
Abstract
The majority of blood vessels are surrounded by adipocytes which have been labelled as perivascular adipose tissue (PVAT). It was originally assumed that this provided a structural support to the circulation, but in recent years it has been clear that these fat cells are highly metabolically active and produce a variety of adipokines and cytokines that can influence local vascular tone. The profile of these secreted factors is very much dependent upon the phenotype of the individual. The PVAT of healthy lean humans largely secretes vasodilator adipokines which will act in a paracrine fashion to reduce peripheral resistance and improve nutrient uptake into tissues, thereby ameliorating the risk of hypertension and diabetes. In obesity PVAT becomes enlarged and inflamed, and the bioavailability of vasodilator adipokines is reduced. The inevitable consequence is a rise in peripheral resistance with a higher blood pressure and a decrease in glucose uptake into tissues such as skeletal muscle leading to insulin resistance and the ultimate development of type 2 diabetes. This chapter will give a brief overview into what has been discovered in terms of PVAT in health and disease.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
15.1 PVAT Structure
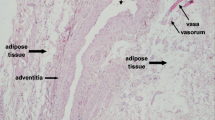
The PVAT that surrounds small blood vessels comprises white adipocytes, inflammatory immune cells, stem cells, nerves and stromal tissue plus microfeeding vessels [1,2,3,4,5]. In the aorta whilst many of the cellular constituents will be similar, the adipocytes are brown in nature [6, 7]. Such adipocytes have a role in thermogenesis, whereas white adipocytes are regarded as energy stores [8, 9]. For many years physiological studies of isolated blood vessels in vitro skeletonised the artery in advance of pharmacological testing. The removal of the PVAT meant that any putative functional contribution to arterial tone was lost. It was not until 1991 that Soltis and Cassis tested the hypothesis that PVAT might play a role in vascular tonal regulation [10]. Using isolated segments of rat aorta, they demonstrated the presence of a diminished response to the exogenous application of noradrenaline when the PVAT was retained, in comparison with arteries that had been denuded of fat tissue. This effect was attributed to the dense sympathetic innervation in PVAT. Also, there was an assumption that externally applied vasoconstrictor substances might have more difficulty in gaining access to the outside of the blood vessel because of the barrier posed by the PVAT [10, 11]. Ten years later Lohn and colleagues demonstrated that the vasoconstrictor responses to angiotensin II (AngII), serotonin and phenylephrine were 95%, 80% and 30% lower, respectively, when PVAT was retained [12]. Furthermore this effect was found to be independent of nitric oxide (NO) formation. It appeared to be mediated by a substance released from the fat, as transfer of the organ bath solution from vessels with PVAT or cultured rat adipocytes, on to arteries without PVAT, resulted in a rapid reduction in the constrictor response [13, 14]. Further data suggested that PVAT released a soluble factor that induced vasodilatation, which was calcium dependent and regulated by tyrosine kinase acting via ATP-dependent potassium channels [15, 16]. The exact identity was left uncertain at this time. It seems clear now that there is a variety of vasodilator adipokines responsible for relaxation which may also be dependent upon the spasmogen used. Several candidates have been suggested including angiotensin 1–7, free fatty acids such as methyl palmitate and hydrogen sulphide, as well as inflammatory cytokines such as interleukin-6 and tumour necrosis factor [1]. There is also an emerging body of evidence that the predominating factor is adiponectin [13, 17]. In this regard it is clear that adiponectin can act as a vasodilator by activating endothelial nitric oxide synthase (NOS) with the consequent production of NO. In addition, adiponectin has anti-inflammatory properties and can inhibit macrophage activation and reduce the proliferation of vascular smooth muscle cells [18, 19]. Adiponectin gene knockout mice exhibit a loss of anticontractile function from PVAT, and their phenotype demonstrates hypertension and glucose intolerance [13]. Pathological states, such as obesity, hypertension, atherosclerosis, type 2 diabetes and myocardial infarction, are associated with endothelial dysfunction and a reduction in plasma adiponectin levels [20,21,22]. Recent studies have demonstrated adiponectin-mediated anticontractile activity in human small arteries [3]; however, the exact mechanism by which healthy PVAT induces vasodilation is difficult to identify, as there appears to be variation dependent upon anatomical location and animal species [2, 3, 5, 17]. For example, in the rat aorta, PVAT appears to cause vasorelaxation by opening ATP-dependent K+ channels in vascular smooth muscle cells, whereas in rat mesenteric arteries voltage-dependent potassium channels seem to be more important [14,15,16]. There is evidence also of modulation of venous function by PVAT and a recent report suggesting that the contractile responses of the inferior vena cava to various agonists can be diminished by activation of KV channels by adipokines released from PVAT [23]. Clearly PVAT can play an important role as a dual modulator of vascular tone. The fine balance between PVAT-derived vasodilator and vasoconstrictor mediators may be crucial for the maintenance of local vascular tone.
15.2 The Role of Inflammation and Oxidative Stress on PVAT Phenotype
Recently, a review from Huang Cao and colleagues discussed the importance of PVAT regulation of vascular tone and underlined the significance of the functional integrity of PVAT, rather than the amount of PVAT itself [24]. In the healthy lean state, PVAT-derived adipokines and cytokines can act on endothelial cells through a NO-dependent mechanism and directly on smooth muscle cells to induce vasorelaxation of the vessel wall [3, 16]. In contrast, in obesity, PVAT shifts its phenotype, demonstrating an increase in oxidative stress, pro-inflammatory cytokines/adipokines release, immune cells migration and the secretory profile of PVAT changes with a reduction in the expression of vasorelaxing factors and an increase in vasoconstrictor factors with, in particular, macrophage infiltration [24]. There is compelling evidence for an increased level of adipokines in the inflammatory state, such as in obesity, and of a decreased cardiometabolic protective role of adiponectin. These obesity-related changes in adipose tissue lead to a significant decrease in the anticontractile effect of PVAT even though its mass is increased. This shift is probably linked to the development of insulin resistance, type 2 diabetes mellitus and hypertension [25,26,27]. In summary, in obesity, the anticontractile effect is significantly decreased, despite an increase in PVAT mass [3].
