
Abstract
The endocannabinoid system (ECS) is composed of lipid signalling ligands, their G-protein coupled receptors and the enzymes involved in ligand generation and metabolism. Increasingly, the ECS is emerging as a critical agent of energy metabolism regulation through its ability to modulate caloric intake centrally as well as nutrient transport, cellular metabolism and energy storage peripherally. Visceral obesity has been associated with an upregulation of ECS activity in several systems and inhibition of the ECS, either pharmacologically or genetically, results in decreased energy intake and increased metabolic output. This review aims to summarize the recent advances that have been made regarding our understanding of the role the ECS plays in crucial peripheral systems pertaining to energy homeostasis: adipose tissues, the liver and skeletal muscle.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 The endocannabinoid system and energy homeostasis
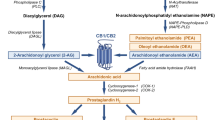
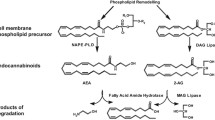
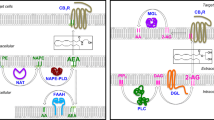
The regulation of energy balance in mammals consists of a complex network of feedback systems involving hormonal and neural control of energy input and energy output. Caloric imbalance leads to rapid changes in adipocyte metabolism, with the relative energy balance (i.e., energy intake vs. energy expenditure) determining whether triglyceride is mobilized or deposited, and how efficiently glucose is oxidized or converted into fatty acids or glycogen. The endocannabinoid system (ECS) consists of two G-protein-coupled receptors (the cannabinoid CB1 and CB2 receptors), their ligands (the two major endocannabinoids, anandamide [AEA] and 2-arachidonoylglycerol [2-AG]), and proteins regulating cannabinoid receptor activity and endocannabinoid tissue levels (see [1], for a comprehensive review), and is emerging as one of the key players in the regulation of energy homeostasis and a major determinant, through its dysfunctions, of metabolic disorders. Whilst the role of endocannabinoids and CB1 receptors in the stimulation of both the homeostatic and hedonic aspects of food-intake is now well established (although it still requires the clarification of several details), the importance of the function of both cannabinoid receptors in peripheral organs involved in the control energy metabolism has been appreciated only during the last 5 years. In this article, we review some of the most recent aspects of endocannabinoid control of metabolism in the four major organs that, in mammals, determine the fate of energy utilization vs. storage, and the malfunctioning of which is at the basis of obesity, dyslipidemia and type 2 diabetes: the white and brown adipose tissues, the liver and the skeletal muscle (Fig. 1).

Summary of some of the major effects of endocannabinoid signalling in peripheral organs controlling fat storage and energy expenditure. AMPK AMP kinase, ECS endocannabinoid system, FA fatty acid, TG triglyceride, VLDL very low density lipoproteins, UCP-1 uncoupling protein-1
1.1 Adipose tissue
Originally considered a passive reservoir for energy storage, this tissue is now considered a complex and highly active metabolic endocrine organ consisting of a wide range of components (adipocytes, connective tissue matrix, nerve tissue, stromovascular cells and immune cells) functioning together as an integrated unit [2]. The impact of the adipose tissue in the control of energy balance is due to its capability not only of lipid-storing and -mobilizing tissue, but also of functionally producing heat (in brown adipose tissue) and releasing adipocytokines [3]. Since the occurrence in the adipose tissue of the ECS and all its constituents, including the two endogenous ligands, AEA and 2-AG, their corresponding selective receptors, and the machinery of proteins and enzymes that is involved in their biosynthesis, release, transport, and degradation, has been already extensively reported [4–6], in this article we will focus purely on the adipocyte fraction of the adipose tissue and on the molecular role of adipose cannabinoid signalling in white (WAT) and brown (BAT) adipose tissue.
1.1.1 WAT
White adipose tissue, with all its fat droplets, is the main store of energy in humans and in part acts as a thermal insulator, helping to maintain body temperature. It is under control of insulin and glucagon, both released from the pancreas, which, upon receptor activation, initiate phosphorylation cascades that regulate hormone-sensitive lipase, the enzyme that catalyzes the breakdown of the stored fat to fatty acids, which are then exported into the blood (as such, bound to albumin, or as triglycerides, covalently bound to glycerol), and subsequently delivered to the liver and other tissues. After the discovery that the ECS has a relevant role in brain areas involved in the regulation of both the homeostatic and hedonic aspects of food intake [6–9], a significant number of experimental reports have emerged on the importance of the ECS also in fat cells and other peripheral tissues implicated in energy metabolism [10–12]. Apart from the identification of human adipocytes as a source of endocannabinoids [13] and expressing the enzymatic machinery not only to produce but also degrade endogenous cannabinoids [5, 14], a relevant number of papers have been published on the function of cannabinoid receptors in the peripheral regulation of lipid metabolism. The presence of CB1 in mature adipocytes, but not in pre-adipocytes, was demonstrated in both human primary fat cells and rodent primary cells and cell lines [15, 16], while the expression of CB2 in mature adipocytes is still a controversial issue [16, 17]. Nevertheless, the consequence of CB2 deficiency has very recently been examined in Cb2 (−/−) and wild-type mice treated with a selective CB2 antagonist or fed a high-fat diet (HFD). Despite the increase in food intake and adiposity with age, Cb2 (−/−) mice maintain insulin sensitivity and do not show signs of obesity-induced inflammation [18].
The large majority of reports has dealt so far with the function in the WAT of CB1 receptors, the modulation of which still remains a promising strategy for the treatment of obesity. Activation of CB1 in adipocytes increases formation (via up-regulation of lipogenic enzymes) and storage of triglycerides, and facilitates the uptake of glucose necessary for de novo lipogenesis of triglycerides. This latter effect is exerted by stimulating solute carrier family 2 (facilitated glucose transporter), member 4 (Slc2a4 aka GLUT4) translocation by increasing influx of extracellular calcium and phosphatidylinositol 3-kinase (PI3K) activity [5, 6, 10]. CB1 receptor stimulation was shown to alter the expression of some glucostatic adipokines in white adipocytes. In particular, visfatin (a novel adipokine with insulin-mimetic actions and increased levels in obesity) is up-regulated after acute WIN 55212-2 stimulation. Conversely, adiponectin (the adipocyte-derived factor with insulin-sensitizing properties and decreased levels in obesity) is downregulated under the same conditions [19], indicating a CB1-mediated positive induction of energy balance and reinforcing the concept of beneficial actions of peripheral CB1 blockade in obesity. Accordingly, treatment with a low-concentration of the CB1 antagonist/inverse agonist Rimonabant, while inhibiting mouse pre-adipocyte proliferation, induces the expression of the adipo-hormone Acrp30 (adiponectin) as well as the enzyme glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and reduces mitogen-activated protein kinase (MAPK) activity without affecting the induction of lipid accumulation [20]. The use of CB1 blockers exerted a direct effect on adipocytes via stimulation adiponectin mRNA expression in obese fa/fa rats [15]. In addition, Rimonobant-induced increased adiponectin release from adipocytes might also activate hepatic 5′-AMP-activated protein kinase (AMPK) to increase fatty acid entry into mitochondria and subsequently β-oxidation [15, 21, 22] (see also Section 1.2). Moreover, Rimonabant induced eNOS activity in mouse primary adipocytes leading to increased mitochondrial biogenesis [23]. Conversely CB1 activation has reverse effects as result of increased p38 (aka mitogen-activated protein kinase 14; Mapk14) phosphorylation downstream of decreased eNOS activity, not only in WAT but in liver and skeletal muscle as well [24].
