
Abstract
Light-harvesting complex II (LHCII) is the major antenna complex in higher plants and green algae. It has been suggested that a major part of the excited state energy dissipation in the so-called “non-photochemical quenching” (NPQ) is located in this antenna complex. We have performed an ultrafast kinetics study of the low-energy fluorescent states related to quenching in LHCII in both aggregated and the crystalline form. In both sample types the chlorophyll (Chl) excited states of LHCII are strongly quenched in a similar fashion. Quenching is accompanied by the appearance of new far-red (FR) fluorescence bands from energetically low-lying Chl excited states. The kinetics of quenching, its temperature dependence down to 4 K, and the properties of the FR-emitting states are very similar both in LHCII aggregates and in the crystal. No such FR-emitting states are found in unquenched trimeric LHCII. We conclude that these states represent weakly emitting Chl–Chl charge-transfer (CT) states, whose formation is part of the quenching process. Quantum chemical calculations of the lowest energy exciton and CT states, explicitly including the coupling to the specific protein environment, provide detailed insight into the chemical nature of the CT states and the mechanism of CT quenching. The experimental data combined with the results of the calculations strongly suggest that the quenching mechanism consists of a sequence of two proton-coupled electron transfer steps involving the three quenching center Chls 610/611/612. The FR-emitting CT states are reaction intermediates in this sequence. The polarity-controlled internal reprotonation of the E175/K179 aa pair is suggested as the switch controlling quenching. A unified model is proposed that is able to explain all known conditions of quenching or non-quenching of LHCII, depending on the environment without invoking any major conformational changes of the protein.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
LHCII, the light-harvesting antenna complex of photosystem (PS) II, in higher plants and green algae is the most abundant membrane protein complex in nature, accounting for about 80% of the light absorbed by PSII. The LHCIIb monomer contains eight chlorophyll (Chl) a, six Chl b, two luteins (Lut), one neoxanthin (Nx), and one violaxanthin (Vx) chromophore as cofactors and the X-ray structure of the native trimers has been determined to high resolution in two different crystal forms (Liu et al. 2004; Standfuss et al. 2005). The trimeric LHCII and the monomeric (minor) LHCII complexes (CP24, CP26, and CP29) together with the PSII core form the PSII supercomplex (Yakushevska et al. 2003; van Amerongen and Croce 2013) whose structure has been determined recently to high resolution (Wei et al. 2016; Su et al. 2017).
All photosynthetic organisms have to cope with largely varying light intensities changing over several orders of magnitude in a diurnal cycle but even within much shorter times (e.g., within seconds in sunflecks). These widely and quickly varying light intensities provide a pronounced challenge to the organisms since the high intensities could easily exceed their maximal photosynthetic capacity. In the absence of effective and rapid regulation and photoprotection mechanisms, highly destructive states such as Chl triplets, singlet oxygen, and other reactive oxygen species would develop (Matsubara et al. 2012; Demmig-Adams et al. 2012; Jahns and Holzwarth 2012; Ruban 2015). One of the most effective regulation mechanisms in photosynthetic organisms that prevents photo-destruction is the so-called “non-photochemical quenching” (NPQ) which converts Chl excited states in the antenna efficiently into thermal energy. While overall NPQ in plants and green algae consists of several different components and occurs in different antenna complexes, it is believed that the major part of the NPQ, the rapidly forming and rapidly relaxing so-called qE-quenching, is located in the LHCII complex (Horton et al. 1996; Holzwarth et al. 2009; Miloslavina et al. 2011; Ruban 2019; Nicol et al. 2019). In vivo it requires the presence of a small protein, PsbS, which is activated by a ΔpH across the membrane (Li et al. 2009). The molecular mechanism of NPQ in LHCII is, however, a matter of intense debate (Jahns and Holzwarth 2012; Holzwarth and Jahns 2014; Ruban 2016, 2019; Bennett et al. 2019).