The mechanism by which adipose inflammation is related to obesity remains unknown, although hypoxia may be key: adipocytes become hypertrophic leading to inadequate perfusion and consequent local hypoxia that is mediated mainly by hypoxia-inducible factor alpha (HIF-1 α) [28,29,30]. HIF-1 α levels are higher in the adipose tissue of obese subjects, and it reverts to normal after weight loss [31]. HIF-1 α is a main hypoxia-inducible transcription factor linked to increases in inflammatory cytokines like TNF-α and IL-6 and reduces the bioavailability of adiponectin [32]. Greenstein et al. have shown that the anticontractile function of PVAT is lost in obese patients and that the experimental application of TNF-α and IL-6 to intact PVAT around healthy rat blood vessels decreased the dilator effect of PVAT [3]. Similarly, it has been shown that the induction of experimental hypoxia for 2.5 h causes PVAT inflammation and loss of its anticontractile function. These changes were reversed, and the anticontractile property rescued by catalase and superoxide dismutase [3]. In addition, using preincubation with the cytokine antagonist infliximab in the organ bath can restore normal PVAT function. Furthermore, after induced hypoxia, the incubation of the vessels with aldosterone can reproduce the inflammatory phenotype in adipose tissue, with loss of anticontractile property, causing PVAT resident macrophage activation, and the uses of free radical scavengers are able to rescue the anticontractile function [33].
In macrophage deficient CD11b-DTR mice during hypoxic and inflammatory conditions [33, 34], macrophages are a key modulator of oxidative stress and systemic inflammation in adipose tissue. The important role of IL-6 and MCP-1 in the recruitment of inflammatory cells, in particular monocytes and macrophages during obesity, is described in a recent review [24]. In particular, it has been demonstrated that 2 weeks of high-fat diet feeding caused an increase in MCP-1 and IL-6 expression in PVAT which was not seen with other adipose tissue depots [35]. Furthermore, animal models with MCP-1 gene deletion show a significantly reduced infiltration of inflammatory cells, and lipid deposition in the arterial wall and IL-6 inhibition reduce intrinsic mechanical stiffness in low-density lipoprotein receptor knockout mice [36, 37]. From such studies we can conclude that both these cytokines are directly involved in the development of arterial stiffness and increased cardiovascular disease risk.
In addition to involvement of the macrophages in PVAT function, it has been demonstrated that eosinophils also play an important role in the loss of PVAT anticontractile function [38]. Withers et al. have shown that the anticontractile effect of PVAT was lost in eosinophil-deficient mice and could be rescued after eosinophil reconstitution with an increase of adiponectin and adipocyte-derived NO bioavailability. In addition, these eosinophil-deficient mice exhibited hypertension and hyperglycaemia [38]. Mechanistically, it appears that eosinophils are a source of catecholamines and activate adipocyte β3-adrenoceptors to release the vasodilators adiponectin and NO. This study and others [39] suggest that targeting eosinophil number in obesity may be a useful therapeutic target. Wu et al. have demonstrated that hypereosinophilia induced by helminth infection improved glucose and insulin tolerance in obesity. Further work is needed to fully elucidate the mechanisms by which eosinophils are involved in PVAT and vascular functionality and their potential as a therapeutic target in obesity.
15.3 The Release of Vasoactive Factors from PVAT
Whilst there was a school of thought that PVAT was only providing mechanical support for the vasculature, there is an accumulating body of evidence that it is highly metabolically active and can regulate vascular tone in a paracrine and endocrine fashion [1,2,3,4]. The exact nature of this regulation depends on the species being tested and the vascular beds that are studied. In addition, the distinctive profile of PVAT activity is dependent certainly in vitro on the spasmogen used to stimulate the arterial tissue. Against this background it is clear that in a variety of vascular beds including skeletal muscle and mesenteric small arteries, as well as larger conduit arteries such as the superior mesenteric and the thoracic aorta, there is a significant contribution to an anticontractile effect in a response to a variety of vasoconstrictor stimulae. Locally, this could play a role in peripheral vascular resistance and in consequence blood pressure as well as nutrient delivery to skeletal muscle. The loss of this latter property could lead to a decrease in glucose uptake and the development of type 2 diabetes. The vasorelaxant activity can be ascribed to a number of adipokines that can be produced and secreted from PVAT, as well as a contribution from PVAT in terms of catecholamine reuptake into the adipocytes. Below is a summary of some of the principle dilator adipokines that may be responsible for the anticontractile function in healthy lean individuals.
-
1.
Adiponectin. This adipokine is secreted in a number of polymeric forms with varying molecular weights [40,41,42]. The ligand binds to two types of adiponectin receptor (type 1 and type 2) [43,44,45]. Type 1 (adiopoR1) is located on both endothelial and vascular smooth muscle cells, and it is activation of this receptor which will stimulate a number of pathways including smooth cell muscle differentiation and growth. With regard to vasorelaxation, activation of the receptor stimulates the production of AMPK which in turn will increase the phosphorylation of endothelial NOS leading to an increased bioavailability of NO in both the endothelium and smooth muscle cells [46]. In addition, in vascular smooth cells, AMPK regulates the opening of large conductance Ca2+-activated K+ channels [47, 48]. In hypertension circulating levels of adiponectin are decreased, and there is restoration to normality when blood pressure is lowered [49]. Low plasma adiponectin is also observed in obesity [20]. The adiponectin gene knockout mouse develops a hypertensive and diabetic phenotype, and the noradrenaline and electrical field stimulation-induced anticontractile effects of PVAT are lost [13].
-
2.
Leptin. This adipokine is recognised to play a vital role in regulating satiety and body weight [50]. Whilst it has central actions, leptin can induce vasodilatation by both endothelium-dependent and endothelium-independent mechanisms [51]. It is of interest that in the spontaneously rat where there is loss of PVAT anticontractile activity, there is reduced expression of leptin in the PVAT with reduced eNOS activation [52].
-
3.
Nitric oxide. The ubiquitous nature of NO means that it is widely recognised as an important vasodilator [53]. It is synthesised by three NOS enzymes: endothelial (eNOS), neuronal (nNOS) and inducible (iNOS). We have demonstrated that the anticontractile effect of PVAT stimulated by noradrenaline in small mesenteric arteries is NOS-dependent [3, 16]. When the PVAT is incubated with a nonspecific NOS inhibitor, the anticontractile effect is completely abolished. It is thought that early in obesity, there is an adaptive increase in NO which may preserve vascular function [54]. In a high-fat fed mouse model of obesity, insulin and leptin which are elevated stimulate NO production from mesenteric PVAT. However, in chronic obesity increased superoxide production results in a reduction in NO bioavailability [55].
-
4.
Hydrogen sulphide. There is evidence in aortic PVAT that hydrogen sulphide (H2S) is produced and that it can induce vasodilation of vascular smooth muscle cells by opening of ATP-sensitive K+ channels or by activation of endothelial intermediate conductance Ca2+-activated K+ channels [15, 56]. A number of vasoconstrictors including phenylephrine, 5HT and angiotensin II stimulate the production of H2S from aortic PVAT, and plasma H2S is reduced in the spontaneously hypertensive rat and in a pharmacologically reduced mouse model of hypertension using oral administration of a NOS inhibitor [57, 58].