As far as in vivo studies are concerned, the peripheral treatment of rats exhibiting high fat diet induced obesity (DIO) with Rimonabant (ip 10 mg/kg/day) resulted in a food-intake-independent reduction of stearoyl-Coenzyme A desaturase 1 (SCD-1) and increase in carnitine palmitoyltransferase 1 (CPT-1) expression, indicating a decrease in lipogenesis and increase in lipolysis [25]. Moreover, Rimonabant was shown to enhance carnitine acetyltransferase (CAT) and carnitine palmityltransferase II (CPT2) expression required for both the β-oxidation pathway and the tricarboxylic acid (TCA) cycle, suggesting a reduced fat storage in WAT also in HFD-mice [26]. A recent preliminary report, by showing that adipocyte-specific and tamoxifen inducible CB1 knockout mice are totally resistant to high fat diet-induced obesity, insulin resistance and dyslipidemia, shows how, at least in rodents, CB1 receptors in adipocytes may play a central role in lipogenesis, and hence also in obesity and related co-morbidities [27]. Indeed, there is ever increasing evidence that endocannabinoid tone in adipocytes is subject to negative feed-back control by hormones and peroxisome proliferator-activated receptors (PPAR), including leptin [7, 28], insulin [29, 30], PPARγ [5, 10], and, following exercise, PPARδ [31]. This feed-back control then becomes impaired in obesity, which can be accompanied by leptin and insulin resistance, and due in part to physical inactivity (and subsequent down-regulation of PPARδ), thereby leading to ECS dysregulation.
The dysregulation of endocannabinoid/CB1 tone in obesity at both central and peripheral levels, and its participation in obesity, dyslipidemia and insulin resistance, are now well established concepts, although the causes of this phenomenon still need to be fully clarified. Elevated levels of endocannabinoids have been observed in the epididymal fat of diet-induced obese mice [4–6], but reduced levels are instead found in the subcutaneous WAT of these animals [4]. Also in overweight and obese humans, endocannabinoid tone, in terms of either endocannabinoid levels or CB1 expression, or both, seems to be elevated in the abdominal WAT and reduced in the subcutaneous WAT [5, 32, 33]. In view of: 1) the postulated protective role of subcutaneous fat as, among others, a buffer preventing ectopic fat deposition, 2) the strong association between abdominal-visceral fat with dyslipidemia, dyslipoproteinemia and insulin resistance [34]; and 3) the lipogenic action of CB1 receptors, the unbalance of the ECS between subcutaneous and visceral WAT might eventually contribute to accumulation of the latter at the expenses of the former, and, hence, to the several metabolic disorders associated with abdominal obesity. Accordingly, a strong positive correlation has been reported between plasma 2-AG levels and the volume of abdominal, but not subcutaneous, WAT [14, 35].
In fact, the role of the peripheral endocannabinoid system in human obesity has been extensively investigated also by measuring circulating endocannabinoid concentrations and the expression of the entire ECS in adipose tissue of lean and obese women before and after a 5% weight loss. A strong negative correlation was found between the adipose tissue expression (strongly reduced) of fatty acid amide hydrolase (FAAH), one of the enzymes that degrades the endocannabinoids, and circulating levels of the latter (proficiently increased), both parameters being unaffected by 5% weight loss [36]. These data are in agreement with the upregulation of the peripheral endocannabinoid system later reported in Zucker vs. lean rats following food deprivation and re-feeding [37]. On the other hand, in abdominally obese men, a 1-year life style intervention, consisting of reduced caloric intake and exercise, and leading to a waist reduction of over 8 cm, was found to be accompanied by a strong decrease of plasma 2-AG levels and a lesser, although still statistically significant , decrease in AEA levels. Importantly, the reduction of plasma 2-AG, but not AEA, levels correlated with the reduction in visceral, but not subcutaneous, WAT, with the decrease in plasma triglyceride and the increase in High-density lipoprotein (HDL)-cholesterol [38].
Finally, an interesting study was recently carried out to evaluate EC levels in the subcutaneous adipose tissue (SAT) of subjects with both obesity (OB) and type 2 diabetes (OBT2D), two conditions characterised by similar adiposity and whole body insulin resistance and lower plasma leptin levels. As compared to normal weight, some alterations were found in the levels of ECs. In particular, AEA and its metabolically related congener (OEA and PEA) levels in the SAT were elevated, while 2-AG levels were reduced in OBT2D but not to a statistically significant extent in OB subjects [39]. Overall, all these findings clearly indicate how diversified the potential role of the ECS is in the various WAT depots, in particular with relation to obesity and type 2 diabetes.
1.1.2 BAT
Brown adipose tissue has a unique ability to generate heat via a non-shivering process for thermoregulation and the utilization of excess caloric intake. The heat generation is related to mitochondrial metabolism, which usually produces the energy-rich storage compound ATP through respiration, but that, in the BAT, is diverted to heat production via a higher-than-normal concentration of the mitochondrial inner membrane protein uncoupling protein-1 (UCP1) [40]. The exact role of UCP1 in the energy expenditure of adult humans is a controversial issue, however it is generally accepted that UCP1 facilitates the leak of protons from the mitochondrial inter-membrane space to the matrix, thus uncoupling the proton gradient from ATP production [41]. The study of human BAT mass is complicated by the fact that the tissue is somewhat inactive normally; making it nearly impossible to distinguish visually between BAT and WAT. Also, in adult humans, brown fat is very scarce and brown adipocytes are dispersed between white adipocytes, the reason for their co-occurrence being explained by the essential plasticity of the adipose organ; if needed, the brown component of the organ can increase at the expense of the white component and vice versa [42]. In recent years, the participation of the ECS in the regulation of energy balance, and hence its possible implication in the modulation of energy expenditure, has prompted the investigation of the role of the ECS within the BAT. Both physiological and pharmacological stimuli appear capable of reactivating and trans-differentiating “dormant” brown fat cells into white adipocytes [3]; on the other hand, inhibition of the cannabinoid CB1 receptor may be capable of an eventual increase in energy expenditure by producing a functional “trans-differentiation” of white into brown adipocytes. Direct exposure (both acute and chronic) to the CB1 antagonist/inverse agonist Rimonabant, as well as siRNA-mediated knock down of CB1, increases uncoupling protein 1 (mitochondrial, proton carrier) (Ucp1) and peroxisome proliferator-activated receptor gamma, coactivator 1 alpha (Ppargc1a aka PGC-1) expression, mitochondrial biogenesis, as well as AMPK phosphorylation in white adipocytes [43]. In contrast, treatment of differentiated brown adipocytes with a CB1 agonist decreased Ucp1 expression and protein product levels in as little as 2 h, without impairing lipid accumulation or terminal adipocyte differentiation, suggesting an acute and differentiation-independent negative effect of cannabinoid signaling on thermogenic UCP1 [19]. In a diet-induced obesity (DIO) model, mice treated with Rimonabant showed decreased lumbar WAT and BAT weights and an induction of genes encoding adipocyte-secreted proteins involved in thermogenesis and mitochondrial β-oxidation [26]. According to this study, Rimonabant reversed the phenotype of obese adipocytes by reversing DIO-induced gene expression changes in both WAT and BAT. These modulations contributed to limiting fat energy storage and, in the BAT, tended to encourage energy dissipation through mitochondrial heat production [26]. Several other reports have also documented a direct role of the ECS in the modulation of proteins involved in thermogenesis [26, 44]. The effects of Rimonabant on brown adipose tissue and its implication for energy expenditure has also been investigated in rats surgically implanted with biotelemetry devices to measure BAT temperature as a putative measure of thermogenesis. Physical activity, body weight, food intake, as well as changes in Ucp1 messenger RNA (mRNA) and protein were measured as well. Rimonabant significantly reduced body weight throughout the entire treatment (showing only a transient decrease in food intake) and resulted in an increase in UCP1 levels with a corresponding intense increase in BAT temperature. All these effects were notably mitigated following denervation, suggesting that the long-term weight loss observed through elevation in energy expenditure is largely mediated by the ECS in the sympathetic nervous system [45]. In full agreement with this hypothesis, Quarta et al very recently showed that conditional mutant mice lacking CB1 expression in central and sympathetic neurons, but not in non-neuronal peripheral organs, display a lean phenotype and resistance to diet-induced obesity. This phenotype results from an increase in lipid oxidation and thermogenesis as a consequence of an enhanced sympathetic tone and decreased energy absorption [46].