The LHCII aggregation model as an explanation for NPQ in vivo—as proposed by Horton and coworkers (Horton et al. 1991; Ruban et al. 1991)—has found support in a wide range of experiments (reviewed in (Horton et al. 2005; Holzwarth and Jahns 2014)). While NPQ in general is a property of the intact system, it is of particular interest for the present work that the excited state quenching of LHCII can be studied—as a model system—in vitro, which allows for the application of a wider range of spectroscopic techniques than are generally applicable to intact chloroplasts or leaves. As a membrane protein in the isolated form LHCII is usually surrounded by a detergent micelle, and in this parent solubilized form the excited states are not quenched efficiently. Aggregation of the isolated LHCII complex in vitro, effected in various ways by removing the detergent, causes strong Chl excited state quenching that is accompanied—in particular at low temperatures—by a pronounced red-shift of the fluorescence (Ruban and Horton 1992; Mullineaux et al. 1993; Pieper et al. 1999a, b; Ostroumov et al. 2007; Miloslavina et al. 2008; Ilioaia et al. 2008; Tutkus et al. 2018; Crisafi et al. 2018), a Raman-detected modification in the conformation of Neoxanthin (Nx) (Ruban and Horton et al. 1995; Robert et al. 2004; Pascal et al. 2005), the carotenoid (Car) that is the most peripheral to the LHCII trimer. Nx is actually protruding from the LHCII complex with one end (Liu et al. 2004; Standfuss et al. 2005; Wei et al. 2016; Su et al. 2017). Interestingly the strong fluorescence red-shift and the specific changes in the Nx conformation have also been observed in LHCII crystals whose Chl excited states have been shown to be also strongly quenched (Pascal et al. 2005). More recently a PsbS-dependent red-shift of the fluorescence maximum and a strongly enhanced FR fluorescence intensity, as compared to isolated trimeric LHCII, have been found at r.t.—and even more pronounced at low temperatures (Lambrev et al. 2010)—in a fluorescence component appearing in intact leaves of Arabidopsis upon induction of NPQ (Holzwarth et al. 2009; Miloslavina et al. 2011). Also the fluorescence lifetimes of the major FR fluorescence component (ca. 200–400 ps in w.t. Arabidopsis) were close to the ones found in the aggregated quenched LHCII complex in vitro (Miloslavina et al. 2008; Holzwarth et al. 2009; Müller et al. 2010; Miloslavina et al. 2011). These remarkable similarities in the spectroscopic properties of quenched LHCII in vitro and in vivo further strengthened the view that qE-quenching may indeed be related to a functional detachment and structural modification of LHCII similar to the aggregation processes in vitro (Holzwarth et al. 2009; Betterle et al. 2009; Lambrev et al. 2010; Johnson et al. 2011), possibly accompanied by some controlled structural change within LHCII (Ruban et al. 2007; Liguori et al. 2015).
So far four alternative photophysical/photochemical mechanisms have been discussed for the quenching of Chl excited states in Chl-protein antenna complexes, three of them involving modified interactions with Cars: (1) Energy transfer from a Chl excited state to the S1 state of a nearby Car, based on the “gear-shift-mechanism” (Frank et al. 1994). The quenching mechanism proposed by Ruban et al. (2007) is a special case of this energy transfer to Car, however, without implying the Car gear-shift but rather a protein conformational change affecting the Chl–Car interaction with Lut. (2) A “carotenoid-induced internal conversion” (CIC) of a Chl excited state by increased coupling to the S1 state of a nearby carotenoid. This mechanism has been proposed initially as an entirely theoretical model (Naqvi 1998; van Amerongen and van Grondelle 2001) but was later invoked for the explanation of some experimental observations (Bode et al. 2008, 2009; Liao et al. 2010; Ilioaia et al. 2011; Chmeliov et al. 2016; Mascoli et al. 2019) and theoretical calculations (Chmeliov et al. 2015). This mechanism differs, however, only slightly from the one originally proposed by Ruban et al. (2007). (3) Chl–Chl exciton coupling enabling the rapid formation of a fluorescent Chl–Chl charge-transfer (CT) state as an intermediate in the excited state quenching (Miloslavina et al. 2008; Müller et al. 2010; Wahadoszamen et al. 2012; Kell et al. 2014; Yang et al. 2014). 4) The formation of a Chl–Car CT state by electron transfer from a Car to a nearby Chl (Dreuw et al. 2003; Holt et al. 2005; Wormit et al. 2009; Dall'Osto et al. 2017; Leuenberger et al. 2017). Experimentally the latter mechanism has been proposed only for quenching in the monomeric minor LHCII antenna complexes (Avenson et al. 2008) and there seems to exist agreement in the literature that it can be excluded as the quenching mechanism for the major LHCIIb aggregates since no associated Chl anion/Car cation states were observed (Ruban et al. 2007; Avenson et al. 2008; Müller et al. 2010). Nevertheless it has been suggested as the quenching mechanism in a recent theoretical study (Cupellini et al. 2020). Double pulse experiments suggested that the Car cation formation was a two-photon process (Amarie et al. 2008, 2009). This is supported by recent experiments showing that it could indeed be an artifact of using high-intensity pulses (van Oort et al. 2018).