-
5.
Hydrogen peroxide. Hydrogen peroxide (H202) is a known vasodilator in endothelium-denuded vessels by the activation of soluble guanylate cyclase in vascular smooth muscle [59]. Some researchers have advanced the hypothesis that H202 plays an important role in this pathway, and Gao et al. found that phenylephrine-induced PVAT anticontractile activity could be diminished using an H202 scavenger.
-
6.
Palmitic acid methyl ester. Palmitic acid methyl ester (PAME) is released from PVAT in the presence of Ca2+ and induces vasodilation of the aorta via Kv channels in a NO-independent and endothelial-dependent manner [60]. In the spontaneously hypertensive rat, PAME secretion from PVAT is reduced, and the vasodilator effect of exogenous PAME in these animals is significantly less than in wild-type normotensive rats.
-
7.
Angiotensin 1–7. PVAT is able to produce angiotensin 1–7, and antagonists ameliorate the PVAT anticontractile effect in aorta [61]. Its physiological role is uncertain, and its contribution to hypertension and metabolic disorders such as type 2 diabetes is still not fully elucidated.
15.4 Autonomic Innervation of Adipose Tissue
There is clear evidence that adipocytes contain a complete adrenergic system including β3-adrenergic receptors, uptake transporters and metabolic enzymes, and this system may modulate the anticontractile function of PVAT [5, 13, 62, 63]. It is still uncertain whether there is precise autonomic innervation of PVAT. Certainly there is clear histopathological proof that sympathetic nerve fibres run through PVAT and innervate the vasculature [5, 64, 65]. It is attractive to postulate that direct stimulation of sympathetic nerve fibres could cause vasoconstriction by activation of α1-adrenoreceptors on the vasculature and that this can be modulated by anticontractile function as a result of the same nerve fibre causing discharge of catecholamines that will bind to adipocyte β3-adrenergic receptors. There are reports that the use of β-adrenoreceptor agonists causes hypotension in rodents and dogs and that this has been shown to induce PVAT-dependent relaxation in pre-constricted mesenteric resistance arteries [66]. Our most recent studies have demonstrated that adipocyte located β3-adrenoreceptors do play a vital role in the anticontractile effect of PVAT, and activation of these receptors by sympathetic nerve-derived noradrenaline triggers the release of the vasodilator adiponectin [13]. In addition, we have shown that the anticontractile function is, at least in part, a result of PVAT sequestering sympathetic nerve-derived noradrenaline via organic cation transporter 3, thereby preventing the noradrenaline from reaching the α1-adrenoreceptors on the vasculature.
15.5 Autonomic Dysfunction in Obesity
It is clear that the sympathetic nervous system becomes pathologically overactive in obesity, and increased neural activity has been shown to correlate with increases in body mass index and the percentage of body fat [67, 68]. Sympathetic over-activity is observed after only 12 days of high-fat feeding in rats and is independent of weight gain [69]. Again our studies indicate that in obesity there is a loss of PVAT anticontractile function [3, 55], which may be a result of adrenoceptor desensitisation similar to that which occurs in heart failure [70]. Inevitably, as described above, this will lead to an increase in peripheral vascular resistance and a decrease in the tissue uptake of glucose. The obese phenotype is associated with adipocyte hypertrophy in PVAT depots with intense inflammation, and the activation of macrophages with a decreased in PVAT located eosinophils [3]. We have demonstrated that the macrophage is central to PVAT function, and without macrophages, even in acute hypoxic states which are known to reduce anticontractile activity of PVAT, there is no loss of the anticontractile effect which is mediated by the preservation of adiponectin bioavailability [33]. Furthermore, the increase in PVAT eosinophils correlates with the loss of PVAT anticontractile function, and the restoration of eosinophil donation is associated with a rescuing of the PVAT function, accompanied by a normalisation of the obesity-associated hypertension and diabetes [38]. The mechanism is still unclear although originally it was assumed that the eosinophil was operating by the transformation of macrophages to a classically active phenotype which is pro-inflammatory, and in doing so this reduced the bioavailability of adiponectin. However, our detailed studies suggest that this is not the case, and it may well be that the catecholamines such as noradrenaline contained within eosinophils are locally released thereby stimulating β3-adrenergic receptors, and promoting the secretion of adiponectin is the most likely explanation [38].
15.6 Involvement of Renin-Angiotensin-Aldosterone System on PVAT Function
The renin-angiotensin-aldosterone system (RAAS) is involved in systemic blood pressure regulation and in renal electrolyte homeostasis. Several studies have demonstrated the presence of local RAS activity in both white and brown perivascular adipose tissue [71]. Angiotensin II (Ang II) is the major component of RAAS with a variety of physiological actions [72]; in the adipose tissue, Ang II may play a role in adipocyte growth and differentiation, stimulating lipogenesis, pre-adipocytes recruitment and their differentiation in mature adipocytes [73]. The effects of Ang II are mediated by two main membrane receptors: the angiotensin type 1 receptor (AT1R) and the angiotensin type 2 receptor (AT2R). The AT1R is responsible for most biological effects of Ang II, including blood pressure control, trophic and pro-inflammatory effects, whereas the AT2R antagonises several AT1R-mediated effects. It has been demonstrated that Ang II through AT1R stimulates transcription factor expression, which may promote adipocytes differentiation and increase of triglycerides content in human adipocytes culture and also in 3 T3-L1 cells, thus leading to adipocyte hypertrophy [74]. The crucial role of AT1R is confirmed by the demonstration that in essential hypertensive patients, in adipocytes in culture and in a rat model of type 2 diabetes, AT1R antagonist is able to increase adiponectin concentrations and to improve insulin sensitivity [75]. Conversely, the binding between Ang II and its AT2R may determine tissue regeneration [76, 77] and upregulation of adiponectin production in neonatal rat ventricular myocytes [77].
Oxidative stress is closely correlated to RAAS; Ang II is a potent inductor of reactive oxygen substances (ROS), and during obesity systemic and related adipose tissue, Ang II levels are increased. In addition to Ang II, aldosterone is also an important mediator of RAAS effects. A review of 2011 discusses the known interaction between adiponectin and aldosterone, demonstrating an inverse relationship [78]. Previous data demonstrate the presence of crosstalk between aldosterone and Ang II which modulates and stimulates the signalling transduction such as the Ang II effects on PVAT [79].
The local RAAS system of PVAT may operate in combination or independently of circulating RAAS components. The precise function of RAAS in perivascular adipose tissue is still controversial but may contribute to vascular tone, leading to structural and functional alterations, especially in pathological conditions such as hypertension and obesity, amplifying the effect of systemic RAAS. It has been hypothesised a crosstalk between RAAS of smooth muscle cells and of endothelial cells which could explain at least in part the implication of tissue RAAS in the perivascular adipose tissue on structural alterations of small resistance arteries. In view of the relevant role of the RAAS system and PVAT on vascular function, it is interesting to understand the possible relationship between them.