1.2 Liver
The liver is a major organ involved in energy homeostasis, being crucial for the metabolism of carbohydrates, proteins and fats [47]. Non-alcoholic fatty liver disease (NAFLD) is a major manifestation of obesity associated metabolic syndrome leading to insulin resistance within hepatocytes, having significant negative effects on lipid and glucose metabolism [48], and ultimately culminating in hepatocellular carcinoma and/or end-stage liver disease-related mortality [49]. It is believed that the high triglyceride levels of steatotic livers, in addition to being a result of the incorporation of free fatty acids from increased adipocyte lipolysis, is significantly contributed to by de novo fat synthesis within the liver itself [50]. Several studies have detected cannabinoid receptor 1 (Cnr1) transcripts and protein products in hepatocytes and hepatic stellate cells, as well as significant levels of AEA and 2-AG, indicating that a significant basal ECS tone exists within the liver [51]. Increasingly, it is becoming clear that antagonism of CB1 signalling ameliorates hepatic insulin resistance and steatosis, having significant effects on lipid metabolism regulation [52]. Genetically obese (Zucker fa/fa) rats, and mice with diet induced hepatic steatosis, treated with the systemic CB1 antagonist/inverse agonist Rimonabant or the largely peripheral CB1 antagonist AM6545 show a reversal of hepatic steatosis and dyslipidemia [53–55], similar to results observed in mice deficient for global or hepatic Cnr1 expression, which are resistant to diet- or alcohol-induced steatosis [11, 56, 57]. Hepatocyte-specific knock out of Cnr1, whilst not protecting against diet-induced obesity per se, improved hepatic steatosis, dyslipidemia, insulin and leptin resistance as compared to wild type mice when fed a high fat diet, indicating a direct role for the ECS within the liver [56].
Mice on high sugar and/or high fat diets exhibit increased hepatic CB1 levels as well as increased hepatic AEA, but not 2-AG, levels [11, 54], supporting the idea that conditions which induce hepatic steatosis also increase ECS tone in the liver. Indeed, hepatic steatosis can also be induced by alcohol (alcoholic fatty liver), which also increases liver CB1 levels and stimulates paracrine CB1 signalling in hepatocytes via 2-AG produced in hepatic stellate cells [57]. Increased Cnr1 expression in this model is mediated by 2-AG indirectly as it is dependent on retinoic acid signalling through the activation of Retinoic Acid Receptor gamma (RARγ), which was found to bind to, and activate, a 5′ enhancer element of Cnr1 [58]. Interestingly, both alcohol and high fat diets upregulate RARγ levels and hepatic CB1 levels in a CB1-dependent manner [54, 58], indicating the possibility that feed-forward loops may be a common mechanism resulting in increased ECS tone in response to various hepatosteatotic stimuli. Interestingly, one of the very early in vitro studies on the effects of the ECS on hepatic fatty acid metabolism concluded that AEA inhibited de novo fatty acid synthesis through its metabolite arachidonic acid [59]. However, diet-induced obesity results in decreased FAAH activity, but not expression, in the liver, resulting in increased AEA levels [11] and presumably concomitantly decreasing the amount of arachidonic acid. Therefore, steatotic conditions appear to hyper-activate the ECS in the liver at both the receptor and ligand levels, which may normally act as a modulator of fat metabolism through the tight regulation of CB1 activity and AEA conversion to arachidonic acid ratio. Increased AEA levels within the liver are also likely to be deleterious as they may stimulate the progression of NAFLD due to the induction of necroinflammatory changes. While both 2-AG and AEA result in hepatic stellate cell death, the susceptibility of hepatocytes to AEA-induced necrosis is dependent on reduced FAAH activity, potentially stimulating inflammation and fibrogenesis [60–62].
The molecular mechanisms by which the ECS modulates metabolism within hepatocytes remain to be elucidated, although several studies have shown that they impinge upon lipogenic and lipolytic pathways. For instance, hepatic CB1 activation results in increased lipogenesis via the upregulation of the lipogenic transcription factor sterol regulatory element binding transcription factor 1 (Srebf1) and subsequently the expression of key enzymes involved in lipogenesis (acetyl-Coenzyme A carboxylase alpha [Acaca, aka Acc1] and fatty acid synthase [Fasn]) [11, 57], whereas CB1 blockade decreased the expression of these same lipogenic enzymes as well as stearoyl-Coenzyme A desaturase 1 (Scd1) [55]. Further, like in the adipose tissue, liver 5′-AMP-activated protein kinase (AMPK) activity was markedly decreased in rats treated with THC, presumably through CB1 stimulation [63]. AMPK is utilized in several tissues (both central and peripheral) as a fuel-level sensor, and in the liver regulates the expression, as well as enzymatic activity, of several key factors of lipid metabolism [21, 22]. AMPK activation, for instance, decreases the expression of Srebf1 and phosphorylates Acaca/ACC1, thus inactivating it and resulting not only in decreased synthesis, but also in increased mitochondrial β-oxidation of fatty acids, the latter due to the reduction of malonyl CoA, an inhibitor of carnitine palmitoyltransferase 1a (Cpt1a), which is critical for the introduction of fatty acids into the mitochondria [21, 22]. Consistent with the above, hepatic Cnr1 knock-out resulted in increased hepatic AMPK phosphorylation and subsequently increased Cpt1a levels and activity [56, 57], perhaps in part through inhibition of Acaca activity. In support of this, Rimonabant treatment significantly decreased the levels of manonyl CoA in the livers of mice on a high sugar, high fat diet [54]. This increase in Cpt1a activity in turn provides a plausible explanation for the observed increase in liver mitochondrial oxygen consumption and β-oxidation of Rimonabant-treated mice [64]. Accordingly, CB1 pharmacological activation was very recently associated with reduced AMPK phosphorylation and endothelial nitric oxide synthase (eNOS) expression resulting in increased p38 phosphorylation [24] in the livers (as well as fat and muscles) of mice on a high fat, but not regular chow diet [24]. This decrease, in turn, led to reduced mitochondrial biogenesis. It must be noted, however, that through the increase of adiponectin expression in adipocytes (see Section “1.1.1”) [15], Rimonabant may also activate hepatic AMPK to increase fatty acid entry into mitochondria and subsequently β-oxidation [65].
