 1974;8(2):164–71.
Boivin A, Brochu G, Marceau S, Marceau P, Hould FS, Tchernof A. Regional differences in adipose tissue metabolism in obese men. Metabolism. 2007;56(4):533–40.
Jensen MD, Sarr MG, Dumesic DA, Southorn PA, Levine JA. Regional uptake of meal fatty acids in humans. Am J Physiol Endocrinol Metab. 2003;285(6):E1282–8.
Marin P, Lonn L, Andersson B, Oden B, Olbe L, Bengtsson BA, et al. Assimilation of triglycerides in subcutaneous and intraabdominal adipose tissues in vivo in men: effects of testosterone. J Clin Endocrinol Metab. 1996;81(3):1018–22.
Koutsari C, Ali AH, Mundi MS, Jensen MD. Storage of circulating free fatty acid in adipose tissue of postabsorptive humans: quantitative measures and implications for body fat distribution. Diabetes. 2011;60(8):2032–40.
de Luca C, Olefsky JM. Inflammation and insulin resistance. FEBS Lett. 2008;582(1):97–105.
Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol. 2010;72:219–46.
Barbarroja N, Lopez-Pedrera R, Mayas MD, Garcia-Fuentes E, Garrido-Sanchez L, Macias-Gonzalez M, et al. The obese healthy paradox: is inflammation the answer? Biochem J. 2010;430(1):141–9.
Kloting N, Fasshauer M, Dietrich A, Kovacs P, Schon MR, Kern M, et al. Insulin-sensitive obesity. American journal of physiology. Endocrinol Metab. 2010;299(3):E506–15.
Ruderman NB, Xu XJ, Nelson L, Cacicedo JM, Saha AK, Lan F, et al. AMPK and SIRT1: a long-standing partnership? Am J Physiol Endocrinol Metab. 2010;298(4):E751–60.
Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112(12):1821–30.
Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Invest. 2005;115(5):1111–9.
De Boer MP, Meijer RI, Wijnstok NJ, Jonk AM, Houben AJ, Stehouwer CD, et al. Microvascular dysfunction: a potential mechanism in the pathogenesis of obesity-associated insulin resistance and hypertension. Microcirculation. 2012;19(1):5–18.
Ye J. Adipose tissue vascularization: its role in chronic inflammation. Curr Diab Rep. 2011;11(3):203–10.
Halberg N, Khan T, Trujillo ME, Wernstedt-Asterholm I, Attie AD, Sherwani S, et al. Hypoxia-inducible factor 1alpha induces fibrosis and insulin resistance in white adipose tissue. Mol Cell Biol. 2009;29(16):4467–83.
Ellis A, Cheng ZJ, Li Y, Jiang YF, Yang J, Pannirselvam M, et al. Effects of a Western diet versus high glucose on endothelium-dependent relaxation in murine micro- and macro-vasculature. Eur J Pharmacol. 2008;601(1–3):111–7.
Booth G, Stalker TJ, Lefer AM, Scalia R. Mechanisms of amelioration of glucose-induced endothelial dysfunction following inhibition of protein kinase C in vivo. Diabetes. 2002;51(5):1556–64.
Kosteli A, Sugaru E, Haemmerle G, Martin JF, Lei J, Zechner R, et al. Weight loss and lipolysis promote a dynamic immune response in murine adipose tissue. J Clin Invest. 2010;120(10):3466–79.
Shimabukuro M, Chinen I, Higa N, Takasu N, Yamakawa K, Ueda S. Effects of dietary composition on postprandial endothelial function and adiponectin concentrations in healthy humans: a crossover controlled study. Am J Clin Nutr. 2007;86(4):923–8.
Ley K. Leukocyte recruitment as seen by intravital microscopy. In: Ley K, editor. Physiology of inflammation. New York: Oxford University Press; 2001. p. 303–37.
Krieglstein CF, Granger DN. Adhesion molecules and their role in vascular disease. Am J Hypertens. 2001;14(6 Pt 2):44S–54.
Jung U, Ley K. Regulation of E-selectin, P-selectin, and intercellular adhesion molecule 1 expression in mouse cremaster muscle vasculature. Microcirculation. 1997;4(2):311–9.
Klein LM, Lavker RM, Matis WL, Murphy GF. Degranulation of human mast cells induces an endothelial antigen central to leukocyte adhesion. Proc Natl Acad Sci U S A. 1989;86(22):8972–6.
Gotsch U, Jager U, Dominis M, Vestweber D. Expression of P-selectin on endothelial cells is upregulated by LPS and TNF-alpha in vivo. Cell Adhes Commun. 1994;2(1):7–14.