In 2006, Gálvez and colleagues observed several changes in in the function and in the mass of PVAT in spontaneously hypertensive rats (SHR) and suggested an interrelation with increased vascular resistance [80]. In the SHR model, they observed a reduced anticontractile effect and mass of mesenteric PVAT compared with control mice, presenting smaller adipocytes and lower leptin content. Subsequently, Lee demonstrated that the changes on PVAT structure and function were observed also in Ang II-induced hypertension in adult male Wistar rats [61]. In addition, it has been shown that Ang II is able to mediate PVAT-associated increase of contractile response to perivascular neuronal excitation by electrical field stimulation, possibly through superoxide production [81]. Recently, it has been sown that angiotensin [1,2,3,4,5,6,7] could mediates the reduced anticontractile function in aorta PVAT of SHR rats), as angiotensin [1,2,3,4,5,6,7] receptor blockade was able to inhibit the anticontractile effect of PVAT [82, 83]. Additionally, angiotensin 1–7 has been also implicated in PVAT anticontractile function of inferior vena cava [23].
Using a mouse model, we have demonstrated that small mesenteric arteries PVAT loses its anticontractile effect after two different inflammatory stimulation: aldosterone and hypoxia [33]. Among multiple factors that may be involved in the control of obesity and hypertension vascular consequences, we have shown that eplerenone reduces the inflammatory effects caused by both aldosterone and hypoxia, and this property of mineralocorticoid-receptors blockers may be of potential therapeutic interest [33]. Further, in a recent study, we demonstrated that blocking ACE-inhibitor and also AT1R have similar effects improving the anticontractile properties of the PVAT following hypoxia. In our study in vitro induction of a hypoxic environment could stimulate the loss of anticontractile perivascular adipose tissue function seen in obese patients that could be prevented using inhibitors or the renin-angiotensin cascade [84].
In conclusion, RAAS system seems to be important in modulating PVAT effects in vascular physiology, and changes in its components production can lead to vascular impairment. Therefore RAAS blockade may protect the vasculature and may reverse the lack of PVAT anticontractile function in vascular diseases.
15.7 Clinical Translation: Melatonin
Several evidences have demonstrated that obesity is involved in pathogenesis and progression of cardiovascular disease (Fig. 15.1). The weight loss represents the obvious treatment for obesity through the lifestyle changes as a first-line treatment and the surgical intervention (as bariatric surgery). Recently it has been shown that there is reduction of adipose tissue inflammation and increase in local adiponectin, and NO bioavailability in obese subjects 6 months after bariatric surgery [85]. In this study, although patients remained morbidly obese, it was observed a significant reduction in body mass index and blood pressure, an improvement in lipid profile and blood glucose concentration and a restoration of anticontractile function of PVAT. These findings are very interesting because they could explain the cases of obese people that do not present complications as hypertension or type 2 diabetes mellitus. More recently, it has been demonstrated that in diet-induced obese rats, sustained weight loss improves PVAT function with restoration of its anticontractile property through reduction in local TNFα and increased NO production mainly by endothelial NOS (NOS-3). These findings are associated with reduction of adipocytes size and infiltration of eosinophils, decrease in macrophage infiltration, normalisation of plasma leptin and insulin and also with reduction in blood pressure, suggesting that the inflammation and dysfunction in PVAT play a crucial role in the pathogenesis of obesity and metabolic syndrome [86].

Hypothetical link between adipose tissue inflammation in obesity, oxidative stress and vascular disease
Melatonin (N-acetyl-5-methoxytryptamine) is an endogenous hormone that presents antioxidant, anti-inflammatory, anti-hyperlipidemic and anti-hypertensive properties, and it is also being seen to participate on glucose homeostasis [87, 88]. All these properties can be corroborated by the fact that melatonin therapy is able to improve blood pressure, lipid profile and parameters of oxidative stress in patients with metabolic syndrome [88]. Furthermore, chronic administration of melatonin in rats fed a normal or high-fat diet resulted in significant reduction in body weight, in circulating insulin, glucose and triglyceride levels, and was able to modulate the normal circadian pattern of plasma adiponectin [89]. Recently, in our animal model of obesity (B6.V-Lepob/OlaHsd), we have observed the effects of melatonin on the anticontractile properties of PVAT. It has been demonstrated that prolonged administration of melatonin is able to reduce hyperglycemia associated with obesity in hyperphagic mice and to ameliorate the inflammation in the perivascular environment. We saw that ob/ob mice treated with melatonin showed a marked reduction in the expression of endothelin-1 (ET-1), interleukin-6 (IL-6) and metalloproteases 2 and 9. In addition, we observed that the increased expression of both TNF-α and CD68 in visceral fat sections from ob/ob mice was significantly reduced after melatonin treatment. The anticontractile function of PVAT, partially lost in our animal model of obesity, may be restored by melatonin treatment only in presence of an intact PVAT, indicating the importance of PVAT oxidative stress in vascular dysfunction of obese animals [90].
Aging represents another condition associated with a progressively decrease of the nighttime peak of melatonin concentrations. During aging, structural and functional changes have been observed. Particularly, the effect of aging on vascular endothelium and small muscle cells (SMC) has been widely investigated, but less is known about the changes of PVAT. A senescence-accelerated prone mouse (SAMP8) is a model of age-related vascular dysfunction with associated increase in blood pressure and cognitive decline. It has been demonstrated that there was overexpression of endothelin-1 (ET-1), inducible NOS (iNOS) and cyclooxygenase-2 (COX-2) in the vasculature of these animals. All of these markers associated with oxidative stress showed a reduction in the level of vascular eNOS, cyclooxygenase-1 (COX-1) and adiponectin. In addition, in SAMP8 mice, the PVAT had lost its protective anticontractile effect, but the long-term treatment with melatonin was able to increase some vasculoprotective markers, to decrease oxidative stress and inflammation and to restore the anticontractile effect of perivascular adipose tissue. Decreased expression of adiponectin and adiponectin receptor 1 was also observed in visceral fat of untreated aging mice, whereas a significant increase was observed after melatonin treatment. The increased production/activity of adiponectin might contribute to explain the improvement of the anticontractile action of perivascular fat observed in the mesenteric small resistance arteries of SAMP8 mice after chronic treatment with melatonin [91].