Finally, a role in high fat-induced hepatosteatosis was recently suggested also for CB2 receptors. An early report had indicated that these receptors are over-expressed in the livers of patients with NAFLD [66]. More recently, it was shown that in the liver of obese mice, Cnr2 mRNA was weakly induced, and that high fat diet—induced hepatic steatosis was enhanced in WT mice treated with JWH-133, a selective CB2 agonist, and blunted in Cnr2 −/− mice [67]. Finally, De Gottardi and colleagues found that both CB1 and CB2-selective agonists can induce lipogenesis in immortalized human hepatocytes in vitro [68]. In particular, CB1 and CB2 agonists increased the degree of steatosis of oleic acid-treated fatty he patocytes, and the CB2 agonist increased the expression of CB1 receptors. CPT-1 was significantly overexpressed and SREBP-1c (aka SREBF1), FAS and lecithin cholesterol acyltransferase (LCAT) were downregulated in fatty immortalized human hepatocytes. Treatment with the CB agonists ACEA and AM1241 partially reversed these changes, except for SREBP-1c. Finally, CB2, but not CB1, agonism decreased the expression of apolipoprotein B [68].
1.3 Muscle
Muscle is a contractile tissue which is key to energy homeostasis due to the large amounts of energy it requires to perform its many functions. This energy is derived from the metabolism of fats and carbohydrates; as such, muscle is a major site of action for insulin, which regulates the entry of glucose into cells, and impairment of glucose uptake as well as glucose transport, and of the subsequent glucose oxidation and/or glycogen synthesis in muscles, is a major determinant of the severity of type 2 diabetes mellitus [69]. Recently several research groups have confirmed the expression of CB receptors as well as other components of the ECS at both the nucleotide and protein level in a variety of human and rodent muscle cells [70–77]. Similar to results obtained in the liver, mice on a high fat diet show increased levels of Cnr1 expression in soleus muscle tissue [78]. Furthermore, the soleus muscle from DIO-mice exhibited levels of 2-AG that were significantly elevated as compared to that of mice fed a normal chow, although the extent of this effect depended on the type of the high fat diet used and on its duration [79]. Conversely, skeletal muscle obtained from rats fed a high fat diet had decreased levels of EC receptors and the 2-AG biosynthetic enzyme diacylglycerol lipase, alpha (DAGLα) and increased levels of the 2-AG degrading enzyme monoglyceride lipase (MGLL aka MAGL) [77], whereas genetically obese (fa/fa) Zucker rats showed decreased CB1 levels in soleus and myocardial tissue [80]. Finally, human primary skeletal muscle myotubes collected from lean and obese subjects showed no differences in Cnr1 gene expression [70]. Further investigations into the apparent species-, diet- and age-dependent effects of obesity on ECS tone in the skeletal muscle are clearly warranted in light of these apparently contradictory results.
Studies performed in vitro with myotubes from lean and obese donors indicate that the ECS has a profound effect on muscle oxidative pathways [70]. CB1 stimulation with AEA had induced a trend to decrease AMPKa1 levels in both lean and obese myotubes (although this did not reach statistical significance, possibly due to the existence of a high ECS tone), while CB1 blockade resulted in a marked increase of AMPKa1 expression, similar to results obtained in liver and adipose tissue (see above), supporting a negative role for the ECS in muscle fatty acid oxidation [63, 70]. Interestingly, in the same study, treatment of lean myotubes with AEA resulted in a increase in the expression of PPARGC1A and its downstream target pyruvate dehydrogenase kinase, isoenzyme 4 (PDK4), a negative regulator or glucose metabolism, while CB1 antagonism in the same myotubes decreased the expression of these genes, hinting that ECS signalling in myotubes might inhibit glucose uptake and metabolism. However, the authors could not repeat these results in other experimental systems. Most recently CB1 activation has been shown to decrease mitochondrial biogenesis in skeletal muscle (as well as in liver and WAT, see above), indicating that the ECS not only regulates mitochondrial activity, but levels as well [24]. The implications that these data have for the role of the ECS in the development of diabetes is significant, as defective mitochondrial oxidative phosphorylation in the skeletal muscle has been associated with insulin resistance [69].