In this contribution reporting on time-resolved fluorescence experiments as a function of temperature, we focus on a detailed spectroscopic and kinetic characterization of the low-energy FR fluorescent states appearing in quenched aggregated LHCIIb in vitro and on the comparison of their properties to the analogous FR-emitting states showing up also in crystalline LHCII. Comparison to analogous studies in the crystal form is particularly important since this allows to exclude or confirm pronounced interactions between neighboring LHCII trimers as an explanation for the quenching and/or the source of the FR emissions. The kinetics of the quenching has been studied before at low temperature (Mullineaux et al. 1993) and at r.t. (Miloslavina et al. 2008; Müller et al. 2010; Magdaong et al. 2013) in aggregates, in LHCII crystals (Pascal et al. 2005), and also for qE-quenching in vivo (Holzwarth et al. 2009). Study of the pronounced temperature dependence of the kinetics reported before also by Chmeliov et al. (2016) provides, however, novel insights. We are comparing here the temperature-dependent kinetics—measured over a wide temperature range and with very high signal-to-noise ratio—of both LHCII aggregates and crystals with the results of quantum chemical calculations explicitly taking into account the Chl exciton states, the Chl–Chl CT states, and also including their protein surrounding. This combination allows us to propose the detailed mechanism of protein control of quenching on the basis of a Chl–Chl CT state mechanism, in full agreement with previous transient absorption and fluorescence experiments (Miloslavina et al. 2008; Holzwarth et al. 2009; Müller et al. 2010).
Methods
Isolation of LHCII from spinach and preparation of aggregated LHCII
BBY particles were prepared according to the method of van Leeuwen et al. (van Leeuwen et al. 1991) with minor modifications. In the final step the pellet was resuspended in 20 mM Tricine, pH 7.5, 5 mM Na-EDTA and diluted to a final concentration of 0.7 mg Chltot/ml. The BBY suspension was quickly solubilized by mixing with n-dodecyl-β-D-maltoside (β-DM) to 0.7% for 10 min. Unsolubilized material was centrifuged out at 10 000 × g for 15 min. The suspension was laid on a linear sucrose density gradient (0.1 M to 0.6 M sucrose) with 5 mM Tricine (pH 7.5) and 0.06% β-DM and centrifuged at 100 000 × g for 16 h at 4 °C. The LHCII trimer fraction was collected from the middle band of the gradient, concentrated by centrifugation in filter tubes (Amicon Ultra 15 centrifugal filter units, nominal molecular weight limit 30 kDa; Merck Millipore) and frozen for storage. LHCII aggregates were prepared by incubating the trimers at a concentration of 0.2–0.3 mg/ml Chl with 0.3–0.4 g/ml Bio-Beads SM-2 (Bio-Rad) for 2 h. The Bio-Bead treatment was carried out three times and in the last treatment the beads were left in the solution overnight at 2–3 °C. After removing the beads, the aggregates were diluted with buffer (5 mM Tricine (pH 7.5), 5 mM MgCl2). The aggregation process was followed by measuring the fluorescence intensity vs. the fluorescence of the solubilized LHCII trimers. Typically aggregates had a 10–15-fold decreased fluorescence yield. The trimer and aggregate suspensions were shock-frozen by inserting them into a liquid nitrogen bath with the samples included in a thin (0.2 mm pathlength) cuvette without adding cryo-protectant. The OD was kept very low (< 0.02) to prevent self-absorption.