Besides the use of melatonin, the blockade of RAAS system has been seen as a potential target to treat vascular diseases. It has been demonstrated that the use of mineralocorticoid receptor (MR) antagonist, as spironolactone, prevents vascular remodelling of a mice model of type 2 diabetes mellitus [92]. Interestingly, Briones et al. showed that PVAT adipocytes present functional aldosterone synthase which is able to generate aldosterone in a process regulated by Ang II through AT1R [93]. They also showed that the use of eplerenone, another MR antagonist, could improve the acetylcholine-induced relaxation in obese diabetic (db/db) mice, without effect in a control group. Further, in a mice model of specifically adipocyte-MR overexpression (adipo-MROE), animals presented features of metabolic syndrome and impaired vasoconstriction to phenylephrine with preserved endothelial function, showing that activation of adipose-MR could change functional properties of arteries [93]. These finds demonstrate a link between PVAT-RAAS system and vascular dysfunction, mainly through aldosterone action. Although it is necessary to further investigate to understand the effect of chronical treatment with MR antagonists over PVAT, this seems to be a potential target to the treatment for vascular complications of diabetes and metabolic syndrome.
15.8 Conclusions
PVAT depots are highly metabolically active and contribute to a physiologically important paracrine activity of vasodilation by the release of a broad spectrum of vasorelaxing adipokines. Perhaps the most prominent of these is adiponectin. In obesity with the development of localised tissue inflammation, the bioavailability of these PVAT-derived vasorelaxing adipokines is reduced, and blood pressure rises as a result of vasoconstriction with diabetes ensuing as a result of a decrease in glucose uptake. Manipulating the inflammasome PVAT is the next challenge in terms of producing novel ways to prevent the complications of obesity which cause so much mortality and morbidity in a culturated society.
References
Aghamohammadzadeh R, Withers S, Lynch F, Greenstein A, Malik R, Heagerty A. Perivascular adipose tissue from human systemic and coronary vessels: the emergence of a new pharmacotherapeutic target. Br J Pharmacol. 2012;165:670–82.
Szasz T, Webb RC. Perivascular adipose tissue: more than just structural support. Clin Sci (Lond). 2012;122:1–12.
Greenstein AS, Khavandi K, Withers SB, Sonoyama K, Clancy O, Jeziorska M, Laing I, Yates AP, Pemberton PW, Malik RA, Heagerty AM. Local inflammation and hypoxia abolish the protective anticontractile properties of perivascular fat in obese patients. Circulation. 2009;119:1661–70.
Galvez-Prieto B, Dubrovska G, Cano MV, Delgado M, Aranguez I, Gonzalez MC, Ruiz-Gayo M, Gollasch M, Fernandez-Alfonso MS. A reduction in the amount and anti-contractile effect of periadventitial mesenteric adipose tissue precedes hypertension development in spontaneously hypertensive rats. Hypertens Res. 2008;31:1415–23.
Bulloch JM, Daly CJ. Autonomic nerves and perivascular fat: interactive mechanisms. Pharmacol Ther. 2014;143:61–73.
Padilla J, Jenkins NT, Vieira-Potter VJ, Laughlin MH. Divergent phenotype of rat thoracic and abdominal perivascular adipose tissues. Am J Physiol Regul Integr Comp Physiol. 2013;304:R543–52.
Galvez-Prieto B, Bolbrinker J, Stucchi P, de Las Heras AI, Merino B, Arribas S, Ruiz-Gayo M, Huber M, Wehland M, Kreutz R, Fernandez-Alfonso MS. Comparative expression analysis of the renin-angiotensin system components between white and brown perivascular adipose tissue. J Endocrinol. 2008;197:55–64.
Coelho M, Oliveira T, Fernandes R. Biochemistry of adipose tissue: an endocrine organ. Arch Med Sci. 2013;9:191–200.
Florian W Kiefer, Paul Cohen, Jorge Plutzky. Fifty Shades of Brown: Perivascular Fat, Thermogenesis, and Atherosclerosis. Circulation. 2012;126(9):1012–5.
Soltis EE, Cassis LA. Influence of perivascular adipose tissue on rat aortic smooth muscle responsiveness. Clin Exp Hypertens A. 1991;13:277–96.
Weston AH, Egner I, Dong Y, Porter EL, Heagerty AM, Edwards G. Stimulated release of a hyperpolarizing factor (ADHF) from mesenteric artery perivascular adipose tissue: involvement of myocyte BKCa channels and adiponectin. Br J Pharmacol. 2013;169:1500–9.
Lohn M, Dubrovska G, Lauterbach B, Luft FC, Gollasch M, Sharma AM. Periadventitial fat releases a vascular relaxing factor. FASEB J. 2002;16:1057–63.
Saxton SN, Ryding KE, Aldous RG, Withers SB, Ohanian J, Heagerty AM. Role of sympathetic nerves and adipocyte catecholamine uptake in the vasorelaxant function of perivascular adipose tissue. Arterioscler Thromb Vasc Biol. 2018;38:880–91.
Lynch FM, Withers SB, Yao Z, Werner ME, Edwards G, Weston AH, Heagerty AM. Perivascular adipose tissue-derived adiponectin activates BK(Ca) channels to induce anticontractile responses. Am J Physiol Heart Circ Physiol. 2013;304:H786–95.
Fang L, Zhao J, Chen Y, Ma T, Xu G, Tang C, Liu X, Geng B. Hydrogen sulfide derived from periadventitial adipose tissue is a vasodilator. J Hypertens. 2009;27:2174–85.
Bussey CE, Withers SB, Saxton SN, Bodagh N, Aldous RG, Heagerty AM. β3-Adrenoceptor stimulation of perivascular adipocytes leads to increased fat cell-derived NO and vascular relaxation in small arteries. Br J Pharmacol. 2018;175:3685–98.
Withers SB, Bussey CE, Saxton SN, Melrose HM, Watkins AE, Heagerty AM. Mechanisms of Adiponectin-Associated Perivascular Function in Vascular Disease. Arterioscler Thromb Vasc Biol. 2014;34(8):1637–42.
Yokota T, Oritani K, Takahashi I, Ishikawa J, Matsuyama A, Ouchi N, Kihara S, Funahashi T, Tenner AJ, Tomiyama Y, Matsuzawa Y. Adiponectin, a new member of the family of soluble defense collagens, negatively regulates the growth of myelomonocytic progenitors and the functions of macrophages. Blood. 2000;96:1723–32.
Tsao TS, Lodish HF, Fruebis J. ACRP30, a new hormone controlling fat and glucose metabolism. Eur J Pharmacol. 2002;440:213–21.
Arita Y, Kihara S, Ouchi N, Takahashi M, Maeda K, Miyagawa J, Hotta K, Shimomura I, Nakamura T, Miyaoka K, Kuriyama H, Nishida M, Yamashita S, Okubo K, Matsubara K, Muraguchi M, Ohmoto Y, Funahashi T, Matsuzawa Y. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem Biophys Res Commun. 1999;257:79–83.