Support for the role of the ECS in modulating glucose transport into muscle cells is rapidly increasing. An early study utilizing isolated soleus muscle from ob/ob Leptin mutant mice treated with Rimonabant showed significant increases in skeletal muscle glucose uptake [81]. This study however did not provide evidence for a direct role of the ECS in skeletal muscle. Indeed the effects of Rimonabant could have been explained by the fact that CB1 signalling decreases adiponectin and increases visfatin production in adipocytes, leading to insulin-insensitivity and decreased muscular glucose uptake [19]. Further, it was reported that Rimonabant-mediated induction of glucose-uptake in a variety of muscle cells in treated mice was connected to its anorexigenic effect, as similar results were obtained from pair fed animals [25]. Recently however, examination of the role of the ECS in modulating glucose uptake by the skeletal muscle has provided evidence for a direct action of endocannabinoids in this tissue and has led to a greater understanding of the molecular mechanisms involved. Isolated rat soleus muscle explants from both lean, insulin-sensitive (Fa/-) and obese, insulin-insensitive (fa/fa) Zucker rats similary react to Rimonabant treatment; both exhibited increased glucose uptake in the absence or presence of insulin, while hypoxia-dependent glucose import was not affected, showing specificity of the ECS to modulate the insulin-mediated glucose uptake pathway [80]. Conversely, AEA treatment of soleus muscle from lean rats resulted in marked decreases in both basal and insulin-induced glucose uptake [80]. The authors however, were unable to detect any differences in the phosphorylation/activation levels of several components of the insulin signalling pathway in response to either CB1 stimulation or antagonism. Utilizing rat myotubes in vitro, Esposito et al. showed that direct CB1 inhibition with Rimonabant at low concentrations, or Cnr1 knock-down, increased glucose uptake without affecting the expression levels of the glucose transporters GLUT1 or GLUT4 [72]. Rimonabant treatment resulted in protein synthesis-independent induction of both the catalytic and regulatory subunits of PI3K, which was dependent on increased cAMP-dependent PKB/Akt activity, as blockade of either PI3K or PKA activity inhibited the Rimonabant-mediated increase in glucose uptake. Interestingly, high Rimonabant concentrations decreased glucose uptake, which may be due either to Rimonabant acting as a CB1 partial agonist on L6 myotubes or to the fact that high concentrations of this compound induced CB1 expression. On the other hand, CB2 blockade had no effect on glucose uptake [72]. However, Cnr2−/− (CB2 knockout) mice have recently been demonstrated not to develop DIO insulin resistance and to have increased insulin-mediated glucose uptake, indicating that CB2 also plays an important role in skeletal muscle glucose metabolism [18]. In another in vitro study, chronic (24 h), but not acute (30 min), Rimonabant treatment of L6 rat myotubes sensitized both PKB/Akt and extracellular signal-regulated kinases (ERK1/2 aka MAPK3/1) to insulin-dependent activation resulting in increased phosphorylation of the PKB/Akt targets forkhead box O3A (Foxo3a) and glycogen synthase kinase 3 alpha/beta (GSK3α/β). Rimonabant, however, did not affect basal phosphorylation levels of these proteins [75]. Increased PKB/Akt phosphorylation was determined to be not the result of enhanced kinase activity but rather of a delay in PKB/Akt dephosphorylation, presumably due to the inhibition of a phosphatase [75]. CB1 stimulation of L6 myotubes in the same study, whilst inhibiting insulin-dependend activation of the MEK1/2-ERK1/2-CREB pathway, had no affect on PKB/Akt activation. However, in a study by Eckardt et al., utilizing primary human skeletal muscle cells, AEA at high concentrations (10 μM) was able to induce ERK1/2, p38 and insulin receptor substrate 1 (IRS1[S307]) phosphorylation (which is inhibitory to insulin signalling), as well as inhibit insulin-dependent PKB/Akt phosphorylation and glucose uptake [73, 82]. Surprisingly, however, AEA was also able to induce basal glucose uptake on its own, perhaps due to AMPK activation, though this phenomenon was left unstudied, thus underscoring the void that still exists in our understanding of the role the ECS plays in modulating glucose uptake in muscle cells [73]. Perhaps the seminal finding in the work by Eckardt et al. was that conditioned media from adipocytes in culture inhibited insulin-dependent Akt phosphorylation and glucose uptake in skeletal muscle cells in a CB1-dependent manner [73]. This finding is supportive of the idea that skeletal muscle insulin resistance is at least in part due to adipocyte-derived factors, including endocannabinoids. However, clearly more detailed analysis of the effects of the ECS on skeletal muscle energy metabolism and glucose transport is required. For example, it is still not understood exactly how CB1 signalling changes glucose transport into skeletal muscle cells, and it is noticeable that none of the studies mentioned above found any changes in the expression of the glucose transporters GLUT1 or 4. Given the noted effects that the ECS has on posttranslational regulation of proteins in the insulin signalling pathway, it is possible that CB1 alters GLUT activity and/or translocation to the cell membrane, although possibly in a way opposite to that observed in the adipose tissue [10]. It is also possible that, in view of the thrifty phenotype that has been suggested to be associated with the ECS [83], CB1 activation, although capable of inhibiting insulin signalling, might be even more important at reducing glucose uptake following physical exercise, which is controlled by AMPK rather than insulin. In this sense, it would be important to investigate the function and dysregulation of endocannabinoids and CB1 receptors during or following acute and prolonged physical exercise, which might also provide some insights into the possible role of the ECS in skeletal muscle regeneration.
1.4 Conclusions
The ECS appears to be involved in most aspects of energy homeostasis. Its important functions in stimulating maximal food intake following a period of food deprivation, through both homeostatic and hedonic pathways, and in controlling nutrient processing, have been covered in other reviews [84, 85] and were not discussed here. Together with the studies mentioned in our article, they suggest for this system of local mediators, which is normally under the control of both systemic and local signals, a general strategy aimed at maximizing nutrient intake and absorption, optimizing energy storage and reducing energy expenditure. This is likely an evolutionary adaptation in mammals as a response to periods of food deprivation, ensuring that they made the most of their occasional “meal”. In the present time of food abundance, and of little effort to procure it (at least for most of the western population), the ECS, however, seems to become easily deranged, thus possibly contributing to the “diabesity” epidemic. Early efforts to counteract, through pharmacological treatment with CB1 receptor antagonists like Rimonabant, endocannabinoid tone over-activity in obesity were successful in clinical trials, but not devoid of worrisome, when not serious, central averse events when translated into daily clinical practice, and therefore had to be interrupted [86]. Nevertheless, other strategies to curb endocannabinoid tone in obesity and type 2 diabetes are becoming available. First of all, exercise and caloric restriction, that is the oldest and, unfortunately, still very difficult to comply with, strategy to fight obesity, seems to be accompanied by a strong reduction in endocannabinoid levels [38]. However, other possibly “easier” dietary approaches are emerging, such as the use of certain sources of dietary omega-3 fatty acids, which, at least in animals, reduce insulin resistance, hepatosteatosis and systemic inflammation partly by inhibiting the biosynthesis of endocannabinoids via reduction of their ultimate biosynthetic precursors [87]. Finally, a strong effort, based also on the recent reports reviewed in this article, is being made to develop peripherally restricted (i.e. relatively blood–brain-barrier impermeable) CB1 receptor antagonists/inverse agonists. These compounds seem to be devoid of the unwanted central side effects of the first generation of compounds, but are still capable of strongly reducing dyslipidemia, liver fat accumulation and insulin resistance, which are partly independent of body weight loss, at least in animal models of obesity [55, 88, 89]. These compounds now await the challenge of appropriate clinical trials, which, however, rather than being performed on globally obese, and otherwise healthy, individuals, should focus mainly on abdominally obese patients with metabolic co-morbidities [86]. Such studies will tell us if these strategies will also work in the clinic, thus at last efficaciously and safely translating into new treatments what so far has been a very exciting, challenging, and as yet not fully clarified item for basic research on metabolism.
References
Di Marzo V. Endocannabinoids: synthesis and degradation. Rev Physiol Biochem Pharmacol. 2008;160:1–24.
Frayn KN, Karpe F, Fielding BA, Macdonald IA, Coppack SW. Integrative physiology of human adipose tissue. Int J Obes Relat Metab Disord. 2003;27(8):875–88.
Klaus S. Adipose tissue as a regulator of energy balance. Curr Drug Targets. 2004;5(3):241–50.
Starowicz KM, Cristino L, Matias I, Capasso R, Racioppi A, Izzo AA, et al. Endocannabinoid dysregulation in the pancreas and adipose tissue of mice fed with a high-fat diet. Obesity (Silver Spring). 2008;16(3):553–65.
Matias I, Gonthier MP, Orlando P, Martiadis V, De Petrocellis L, Cervino C, et al. Regulation, function, and dysregulation of endocannabinoids in models of adipose and beta-pancreatic cells and in obesity and hyperglycemia. J Clin Endocrinol Metab. 2006;91(8):3171–80.