Fries JW, Williams AJ, Atkins RC, Newman W, Lipscomb MF, Collins T. Expression of VCAM-1 and E-selectin in an in vivo model of endothelial activation. Am J Pathol. 1993;143(3):725–37.
Zweifach BW, Lipowsky HH. Pressure-flow relations in blood and lymph microcirculation. In: Renkin EM, Michel CC, editors. Handbook of physiology. The cardiovascular system: microcirculation. Bethesda: American Physiological Society; 1984. p. 251–307.
Talukdar S, Oh DY, Bandyopadhyay G, Li D, Xu J, McNelis J, et al. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat Med. 2012.
Lorant DE, Topham MK, Whatley RE, McEver RP, McIntyre TM, Prescott SM, et al. Inflammatory roles of P-selectin. J Clin Invest. 1993;92(2):559–70.
Collins T, Read MA, Neish AS, Whitley MZ, Thanos D, Maniatis T. Transcriptional regulation of endothelial cell adhesion molecules: NF-kappa B and cytokine-inducible enhancers. FASEB J. 1995;9(10):899–909.
Russo HM, Wickenheiser KJ, Luo W, Ohman MK, Franchi L, Wright AP, et al. P-selectin glycoprotein ligand-1 regulates adhesive properties of the endothelium and leukocyte trafficking into adipose tissue. Circ Res. 2010;107(3):388–97.
Sato C, Shikata K, Hirota D, Sasaki M, Nishishita S, Miyamoto S, et al. P-selectin glycoprotein ligand-1 deficiency is protective against obesity-related insulin resistance. Diabetes. 2011;60(1):189–99.
Zarbock A, Ley K, McEver RP, Hidalgo A. Leukocyte ligands for endothelial selectins: specialized glycoconjugates that mediate rolling and signaling under flow. Blood. 2011;118(26):6743–51.
Broijersen A, Karpe F, Hamsten A, Goodall AH, Hjemdahl P. Alimentary lipemia enhances the membrane expression of platelet P-selectin without affecting other markers of platelet activation. Atherosclerosis. 1998;137(1):107–13.
Kalsch T, Elmas E, Nguyen XD, Kralev S, Leweling H, Kluter H, et al. Effects of alimentary lipemia and inflammation on platelet CD40-ligand. Thromb Res. 2007;120(5):703–8.
Schneider DJ, Hardison RM, Lopes N, Sobel BE, Brooks MM. Association between increased platelet P-selectin expression and obesity in patients with type 2 diabetes: a BARI 2D (Bypass Angioplasty Revascularization Investigation 2 Diabetes) substudy. Diabetes Care. 2009;32(5):944–9.
De Pergola G, Pannacciulli N, Coviello M, Scarangella A, Di Roma P, Caringella M, et al. sP-selectin plasma levels in obesity: association with insulin resistance and related metabolic and prothrombotic factors. Nutr, Metab, Cardiovasc Dis: NMCD. 2008;18(3):227–32.
Gallistl S, Sudi KM, Borkenstein M, Weinhandl G, Zotter H, Muntean W. Correlation between cholesterol, soluble P-selectin, and D-dimer in obese children and adolescents. Blood Coagul Fibrinolysis. 2000;11(8):755–60.
Kato H, Kashiwagi H, Shiraga M, Tadokoro S, Kamae T, Ujiie H, et al. Adiponectin acts as an endogenous antithrombotic factor. Arterioscler Thromb Vasc Biol. 2006;26(1):224–30.
Johnston GI, Cook RG, McEver RP. Cloning of GMP-140, a granule membrane protein of platelets and endothelium: sequence similarity to proteins involved in cell adhesion and inflammation. Cell. 1989;56(6):1033–44.
Ichimura H, Parthasarathi K, Quadri S, Issekutz AC, Bhattacharya J. Mechano-oxidative coupling by mitochondria induces proinflammatory responses in lung venular capillaries. J Clin Invest. 2003;111(5):691–9.
Takano M, Meneshian A, Sheikh E, Yamakawa Y, Wilkins KB, Hopkins EA, et al. Rapid upregulation of endothelial P-selectin expression via reactive oxygen species generation. Am J Physiol Heart Circ Physiol. 2002;283(5):H2054–61.
Matsushita K, Morrell CN, Cambien B, Yang SX, Yamakuchi M, Bao C, et al. Nitric oxide regulates exocytosis by S-nitrosylation of N-ethylmaleimide-sensitive factor. Cell. 2003;115(2):139–50.
Davenpeck KL, Gauthier TW, Lefer AM. Inhibition of endothelial-derived nitric oxide promotes P-selectin expression and actions in the rat microcirculation. Gastroenterology. 1994;107(4):1050–8.