Imatoh T, Miyazaki M, Momose Y, Tanihara S, Une H. Adiponectin levels associated with the development of hypertension: a prospective study. Hypertens Res. 2008;31:229–33.
Nayak S, Soon SQ, Kunjal R, Ramadoo R, Baptiste O, Persad J, Temull V, Diptee L, Balgobin S. Relationship between adiponectin, inflammatory markers and obesity in type 2 diabetic and non-diabetic Trinidadians. Arch Physiol Biochem. 2009;115:28–33.
Lu C, Zhao AX, Gao YJ, Lee RM. Modulation of vein function by perivascular adipose tissue. Eur J Pharmacol. 2011;657:111–6.
Huang Cao ZF, Stoffel E, Cohen P. Role of perivascular adipose tissue in vascular physiology and pathology. Hypertension. 2017;69:770–7.
Berg AH, Scherer PE. Adipose tissue, inflammation, and cardiovascular disease. Circ Res. 2005;96:939–49.
Lazar MA. How obesity causes diabetes: not a tall tale. Science. 2005;307:373–5.
Gao YJ, Holloway AC, Zeng ZH, Lim GE, Petrik JJ, Foster WG, Lee RM. Prenatal exposure to nicotine causes postnatal obesity and altered perivascular adipose tissue function. Obes Res. 2005;13:687–92.
Clambey ET, McNamee EN, Westrich JA, Glover LE, Campbell EL, Jedlicka P, de Zoeten EF, Cambier JC, Stenmark KR, Colgan SP, Eltzschig HK. Hypoxia-inducible factor-1 alpha-dependent induction of FoxP3 drives regulatory T-cell abundance and function during inflammatory hypoxia of the mucosa. Proc Natl Acad Sci U S A. 2012;109:E2784–93.
Halberg N, Khan T, Trujillo ME, Wernstedt-Asterholm I, Attie AD, Sherwani S, Wang ZV, Landskroner-Eiger S, Dineen S, Magalang UJ, Brekken RA, Scherer PE. Hypoxia-inducible factor 1alpha induces fibrosis and insulin resistance in white adipose tissue. Mol Cell Biol. 2009;29:4467–83.
Lolmede K, Durand de Saint Front V, Galitzky J, Lafontan M, Bouloumie A. Effects of hypoxia on the expression of proangiogenic factors in differentiated 3T3-F442A adipocytes. Int J Obes Relat Metab Disord. 2003;27:1187–95.
Cancello R, Henegar C, Viguerie N, Taleb S, Poitou C, Rouault C, Coupaye M, Pelloux V, Hugol D, Bouillot JL, Bouloumie A, Barbatelli G, Cinti S, Svensson PA, Barsh GS, Zucker JD, Basdevant A, Langin D, Clement K. Reduction of macrophage infiltration and chemoattractant gene expression changes in white adipose tissue of morbidly obese subjects after surgery-induced weight loss. Diabetes. 2005;54:2277–86.
Chen B, Lam KS, Wang Y, Wu D, Lam MC, Shen J, Wong L, Hoo RL, Zhang J, Xu A. Hypoxia dysregulates the production of adiponectin and plasminogen activator inhibitor-1 independent of reactive oxygen species in adipocytes. Biochem Biophys Res Commun. 2006;341:549–56.
Withers SB, Agabiti-Rosei C, Livingstone DM, Little MC, Aslam R, Malik RA, Heagerty AM. Macrophage activation is responsible for loss of anticontractile function in inflamed perivascular fat. Arterioscler Thromb Vasc Biol. 2011;31:908–13.
Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, Nakayama O, Makishima M, Matsuda M, Shimomura I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest. 2004;114:1752–61.
Chatterjee TK, Stoll LL, Denning GM, Harrelson A, Blomkalns AL, Idelman G, Rothenberg FG, Neltner B, Romig-Martin SA, Dickson EW, Rudich S, Weintraub NL. Proinflammatory phenotype of perivascular adipocytes: influence of high-fat feeding. Circ Res. 2009;104:541–9.
Deshmane SL, Kremlev S, Amini S, Sawaya BE. Monocyte chemoattractant protein-1 (MCP-1): an overview. J Interf Cytokine Res. 2009;29:313–26.
Du B, Ouyang A, Eng JS, Fleenor BS. Aortic perivascular adipose-derived interleukin-6 contributes to arterial stiffness in low-density lipoprotein receptor deficient mice. Am J Physiol Heart Circ Physiol. 2015;308:H1382–90.
Withers SB, Forman R, Meza-Perez S, Sorobetea D, Sitnik K, Hopwood T, Lawrence CB, Agace WW, Else KJ, Heagerty AM, Svensson-Frej M, Cruickshank SM. Eosinophils are key regulators of perivascular adipose tissue and vascular functionality. Sci Rep. 2017;7:44571.
Wu D, Molofsky AB, Liang HE, Ricardo-Gonzalez RR, Jouihan HA, Bando JK, Chawla A, Locksley RM. Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science. 2011;332:243–7.
Maeda N, Shimomura I, Kishida K, Nishizawa H, Matsuda M, Nagaretani H, Furuyama N, Kondo H, Takahashi M, Arita Y, Komuro R, Ouchi N, Kihara S, Tochino Y, Okutomi K, Horie M, Takeda S, Aoyama T, Funahashi T, Matsuzawa Y. Diet-induced insulin resistance in mice lacking adiponectin/ACRP30. Nat Med. 2002;8:731–7.
Fruebis J, Tsao TS, Javorschi S, Ebbets-Reed D, Erickson MR, Yen FT, Bihain BE, Lodish HF. Proteolytic cleavage product of 30-kDa adipocyte complement-related protein increases fatty acid oxidation in muscle and causes weight loss in mice. Proc Natl Acad Sci U S A. 2001;98:2005–10.
Kovacova Z, Tencerova M, Roussel B, Wedellova Z, Rossmeislova L, Langin D, Polak J, Stich V. The impact of obesity on secretion of adiponectin multimeric isoforms differs in visceral and subcutaneous adipose tissue. Int J Obes. 2012;36:1360–5.
Lee MH, Klein RL, El-Shewy HM, Luttrell DK, Luttrell LM. The adiponectin receptors AdipoR1 and AdipoR2 activate ERK1/2 through a Src/Ras-dependent pathway and stimulate cell growth. Biochemistry. 2008;47:11682–92.