Cota D, Marsicano G, Tschop M, Grubler Y, Flachskamm C, Schubert M, et al. The endogenous cannabinoid system affects energy balance via central orexigenic drive and peripheral lipogenesis. J Clin Invest. 2003;112(3):423–31.
Di Marzo V, Goparaju SK, Wang L, Liu J, Batkai S, Jarai Z, et al. Leptin-regulated endocannabinoids are involved in maintaining food intake. Nature. 2001;410(6830):822–5.
Di Marzo V, Matias I. Endocannabinoid control of food intake and energy balance. Nat Neurosci. 2005;8(5):585–9.
Soria-Gomez E, Matias I, Rueda-Orozco PE, Cisneros M, Petrosino S, Navarro L, et al. Pharmacological enhancement of the endocannabinoid system in the nucleus accumbens shell stimulates food intake and increases c-Fos expression in the hypothalamus. Br J Pharmacol. 2007;151(7):1109–16.
Pagano C, Pilon C, Calcagno A, Urbanet R, Rossato M, Milan G, et al. The endogenous cannabinoid system stimulates glucose uptake in human fat cells via phosphatidylinositol 3-kinase and calcium-dependent mechanisms. J Clin Endocrinol Metab. 2007;92(12):4810–9.
Osei-Hyiaman D, DePetrillo M, Pacher P, Liu J, Radaeva S, Batkai S, et al. Endocannabinoid activation at hepatic CB1 receptors stimulates fatty acid synthesis and contributes to diet-induced obesity. J Clin Invest. 2005;115(5):1298–305.
Bluher M, Michael MD, Peroni OD, Ueki K, Carter N, Kahn BB, et al. Adipose tissue selective insulin receptor knockout protects against obesity and obesity-related glucose intolerance. Dev Cell. 2002;3(1):25–38.
Gonthier MP, Hoareau L, Festy F, Matias I, Valenti M, Bes-Houtmann S, et al. Identification of endocannabinoids and related compounds in human fat cells. Obesity (Silver Spring). 2007;15(4):837–45.
Bluher M, Engeli S, Kloting N, Berndt J, Fasshauer M, Batkai S, et al. Dysregulation of the peripheral and adipose tissue endocannabinoid system in human abdominal obesity. Diabetes. 2006;55(11):3053–60.
Bensaid M, Gary-Bobo M, Esclangon A, Maffrand JP, Le Fur G, Oury-Donat F, et al. The cannabinoid CB1 receptor antagonist SR141716 increases Acrp30 mRNA expression in adipose tissue of obese fa/fa rats and in cultured adipocyte cells. Mol Pharmacol. 2003;63(4):908–14.
Roche R, Hoareau L, Bes-Houtmann S, Gonthier MP, Laborde C, Baron JF, et al. Presence of the cannabinoid receptors, CB1 and CB2, in human omental and subcutaneous adipocytes. Histochem Cell Biol. 2006;126(2):177–87.
Spoto B, Fezza F, Parlongo G, Battista N, Sgro E, Gasperi V, et al. Human adipose tissue binds and metabolizes the endocannabinoids anandamide and 2-arachidonoylglycerol. Biochimie. 2006;88(12):1889–97.
Agudo J, Martin M, Roca C, Molas M, Bura AS, Zimmer A, et al. Deficiency of CB2 cannabinoid receptor in mice improves insulin sensitivity but increases food intake and obesity with age. Diabetologia. 2010;53(12):2629–40.
Perwitz N, Fasshauer M, Klein J. Cannabinoid receptor signaling directly inhibits thermogenesis and alters expression of adiponectin and visfatin. Horm Metab Res. 2006;38(5):356–8.
Gary-Bobo M, Elachouri G, Scatton B, Le Fur G, Oury-Donat F, Bensaid M. The cannabinoid CB1 receptor antagonist rimonabant (SR141716) inhibits cell proliferation and increases markers of adipocyte maturation in cultured mouse 3T3 F442A preadipocytes. Mol Pharmacol. 2006;69(2):471–8.
Lage R, Dieguez C, Vidal-Puig A, Lopez M. AMPK: a metabolic gauge regulating whole-body energy homeostasis. Trends Mol Med. 2008;14(12):539–49.
Viollet B, Guigas B, Leclerc J, Hebrard S, Lantier L, Mounier R, et al. AMP-activated protein kinase in the regulation of hepatic energy metabolism: from physiology to therapeutic perspectives. Acta Physiol (Oxf). 2009;196(1):81–98.
Tedesco L, Valerio A, Cervino C, Cardile A, Pagano C, Vettor R, et al. Cannabinoid type 1 receptor blockade promotes mitochondrial biogenesis through endothelial nitric oxide synthase expression in white adipocytes. Diabetes. 2008;57(8):2028–36.
Tedesco L, Valerio A, Dossena M, Cardile A, Ragni M, Pagano C, et al. Cannabinoid receptor stimulation impairs mitochondrial biogenesis in mouse white adipose tissue, muscle, and liver: the role of eNOS, p38 MAPK, and AMPK pathways. Diabetes. 2010;59(11):2826–36.
Nogueiras R, Veyrat-Durebex C, Suchanek PM, Klein M, Tschop J, Caldwell C, et al. Peripheral, but not central, CB1 antagonism provides food intake-independent metabolic benefits in diet-induced obese rats. Diabetes. 2008;57(11):2977–91.
Jbilo O, Ravinet-Trillou C, Arnone M, Buisson I, Bribes E, Peleraux A, et al. The CB1 receptor antagonist rimonabant reverses the diet-induced obesity phenotype through the regulation of lipolysis and energy balance. FASEB J. 2005;19(11):1567–9.
Mancini G, Quarta C, Srivastava RK, Klaus S, Pagotto U, Lutz B, editors. Adipocyte-specific cb1 conditional knock-out mice: new insights in the study of obesity and metabolic syndrome. 20th Annual Symposium of the International Cannabinoid Research Society; 2010 July 23–27; Lund, Sweden.
Matias I, Di Marzo V. Endocannabinoid synthesis and degradation, and their regulation in the framework of energy balance. J Endocrinol Investig. 2006;29(3 Suppl):15–26.
Di Marzo V, Verrijken A, Hakkarainen A, Petrosino S, Mertens I, Lundbom N, et al. Role of insulin as a negative regulator of plasma endocannabinoid levels in obese and nonobese subjects. Eur J Endocrinol. 2009;161(5):715–22.
Murdolo G, Kempf K, Hammarstedt A, Herder C, Smith U, Jansson PA. Insulin differentially modulates the peripheral endocannabinoid system in human subcutaneous abdominal adipose tissue from lean and obese individuals. J Endocrinol Investig. 2007;30(8):RC17–21.
Yan ZC, Liu DY, Zhang LL, Shen CY, Ma QL, Cao TB, et al. Exercise reduces adipose tissue via cannabinoid receptor type 1 which is regulated by peroxisome proliferator-activated receptor-delta. Biochem Biophys Res Commun. 2007;354(2):427–33.