Ahluwalia A, Foster P, Scotland RS, McLean PG, Mathur A, Perretti M, et al. Antiinflammatory activity of soluble guanylate cyclase: cGMP-dependent down-regulation of P-selectin expression and leukocyte recruitment. Proc Natl Acad Sci U S A. 2004;101(5):1386–91.
Fisslthaler B, Fleming I. Activation and signaling by the AMP-activated protein kinase in endothelial cells. Circ Res. 2009;105(2):114–27.
Lindholm CR, Ertel RL, Bauwens JD, Schmuck EG, Mulligan JD, Saupe KW. A high-fat diet decreases AMPK activity in multiple tissues in the absence of hyperglycemia or systemic inflammation in rats. J Physiol Biochem 2012.
Wu Y, Song P, Xu J, Zhang M, Zou MH. Activation of protein phosphatase 2A by palmitate inhibits AMP-activated protein kinase. J Biol Chem. 2007;282(13):9777–88.
Mugabo Y, Mukaneza Y, Renier G. Palmitate induces C-reactive protein expression in human aortic endothelial cells. Relevance to fatty acid-induced endothelial dysfunction. Metab Clin Exp. 2011;60(5):640–8.
Koeffler HP, Ranyard J, Pertcheck M. Myeloperoxidase: its structure and expression during myeloid differentiation. Blood. 1985;65(2):484–91.
Rensen SS, Slaats Y, Nijhuis J, Jans A, Bieghs V, Driessen A, et al. Increased hepatic myeloperoxidase activity in obese subjects with nonalcoholic steatohepatitis. Am J Pathol. 2009;175(4):1473–82.
Elgazar-Carmon V, Rudich A, Hadad N, Levy R. Neutrophils transiently infiltrate intra-abdominal fat early in the course of high-fat feeding. J Lipid Res. 2008;49(9):1894–903.
Rossmann C, Rauh A, Hammer A, Windischhofer W, Zirkl S, Sattler W, et al. Hypochlorite-modified high-density lipoprotein promotes induction of HO-1 in endothelial cells via activation of p42/44 MAPK and zinc finger transcription factor Egr-1. Arch Biochem Biophys. 2011;509(1):16–25.
Spallarossa P, Garibaldi S, Barisione C, Ghigliotti G, Altieri P, Tracchi I, et al. Postprandial serum induces apoptosis in endothelial cells: Role of polymorphonuclear-derived myeloperoxidase and metalloproteinase-9 activity. Atherosclerosis. 2008;198(2):458–67.
Malle E, Furtmuller PG, Sattler W, Obinger C. Myeloperoxidase: a target for new drug development? Br J Pharmacol. 2007;152(6):838–54.
Shore SA, Terry RD, Flynt L, Xu A, Hug C. Adiponectin attenuates allergen-induced airway inflammation and hyperresponsiveness in mice. J Allergy Clin Immunol. 2006;118(2):389–95.
Liu M, Liu F. Transcriptional and post-translational regulation of adiponectin. Biochem J. 2010;425(1):41–52.
Ouedraogo R, Gong Y, Berzins B, Wu X, Mahadev K, Hough K, et al. Adiponectin deficiency increases leukocyte-endothelium interactions via upregulation of endothelial cell adhesion molecules in vivo. J Clin Invest. 2007;117(6):1718–26.
Chen H, Montagnani M, Funahashi T, Shimomura I, Quon MJ. Adiponectin stimulates production of nitric oxide in vascular endothelial cells. J Biol Chem. 2003;278(45):45021–6.
Tsuchida A, Yamauchi T, Takekawa S, Hada Y, Ito Y, Maki T, et al. Peroxisome proliferator-activated receptor (PPAR)alpha activation increases adiponectin receptors and reduces obesity-related inflammation in adipose tissue: comparison of activation of PPARalpha, PPARgamma, and their combination. Diabetes. 2005;54(12):3358–70.
Trevaskis JL, Gawronska-Kozak B, Sutton GM, McNeil M, Stephens JM, Smith SR, et al. Role of adiponectin and inflammation in insulin resistance of Mc3r and Mc4r knockout mice. Obesity (Silver Spring). 2007;15(11):2664–72.
Kim JY, van de Wall E, Laplante M, Azzara A, Trujillo ME, Hofmann SM, et al. Obesity-associated improvements in metabolic profile through expansion of adipose tissue. J Clin Invest. 2007;117(9):2621–37.
Acknowledgments
This work was supported by a grant from The American Diabetes Association and NIDDK grant # R01DK64344
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Scalia, R. The microcirculation in adipose tissue inflammation. Rev Endocr Metab Disord 14, 69–76 (2013). https://doi.org/10.1007/s11154-013-9236-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11154-013-9236-x