Ding M, Xie Y, Wagner RJ, Jin Y, Carrao AC, Liu LS, Guzman AK, Powell RJ, Hwa J, Rzucidlo EM, Martin KA. Adiponectin induces vascular smooth muscle cell differentiation via repression of mammalian target of rapamycin complex 1 and FoxO4. Arterioscler Thromb Vasc Biol. 2011;31:1403–10.
Ding M, Carrao AC, Wagner RJ, Xie Y, Jin Y, Rzucidlo EM, Yu J, Li W, Tellides G, Hwa J, Aprahamian TR, Martin KA. Vascular smooth muscle cell-derived adiponectin: a paracrine regulator of contractile phenotype. J Mol Cell Cardiol. 2012;52:474–84.
Almabrouk TA, Ugusman AB, Katwan OJ, Salt IP, Kennedy S. Deletion of AMPKalpha1 attenuates the anticontractile effect of perivascular adipose tissue (PVAT) and reduces adiponectin release. Br J Pharmacol. 2017;174(20):3398–3410.
Fukao M, Mason HS, Britton FC, Kenyon JL, Horowitz B, Keef KD. Cyclic GMP-dependent protein kinase activates cloned BKCa channels expressed in mammalian cells by direct phosphorylation at serine 1072. J Biol Chem. 1999;274:10927–35.
Foller M, Jaumann M, Dettling J, Saxena A, Pakladok T, Munoz C, Ruth P, Sopjani M, Seebohm G, Ruttiger L, Knipper M, Lang F. AMP-activated protein kinase in BK-channel regulation and protection against hearing loss following acoustic overstimulation. FASEB J. 2012;26:4243–53.
Yilmaz MI, Sonmez A, Caglar K, Celik T, Yenicesu M, Eyileten T, Acikel C, Oguz Y, Yavuz I, Vural A. Effect of antihypertensive agents on plasma adiponectin levels in hypertensive patients with metabolic syndrome. Nephrology (Carlton). 2007;12:147–53.
Brennan AM, Mantzoros CS. Drug insight: the role of leptin in human physiology and pathophysiology—emerging clinical applications. Nat Clin Pract Endocrinol Metab. 2006;2:318–27.
Leung YM, Kwan CY. Dual vascular effects of leptin via endothelium: hypothesis and perspective. Chin J Physiol. 2008;51:1–6.
Galvez-Prieto B, Somoza B, Gil-Ortega M, Garcia-Prieto CF, de Las Heras AI, Gonzalez MC, Arribas S, Aranguez I, Bolbrinker J, Kreutz R, Ruiz-Gayo M, Fernandez-Alfonso MS. Anticontractile effect of perivascular adipose tissue and Leptin are reduced in hypertension. Front Pharmacol. 2012;3:103.
Michel T, Feron O. Nitric oxide synthases: which, where, how, and why? J Clin Invest. 1997;100:2146–52.
Gil-Ortega M, Stucchi P, Guzman-Ruiz R, Cano V, Arribas S, Gonzalez MC, Ruiz-Gayo M, Fernandez-Alfonso MS, Somoza B. Adaptative nitric oxide overproduction in perivascular adipose tissue during early diet-induced obesity. Endocrinology. 2010;151:3299–306.
Aghamohammadzadeh R, Unwin RD, Greenstein AS, Heagerty AM. Effects of obesity on perivascular adipose tissue vasorelaxant function: nitric oxide, inflammation and elevated systemic blood pressure. J Vasc Res. 2015;52:299–305.
Mustafa AK, Sikka G, Gazi SK, Steppan J, Jung SM, Bhunia AK, Barodka VM, Gazi FK, Barrow RK, Wang R, Amzel LM, Berkowitz DE, Snyder SH. Hydrogen sulfide as endothelium-derived hyperpolarizing factor sulfhydrates potassium channels. Circ Res. 2011;109:1259–68.
Yan H, Du J, Tang C. The possible role of hydrogen sulfide on the pathogenesis of spontaneous hypertension in rats. Biochem Biophys Res Commun. 2004;313:22–7.
Zhong G, Chen F, Cheng Y, Tang C, Du J. The role of hydrogen sulfide generation in the pathogenesis of hypertension in rats induced by inhibition of nitric oxide synthase. J Hypertens. 2003;21:1879–85.
Gao YJ, Lu C, Su LY, Sharma AM, Lee RM. Modulation of vascular function by perivascular adipose tissue: the role of endothelium and hydrogen peroxide. Br J Pharmacol. 2007;151:323–31.
Lee YC, Chang HH, Chiang CL, Liu CH, Yeh JI, Chen MF, Chen PY, Kuo JS, Lee TJ. Role of perivascular adipose tissue-derived methyl palmitate in vascular tone regulation and pathogenesis of hypertension. Circulation. 2011;124:1160–71.
Lee RM, Ding L, Lu C, Su LY, Gao YJ. Alteration of perivascular adipose tissue function in angiotensin II-induced hypertension. Can J Physiol Pharmacol. 2009;87:944–53.
Pizzinat N, Marti L, Remaury A, Leger F, Langin D, Lafontan M, Carpene C, Parini A. High expression of monoamine oxidases in human white adipose tissue: evidence for their involvement in noradrenaline clearance. Biochem Pharmacol. 1999;58:1735–42.
Ayala-Lopez N, Martini M, Jackson WF, Darios E, Burnett R, Seitz B, Fink GD, Watts SW. Perivascular adipose tissue contains functional catecholamines. Pharmacol Res Perspect. 2014;2:e00041.
Bamshad M, Aoki VT, Adkison MG, Warren WS, Bartness TJ. Central nervous system origins of the sympathetic nervous system outflow to white adipose tissue. Am J Phys. 1998;275:R291–9.
Foster MT, Bartness TJ. Sympathetic but not sensory denervation stimulates white adipocyte proliferation. Am J Physiol Regul Integr Comp Physiol. 2006;291:R1630–7.
Shen YT, Cervoni P, Claus T, Vatner SF. Differences in beta 3-adrenergic receptor cardiovascular regulation in conscious primates, rats and dogs. J Pharmacol Exp Ther. 1996;278:1435–43.
Smith MM, Minson CT. Obesity and adipokines: effects on sympathetic overactivity. J Physiol. 2012;590:1787–801.
Manolis AJ, Poulimenos LE, Kallistratos MS, Gavras I, Gavras H. Sympathetic overactivity in hypertension and cardiovascular disease. Curr Vasc Pharmacol. 2014;12:4–15.
Muntzel MS, Al-Naimi OA, Barclay A, Ajasin D. Cafeteria diet increases fat mass and chronically elevates lumbar sympathetic nerve activity in rats. Hypertension. United States. 2012;60:1498–502.
Post SR, Hammond HK, Insel PA. Beta-adrenergic receptors and receptor signaling in heart failure. Annu Rev Pharmacol Toxicol. 1999;39:343–60.