Sarzani R, Bordicchia M, Marcucci P, Bedetta S, Santini S, Giovagnoli A, et al. Altered pattern of cannabinoid type 1 receptor expression in adipose tissue of dysmetabolic and overweight patients. Metabolism. 2009;58(3):361–7.
Bennetzen MF, Nielsen TS, Paulsen SK, Bendix J, Fisker S, Jessen N, et al. Reduced cannabinoid receptor 1 protein in subcutaneous adipose tissue of obese. Eur J Clin Investig. 2010;40(2):121–6.
Despres JP, Lemieux I. Abdominal obesity and metabolic syndrome. Nature. 2006;444(7121):881–7.
Cote M, Matias I, Lemieux I, Petrosino S, Almeras N, Despres JP, et al. Circulating endocannabinoid levels, abdominal adiposity and related cardiometabolic risk factors in obese men. Int J Obes (Lond). 2007;31(4):692–9.
Engeli S, Bohnke J, Feldpausch M, Gorzelniak K, Janke J, Batkai S, et al. Activation of the peripheral endocannabinoid system in human obesity. Diabetes. 2005;54(10):2838–43.
Izzo AA, Piscitelli F, Capasso R, Aviello G, Romano B, Borrelli F, et al. Peripheral endocannabinoid dysregulation in obesity: relation to intestinal motility and energy processing induced by food deprivation and re-feeding. Br J Pharmacol. 2009;158(2):451–61.
Di Marzo V, Cote M, Matias I, Lemieux I, Arsenault BJ, Cartier A, et al. Changes in plasma endocannabinoid levels in viscerally obese men following a 1 year lifestyle modification programme and waist circumference reduction: associations with changes in metabolic risk factors. Diabetologia. 2009;52(2):213–7.
Annuzzi G, Piscitelli F, Di Marino L, Patti L, Giacco R, Costabile G, et al. Differential alterations of the concentrations of endocannabinoids and related lipids in the subcutaneous adipose tissue of obese diabetic patients. Lipids Health Dis. 2010;9:43.
Cannon B, Nedergaard J. Brown adipose tissue: function and physiological significance. Physiol Rev. 2004;84(1):277–359.
Cioffi F, Senese R, de Lange P, Goglia F, Lanni A, Lombardi A. Uncoupling proteins: a complex journey to function discovery. Biofactors. 2009;35(5):417–28.
Cinti S. Adipocyte differentiation and transdifferentiation: plasticity of the adipose organ. J Endocrinol Investig. 2002;25(10):823–35.
Perwitz N, Wenzel J, Wagner I, Buning J, Drenckhan M, Zarse K, et al. Cannabinoid type 1 receptor blockade induces transdifferentiation towards a brown fat phenotype in white adipocytes. Diabetes Obes Metab. 2010;12(2):158–66.
Bellocchio L, Cervino C, Vicennati V, Pasquali R, Pagotto U. Cannabinoid type 1 receptor: another arrow in the adipocytes’ bow. J Neuroendocrinol. 2008;20 Suppl 1:130–8.
Verty AN, Allen AM, Oldfield BJ. The effects of rimonabant on brown adipose tissue in rat: implications for energy expenditure. Obesity (Silver Spring). 2009;17(2):254–61.
Quarta C, Bellocchio L, Mancini G, Mazza R, Cervino C, Braulke LJ, et al. CB(1) signaling in forebrain and sympathetic neurons is a key determinant of endocannabinoid actions on energy balance. Cell Metab. 2010;11(4):273–85.
Mitra V, Metcalf J. Metabolic functions of the liver. Anaesth Intensive Care Med. 2009;10(7):334–5.
Parekh S, Anania FA. Abnormal lipid and glucose metabolism in obesity: implications for nonalcoholic fatty liver disease. Gastroenterology. 2007;132(6):2191–207.
Ratziu V, Poynard T. Assessing the outcome of nonalcoholic steatohepatitis? It’s time to get serious. Hepatology. 2006;44(4):802–5.
Postic C, Girard J. The role of the lipogenic pathway in the development of hepatic steatosis. Diabetes Metab. 2008;34(6 Pt 2):643–8.
Pacher P, Batkai S, Kunos G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol Rev. 2006;58(3):389–462.
Vettor R, Pagano C. The role of the endocannabinoid system in lipogenesis and fatty acid metabolism. Best Pract Res Clin Endocrinol Metab. 2009;23(1):51–63.
Gary-Bobo M, Elachouri G, Gallas JF, Janiak P, Marini P, Ravinet-Trillou C, et al. Rimonabant reduces obesity-associated hepatic steatosis and features of metabolic syndrome in obese Zucker fa/fa rats. Hepatology. 2007;46(1):122–9.
Jourdan T, Djaouti L, Demizieux L, Gresti J, Verges B, Degrace P. CB1 antagonism exerts specific molecular effects on visceral and subcutaneous fat and reverses liver steatosis in diet-induced obese mice. Diabetes. 2010;59(4):926–34.
Tam J, Vemuri VK, Liu J, Batkai S, Mukhopadhyay B, Godlewski G, et al. Peripheral CB1 cannabinoid receptor blockade improves cardiometabolic risk in mouse models of obesity. J Clin Invest. 2010;120(8):2953–66.
Osei-Hyiaman D, Liu J, Zhou L, Godlewski G, Harvey-White J, Jeong WI, et al. Hepatic CB1 receptor is required for development of diet-induced steatosis, dyslipidemia, and insulin and leptin resistance in mice. J Clin Invest. 2008;118(9):3160–9.
Jeong WI, Osei-Hyiaman D, Park O, Liu J, Batkai S, Mukhopadhyay P, et al. Paracrine activation of hepatic CB1 receptors by stellate cell-derived endocannabinoids mediates alcoholic fatty liver. Cell Metab. 2008;7(3):227–35.
Mukhopadhyay B, Liu J, Osei-Hyiaman D, Godlewski G, Mukhopadhyay P, Wang L, et al. Transcriptional regulation of cannabinoid receptor-1 expression in the liver by retinoic acid acting via retinoic acid receptor-gamma. J Biol Chem. 2010;285(25):19002–11.
Guzman M, Fernandez-Ruiz JJ, Sanchez C, Velasco G, Ramos JA. Effects of anandamide on hepatic fatty acid metabolism. Biochem Pharmacol. 1995;50(6):885–8.
Siegmund SV, Seki E, Osawa Y, Uchinami H, Cravatt BF, Schwabe RF. Fatty acid amide hydrolase determines anandamide-induced cell death in the liver. J Biol Chem. 2006;281(15):10431–8.
Siegmund SV, Qian T, de Minicis S, Harvey-White J, Kunos G, Vinod KY, et al. The endocannabinoid 2-arachidonoyl glycerol induces death of hepatic stellate cells via mitochondrial reactive oxygen species. FASEB J. 2007;21(11):2798–806.
Trauner M, Arrese M, Wagner M. Fatty liver and lipotoxicity. Biochim Biophys Acta. 2010;1801(3):299–310.