LHCII crystallization
Hexagonal plates of pea LHCII were obtained by vapor diffusion at 15–22 °C as described (Standfuss et al. 2005), at 10–20% polyethylene glycol 350 monomethyl ether, 10–20% glycerol, 50 mM MES buffer pH 5.3 − 5.6 and 20 mM sodium chloride. Crystals had a typical size of around 150 × 150 × 15 µm. Thin hexagonal crystals were harvested from the mother liquor and washed at least twice with a chlorophyll-free cryo-protectant solution (polyethylene glycol 350 monomethyl ether and glycerol added to the mother liquor at a final concentration of 20% each). The crystals were frozen immediately after crystal growth in the cryo-loops and the crystals were never thawed up for the measurements but were inserted frozen into the cooling setup of the fluorescence lifetime apparatus. Crystals are optically dense up to about 685 nm which causes self-absorption of the fluorescence (Barros et al. 2009). We have minimized self-absorption by exciting and detecting in a front-face mode. Furthermore most of the fluorescence of interest in this work occurs at wavelengths above 685 nm and is thus not affected by self-absorption.
Time-resolved fluorescence
Fluorescence kinetics as well as steady-state fluorescence has been measured using a single-photon timing (SPT) apparatus as described elsewhere (Müller et al. 1991) with temporal resolution of 2–3 ps under magic angle polarization conditions in a liquid helium cryostat. The measured instrument response function (IRF) of the lifetime spectrometer had a width (FWHM) of 35 ps. It has been demonstrated in the past that lifetimes down to 3 ps can be resolved reliably with that apparatus even for very complex multi-component kinetics. For example the well-known 3 ps kinetics of charge separation in bacterial reaction centers in combination with complex charge recombination kinetics have been resolved (Müller et al.1991, 1992; Holzwarth 1995; Müller et al. 1996). This is possible due to the extremely high signal-to-noise ratio and large linear dynamic range of the SPT technique, which typically exceeds that of streak camera systems by at least two orders of magnitude.
For measurements on aggregates, the solution was frozen without adding a cryo-protectant in order not to disturb the aggregated structure and not to modify the surface properties. Crystals were cooled by a home-built open cold nitrogen jet stream with the crystal mounted in a cryoloop. Aggregated (oligomers) and crystalline LHCII were excited at 663 nm and the fluorescence decays were detected in the 670–780 nm wavelength region as a function of temperature. Signals were recorded at very high signal/noise ratio (typically 20,000–60,000 peak counts in the decay) taking advantage of the very large linear dynamic range of the SPT technique. Time-resolved fluorescence has been analyzed by global and target analysis procedures and results are presented as decay-associated emission spectra (DAES) and species-associated emission spectra (SAES) (Holzwarth 1996). For excellent fits, the total χ2 values should be below 1.1 and the residuals should have only minor deviations from a random distribution around zero. Additionally in the target analysis procedure the free-running SAES should have physically reasonable shapes and the model should result in physically reasonable rate constants which are internally consistent (Holzwarth 1996).
Quantum chemical calculations
Quantum chemical calculations have in the past addressed the locally excited and exciton states of LHCII (see (Konig and Neugebauer 2011; Müh and Renger 2012), while the description of CT states has been a challenge for computational chemistry so far; (see also (Balevicius Jr et al. 2017; Cupellini et al. 2019; Segatta et al. 2019) for reviews).
For the problem at hand, we need to include a cluster of at least three excitonically coupled Chl molecules representing the lowest excited states in the complex (Müh et al. 2010; Novoderezhkin et al. 2011; Magdaong et al. 2013), together with their protein environment. Such extensive super-system calculations for CT states require the use of methods that are both computationally affordable and still provide physically meaningful results. We use here the long-range corrected density functionals CAM-B3LYP (Yanai et al. 2004) and ωB97XD (Chai and Head-Gordon 2008). Our study, to the best of our knowledge, reports for the first time long-range corrected time-dependent density functional theory (TD-DFT) calculations encompassing a cluster of three Chls. The three Chls constitute the minimal unit that is required to understand the mechanisms of CT state formation and quenching in LHCII out of the lowest energy exciton states. The computational effort has been reduced by several simplifications (e.g., removal of the phytyl chains of the Chls and the representation of the protein environment as a point charge field (PCF)), but it retains all essential properties that are required to describe the involved electronic states, including in particular the CT states, and their dependence on environmental charges and different protonation states. We are aware that the absolute excitation energies calculated in this manner do carry some substantial systematic errors, in particular since we ignore multi-electron excitation character in the TD-DFT approach (Grimme 1996). We note, however, that at this stage we are primarily interested in revealing the relative changes of the CT energies upon some very specific changes in the environment, i.e., the transfer of a single proton between adjacent cofactors such as an amino acid (aa) pair in the protein. Such relative changes in the energies of the CT states are much more precise in our calculations than their absolute energies. More precise calculations of the CT state energies would require very costly calculations, and would have to include in particular also a dynamic charge environment, rather than a fixed PCF environment.