Engeli S, Gorzelniak K, Kreutz R, Runkel N, Distler A, Sharma AM. Co-expression of renin-angiotensin system genes in human adipose tissue. J Hypertens. 1999;17:555–60.
Touyz RM, Schiffrin EL. Signal transduction mechanisms mediating the physiological and pathophysiological actions of angiotensin II in vascular smooth muscle cells. Pharmacol Rev. 2000;52:639–72.
Cassis LA, Police SB, Yiannikouris F, Thatcher SE. Local adipose tissue renin-angiotensin system. Curr Hypertens Rep. 2008;10:93–8.
Jones BH, Standridge MK, Moustaid N. Angiotensin II increases lipogenesis in 3T3-L1 and human adipose cells. Endocrinology. 1997;138:1512–9.
Furuhashi M, Ura N, Takizawa H, Yoshida D, Moniwa N, Murakami H, Higashiura K, Shimamoto K. Blockade of the renin-angiotensin system decreases adipocyte size with improvement in insulin sensitivity. J Hypertens. 2004;22:1977–82.
Sowers JR. Endocrine functions of adipose tissue: focus on adiponectin. Clin Cornerstone. 2008;9:32–8. discussion 39-40
Guo B, Li Y, Han R, Zhou H, Wang M. Angiotensin II upregulation of cardiomyocyte adiponectin production is nitric oxide/cyclic GMP dependent. Am J Med Sci. 2011;341:350–5.
Flynn C, Bakris GL. Interaction between adiponectin and aldosterone. Cardiorenal Med. 2011;1:96–101.
Lemarie CA, Paradis P, Schiffrin EL. New insights on signaling cascades induced by cross-talk between angiotensin II and aldosterone. J Mol Med (Berl). 2008;86:673–8.
Galvez B, de Castro J, Herold D, Dubrovska G, Arribas S, Gonzalez MC, Aranguez I, Luft FC, Ramos MP, Gollasch M, Fernandez Alfonso MS. Perivascular adipose tissue and mesenteric vascular function in spontaneously hypertensive rats. Arterioscler Thromb Vasc Biol. 2006;26:1297–302.
Gao YJ, Takemori K, Su LY, An WS, Lu C, Sharma AM, Lee RM. Perivascular adipose tissue promotes vasoconstriction: the role of superoxide anion. Cardiovasc Res. 2006;71:363–73.
Lu C, Su LY, Lee RM, Gao YJ. Alterations in perivascular adipose tissue structure and function in hypertension. Eur J Pharmacol. 2011;656:68–73.
Lee RM, Lu C, Su LY, Gao YJ. Endothelium-dependent relaxation factor released by perivascular adipose tissue. J Hypertens. 2009;27:782–90.
Rosei CA, Withers SB, Belcaid L, De Ciuceis C, Rizzoni D, Heagerty AM. Blockade of the renin-angiotensin system in small arteries and anticontractile function of perivascular adipose tissue. J Hypertens. 2015;33:1039–45.
Aghamohammadzadeh R, Greenstein AS, Yadav R, Jeziorska M, Hama S, Soltani F, Pemberton PW, Ammori B, Malik RA, Soran H, Heagerty AM. Effects of bariatric surgery on human small artery function: evidence for reduction in perivascular adipocyte inflammation, and the restoration of normal anticontractile activity despite persistent obesity. J Am Coll Cardiol. 2013;62:128–35.
Bussey CE, Withers SB, Aldous RG, Edwards G, Heagerty AM. Obesity-related perivascular adipose tissue damage is reversed by sustained weight loss in the rat. Arterioscler Thromb Vasc Biol. 2016;36:1377–85.
Bonnefont-Rousselot D, Collin F, Jore D, Gardes-Albert M. Reaction mechanism of melatonin oxidation by reactive oxygen species in vitro. J Pineal Res. 2011;50:328–35.
Prunet-Marcassus B, Desbazeille M, Bros A, Louche K, Delagrange P, Renard P, Casteilla L, Penicaud L. Melatonin reduces body weight gain in Sprague Dawley rats with diet-induced obesity. Endocrinology. 2003;144:5347–52.
Kozirog M, Poliwczak AR, Duchnowicz P, Koter-Michalak M, Sikora J, Broncel M. Melatonin treatment improves blood pressure, lipid profile, and parameters of oxidative stress in patients with metabolic syndrome. J Pineal Res. 2011;50:261–6.
Agabiti-Rosei C, De Ciuceis C, Rossini C, Porteri E, Rodella LF, Withers SB, Heagerty AM, Favero G, Agabiti-Rosei E, Rizzoni D, Rezzani R. Anticontractile activity of perivascular fat in obese mice and the effect of long-term treatment with melatonin. J Hypertens. 2014;32:1264–74.
Rios-Lugo MJ, Cano P, Jimenez-Ortega V, Fernandez-Mateos MP, Scacchi PA, Cardinali DP, Esquifino AI. Melatonin effect on plasma adiponectin, leptin, insulin, glucose, triglycerides and cholesterol in normal and high fat-fed rats. J Pineal Res. 2010;49:342–8.
Agabiti-Rosei C, Favero G, De Ciuceis C, Rossini C, Porteri E, Rodella LF, Franceschetti L, Maria Sarkar A, Agabiti-Rosei E, Rizzoni D, Rezzani R. Effect of long-term treatment with melatonin on vascular markers of oxidative stress/inflammation and on the anticontractile activity of perivascular fat in aging mice. Hypertens Res. 2017;40:41–50.
Briones AM, Nguyen Dinh Cat A, Callera GE, Yogi A, Burger D, He Y, Correa JW, Gagnon AM, Gomez-Sanchez CE, Gomez-Sanchez EP, Sorisky A, Ooi TC, Ruzicka M, Burns KD, Touyz RM. Adipocytes produce aldosterone through calcineurin-dependent signaling pathways: implications in diabetes mellitus-associated obesity and vascular dysfunction. Hypertension. 2012;59:1069–78.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Agabiti-Rosei, C., Barp, C., Saxton, S.N., Heagerty, A.M. (2020). The Role of Perivascular Adipose Tissue in Arterial Function in Health and Disease. In: Agabiti-Rosei, E., Heagerty, A.M., Rizzoni, D. (eds) Microcirculation in Cardiovascular Diseases. Updates in Hypertension and Cardiovascular Protection. Springer, Cham. https://doi.org/10.1007/978-3-030-47801-8_15
Download citation
DOI: https://doi.org/10.1007/978-3-030-47801-8_15
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-47800-1
Online ISBN: 978-3-030-47801-8
eBook Packages: MedicineMedicine (R0)