Kola B, Hubina E, Tucci SA, Kirkham TC, Garcia EA, Mitchell SE, et al. Cannabinoids and ghrelin have both central and peripheral metabolic and cardiac effects via AMP-activated protein kinase. J Biol Chem. 2005;280(26):25196–201.
Flamment M, Gueguen N, Wetterwald C, Simard G, Malthiery Y, Ducluzeau PH. Effects of the cannabinoid CB1 antagonist, rimonabant, on hepatic mitochondrial function in rats fed a high fat diet. Am J Physiol Endocrinol Metab. 2009.
Yamauchi T, Kamon J, Minokoshi Y, Ito Y, Waki H, Uchida S, et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med. 2002;8(11):1288–95.
Mendez-Sanchez N, Zamora-Valdes D, Pichardo-Bahena R, Barredo-Prieto B, Ponciano-Rodriguez G, Bermejo-Martinez L, et al. Endocannabinoid receptor CB2 in nonalcoholic fatty liver disease. Liver Int. 2007;27(2):215–9.
Deveaux V, Cadoudal T, Ichigotani Y, Teixeira-Clerc F, Louvet A, Manin S, et al. Cannabinoid CB2 receptor potentiates obesity-associated inflammation, insulin resistance and hepatic steatosis. PLoS ONE. 2009;4(6):e5844.
De Gottardi A, Spahr L, Ravier-Dall’Antonia F, Hadengue A. Cannabinoid receptor 1 and 2 agonists increase lipid accumulation in hepatocytes. Liver Int. 2010;30(10):1482–9.
Abdul-Ghani MA, DeFronzo RA. Pathogenesis of insulin resistance in skeletal muscle. J Biomed Biotechnol. 2010;2010:476279.
Cavuoto P, McAinch AJ, Hatzinikolas G, Cameron-Smith D, Wittert GA. Effects of cannabinoid receptors on skeletal muscle oxidative pathways. Mol Cell Endocrinol. 2007;267(1–2):63–9.
Cavuoto P, McAinch AJ, Hatzinikolas G, Janovska A, Game P, Wittert GA. The expression of receptors for endocannabinoids in human and rodent skeletal muscle. Biochem Biophys Res Commun. 2007;364(1):105–10.
Esposito I, Proto MC, Gazzerro P, Laezza C, Miele C, Alberobello AT, et al. The cannabinoid CB1 receptor antagonist rimonabant stimulates 2-deoxyglucose uptake in skeletal muscle cells by regulating the expression of phosphatidylinositol-3-kinase. Mol Pharmacol. 2008;74(6):1678–86.
Eckardt K, Sell H, Taube A, Koenen M, Platzbecker B, Cramer A, et al. Cannabinoid type 1 receptors in human skeletal muscle cells participate in the negative crosstalk between fat and muscle. Diabetologia. 2009;52(4):664–74.
Brighton PJ, McDonald J, Taylor AH, Challiss RA, Lambert DG, Konje JC, et al. Characterization of anandamide-stimulated cannabinoid receptor signaling in human ULTR myometrial smooth muscle cells. Mol Endocrinol. 2009;23(9):1415–27.
Lipina C, Stretton C, Hastings S, Hundal JS, Mackie K, Irving AJ, et al. Regulation of MAP kinase-directed mitogenic and protein kinase B-mediated signaling by cannabinoid receptor type 1 in skeletal muscle cells. Diabetes. 2010;59(2):375–85.
Mukhopadhyay P, Rajesh M, Batkai S, Patel V, Kashiwaya Y, Liaudet L, et al. CB1 cannabinoid receptors promote oxidative stress and cell death in murine models of doxorubicin-induced cardiomyopathy and in human cardiomyocytes. Cardiovasc Res. 2010;85(4):773–84.
Crespillo A, Suarez J, Bermudez-Silva FJ, Rivera P, Vida M, Alonso M, et al. Expression of cannabinoid system in muscle: effects of high fat diet and CB1 receptor blockade. Biochem J. 2010.
Pagotto U, Marsicano G, Cota D, Lutz B, Pasquali R. The emerging role of the endocannabinoid system in endocrine regulation and energy balance. Endocr Rev. 2006;27(1):73–100.
Matias I, Petrosino S, Racioppi A, Capasso R, Izzo AA, Di Marzo V. Dysregulation of peripheral endocannabinoid levels in hyperglycemia and obesity: effect of high fat diets. Mol Cell Endocrinol. 2008;286(1–2 Suppl 1):S66–78.
Lindborg KA, Teachey MK, Jacob S, Henriksen EJ. Effects of in vitro antagonism of endocannabinoid-1 receptors on the glucose transport system in normal and insulin-resistant rat skeletal muscle. Diabetes Obes Metab. 2010;12(8):722–30.
Liu YL, Connoley IP, Wilson CA, Stock MJ. Effects of the cannabinoid CB1 receptor antagonist SR141716 on oxygen consumption and soleus muscle glucose uptake in Lep(ob)/Lep(ob) mice. Int J Obes (Lond). 2005;29(2):183–7.
Aguirre V, Werner ED, Giraud J, Lee YH, Shoelson SE, White MF. Phosphorylation of Ser307 in insulin receptor substrate-1 blocks interactions with the insulin receptor and inhibits insulin action. J Biol Chem. 2002;277(2):1531–7.
Bellocchio L, Cervino C, Pasquali R, Pagotto U. The endocannabinoid system and energy metabolism. J Neuroendocrinol. 2008;20(6):850–7.
Di Marzo V, Ligresti A, Cristino L. The endocannabinoid system as a link between homoeostatic and hedonic pathways involved in energy balance regulation. Int J Obes (Lond). 2009;33 Suppl 2:S18–24.
Izzo AA, Sharkey KA. Cannabinoids and the gut: new developments and emerging concepts. Pharmacol Ther. 2010;126(1):21–38.
Di Marzo V, Despres JP. CB1 antagonists for obesity—what lessons have we learned from rimonabant? Nat Rev Endocrinol. 2009;5(11):633–8.
Batetta B, Griinari M, Carta G, Murru E, Ligresti A, Cordeddu L, et al. Endocannabinoids may mediate the ability of (n-3) fatty acids to reduce ectopic fat and inflammatory mediators in obese Zucker rats. J Nutr. 2009;139(8):1495–501.
Receveur JM, Murray A, Linget JM, Norregaard PK, Cooper M, Bjurling E, et al. Conversion of 4-cyanomethyl-pyrazole-3-carboxamides into CB1 antagonists with lowered propensity to pass the blood–brain-barrier. Bioorg Med Chem Lett. 2010;20(2):453–7.
Son MH, Kim HD, Chae YN, Kim MK, Shin CY, Ahn GJ, et al. Peripherally acting CB1-receptor antagonist: the relative importance of central and peripheral CB1 receptors in adiposity control. Int J Obes (Lond). 2010;34(3):547–56.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Silvestri, C., Ligresti, A. & Di Marzo, V. Peripheral effects of the endocannabinoid system in energy homeostasis: Adipose tissue, liver and skeletal muscle. Rev Endocr Metab Disord 12, 153–162 (2011). https://doi.org/10.1007/s11154-011-9167-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11154-011-9167-3