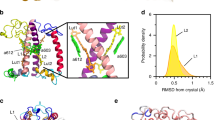
Our present calculations bear some close similarity to previous work on Chl–Car coupling in LHCII (Kröner and Götze 2012). Our model contains the coupled Chl cluster consisting of the three Chl a molecules forming the quenching center (residues number 601, 602 and 607 of chain A in PDB structure 2BHW (Standfuss et al. 2005)—these residues are labeled Chl 610, 612 and 611, respectively, throughout this work in accordance with the previously published structure (Liu et al. 2004); c.f. Fig. S1, electronic Supporting Information (ESI) for the shell of aa residues and lipids embedding the chromophores at a distance up to 4 nm (ONIOM method) (Dapprich et al. 1999) in the crystal structure. A detailed list of the aa residues and cofactors incorporated in the shell and the quantum mechanically treated core are also listed in ESI, Fig. S1. Our choice is consistent with the fact that the excitonically coupled cluster of these three Chls has been identified as providing the lowest energy excited states in the LHCII complex (Müh et al. 2010; Novoderezhkin et al. 2011).
The calculations were performed in the following manner: In a first series of calculations, we explored a wide range of possibly relevant groups (in particular amino acid pairs in different possible (re)protonation states) surrounding the quenching center that might strongly influence CT state energies. Out of all the re-protonizable groups tested two pairs of aas or lipids were identified close to the proposed quenching Chl cluster of interest.Footnote 1 Two groups were found to be the predominant factors controlling the Chl–Chl CT state energies: the E175/K179 pair and the PG/K182 couple, with phosphatidylglycerol (PG) as an integral constituent of the LHCII trimer structure and coordinating Chl 611 (Liu et al. 2004). We subsequently calculated the point charge fields (PCFs) describing the protonation states (i.e., four different PCF configurations in total) using the AMBER99 (Wang et al. 2000) force field Coulomb parameters. PG charge parameters were computed as outlined in (Kröner and Götze 2012). For each of the four PCFs, we then computed the TD-CAM-B3LYP/6-31G(d) excited state energies for all vertical excitations within the Chl Q band excitation region, including the Chl–Chl CT states. For comparison, the Chl cluster spectrum in vacuo has also been calculated. A more detailed description of the computational methods used and the exact sequence of steps taken is presented in the ESI.
Results
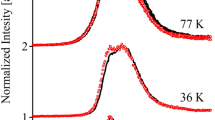
The temperature dependence of the steady-state fluorescence of LHCII aggregates and of LHCII crystals is shown in Fig. 1. The 5 K emission spectrum of the trimeric LHCII in detergent is also shown for comparison. It peaks at − 680 nm and shows the well-known low-intensity vibrational sideband around 740 nm (Pieper et al. 1999a, b, 2001). Already at r.t. (Miloslavina et al. 2008) but much more pronounced at lower temperatures (Fig. 1), LHCII aggregates show a new red-shifted emission maximum (peak around 690–700 nm depending on temperature) and also a substantially enhanced fluorescence tail extending well above 780 nm as compared to trimeric complexes at the same temperature. This FR part of the fluorescence spectrum increases drastically upon lowering the temperature. Below 170 K a long-wave shoulder appears (∼ 710 nm, Fig. 1a) and around 100 K the spectrum shows a plateau in the region 690–710 nm. Lowering the temperature further, this plateau develops into a broad band with a maximum in the region 690–700 nm and with further decrease of the temperature to 5 K this FR maximum shifts hypsochromically to 692 nm and the band narrows pronouncedly. From 170 to 5 K, the total fluorescence intensity increases by a factor of 7–8. Crystalline LHCII (Fig. 1b) shows a similar temperature behavior. At corresponding temperatures the crystal has a slightly narrower emission spectrum than the aggregates with maxima at 700 nm (100 K), 695 nm (50 K), and 687 nm with a pronounced shoulder at 700 nm (5 K). As for aggregates a very strong increase of the fluorescence yield by a factor of ≈10 is observed going from 200 to 5 K for crystals. Overall the temperature dependence of the emissions of both LHCII forms reveals a very complex mixture of different T-dependencies rather than a single uniform T-effect.

Steady-state fluorescence spectra (not normalized) of LHCII aggregates (A) and crystal (B) at different temperatures. The fluorescence spectrum at T = 5 K for LHCII trimers in detergent is also shown for comparison. The trimer spectrum is normalized to the maximal intensity of the aggregates at 5 K. The excitation wavelength was 663 nm
The experimental fluorescence decays of LHCII aggregates and crystals at various wavelengths and temperatures as measured by single-photon counting are given in the electronic Supporting Information (ESI) Fig. S2. These data show that for both aggregates and crystals the normal LHCII emission around 675–680 nm is highly quenched (very short lifetimes). The results from the global lifetime analysis are shown as decay-associated emission spectra (DAES) in Fig. S3 together with the average lifetimes τavg as a function of wavelength. Typically six lifetimes were required to fit the decays over the whole wavelength range in a global fit. Interestingly the τavg of both aggregates and crystals increases strongly towards the longer emission wavelengths and decreases strongly with increasing temperature (Fig. S4), quite in contrast to the more or less wavelength- and T-independent decays for trimers in the same wavelength range (Fig. S2). The spectral shapes of the fluorescence spectra and the plots of the average lifetimes differ largely. While the maxima in the steady-state fluorescence are for most temperatures below 700 nm the average lifetimes have their maximum above 700 nm. This behavior indicates that the fluorescence is not uniform across the emission band but originates from several different species emitting with very different spectra and lifetimes. The increase in τavg is most pronounced at the lower temperatures while at higher temperatures τavg is much less wavelength-dependent (Figs. S3–S5). Near 680 nm τavg is very short in all cases, ranging typically well below 100 ps again drastically different from trimers, which show a uniform long average lifetime of ca. 5 ns also around 680 nm. Generally two kinetic rise terms (negative-amplitude components) are resolved (Fig. S3). For temperatures ≤ 100 K the shortest lifetime components are ca. 8 ps and as short as 4 ps for 170 K with spectral maximum near 680 nm. The longer lifetimes range from 40–60 ps up to 4.3 ns and for each temperature the longest lifetime shows the most bathochromically shifted DAES. Lowering the temperature shifts the longest-lived DAES pronouncedly to the blue. Thus at 5 K the maximum of the slowest component in both aggregates and crystals is located at 690–695 nm but at 170 K the longest-lived component peaks at 715–720 nm. Also the long-wavelength components show the smallest amplitudes in the FR region. At r.t. maximal lifetimes of aggregates are in the range of ca. 500 ps (Miloslavina et al. 2008).
Global target modeling
More insight into the kinetic details and the physical basis of the spectral/kinetic components can be obtained from kinetic target analysis (Holzwarth 1988, 1991, 1996) by testing out different kinetic compartment models on the data in a global fashion. Initially a wide range of different compartment models has been tested on the data, starting with the 5 K data (c.f. Fig. S6 for a selection of the many different kinetic schemes tested). At the lowest temperatures, energy can only flow energetically downhill while all energetic uphill processes are prohibited. Thus at the low temperatures pure sequential kinetic forward models or branched/parallel sequential forward models with zero backward reaction/energy transfer rates should be able to describe the data. In the target analysis, the requirement for the acceptance of a particular kinetic scheme as physically reasonable is not only that the fluorescence decays over the whole spectrum must be fitted very well. It is furthermore required that the resulting SAES must also show physically reasonable shapes (for example all positive amplitudes) (Holzwarth 1996). From comparison of all the tested kinetic schemes, it was found that either a model of two parallel sequential kinetic schemes (6 components) or a branched sequential scheme (also 6 components) populated from the same starting compartment were required for a physically reasonable and numerically satisfactory description of the data in a compartment model. On the basis of the pure mathematical quality of fit criteria alone (residuals, χ2 values, physically reasonable shapes of spectra etc.), these two schemes could not be distinguished unequivocally in all cases (i.e., different temperatures, crystals vs. aggregates etc.). The results of these two models differed only significantly in the rates of the initial fast step which is likely an internal energy transfer process. However, they gave essentially identical results for the spectra and the kinetics of formation and decay of the FR-emitting states, which are the most relevant for the present analysis. We thus present and discuss here only the results for the branched model (c.f. Figs. 2 and 3 and Fig. S6). The time-dependent concentrations of all species within this model are shown in Fig. S7. The excellent quality of the resulting fits is shown for both LHCII forms for selected wavelengths and temperatures in Figs. S8–S11. The residual plots for all emission wavelengths in the respective experiments are also shown in these figures.

SAES (left) and kinetic models with rate constants [ns−1] (right) obtained from the target analysis of the fluorescence kinetics of LHCII aggregates at different temperatures using the branched kinetic scheme. The excitation wavelength was 663 nm. Right hand side: Each compartment for each species (rectangular box) is colored in the same way as the corresponding SAES on the left. The rate constants (ns−1, black numbers) of processes are shown next to the arrows. The assignment to Chl* or CT states as discussed in the text is shown inside of the box. The fast rate constants (rates ≥ 50 ns−1) have an error of ca. ± 10%, while the slower rate constants have an error of ca. ± 5%. Note that all SAES are normalized for better comparison (see text explaining the fact that the CT states have a low radiative rate and thus a smaller area under the curve, corresponding to a smaller amplitude, as compared to ordinary Chl excited states). The corresponding data showing IRF, experimental decays, theoretical fitting function, quality of fit and all the residual plots are given in Figs. S8–S11 of the ESI

SAES (left) and kinetic models with rate constants [ns−1] (right) obtained from the target analysis of the fluorescence kinetics of LHCII crystals at different temperatures using the branched kinetic scheme. The excitation wavelength was 663 nm. Right side: Each compartment (rectangular box) is marked by a number and the box is colored in the same way as the corresponding SAES to the left. The rate constants (ns−1, black numbers) are shown next to the arrows. The assignment to Chl* or CT states as discussed in the text is shown inside of the box. The fast rate constants (rates ≥ 50 ns−1) have an error of ca. ± 10% while the slower rate constants have an error of ca. ± 5%. Note that all SAES are normalized for better comparison (c.f. caption Fig. 2). The corresponding data showing IRF, experimental decays, theoretical fitting function, quality of fit and all the residual plots are given in Figs. S8–S11 of the ESI
There is one additional strong argument favoring the choice of the branched kinetic scheme: Only with this model, it was possible to fit all the data of both LHCII forms over the entire temperature range up to about 220 K in a satisfactory manner. At still higher temperatures, the spectra (SAES) of the FR components become extremely broad and both spectra and kinetics are strongly overlapping, both for crystals as well as for aggregates (data not shown). Notably the data in the higher temperature range are very similar to the previously published r.t. data (Miloslavina et al. 2008; Müller et al. 2010)). Thus a resolution of the kinetics with the branched (or parallel) models was not possible anymore and a simplified sequential single branch scheme is adequate, in agreement with the previous data (Miloslavina et al. 2008; Müller et al. 2010).
Fitting using simpler target models, e.g., the model proposed by Chmeliov et al. (2016) as well as a sequential model with three fluorescing states, was also performed. Exemplary results of such fits for both aggregates as well as the crystal are shown in Figs. S12–S16, with typical examples of resulting SAES shown in Fig. S12. These results show that the kinetic model proposed by Chmeliov et al., involving two fluorescing states and a non-fluorescent (“dark” state), falls far short of adequately describing the experimental data, both for aggregates (total χ2 = 22.1 of the fit for T = 100 K) as well as for LHCII crystals (total χ2 = 13.1 of the fit for T = 100 K). The sequential model (reversible reaction steps) with three fluorescent components results in a better—but still overall inadequate—fit quality (total χ2 in the range of 2–3 for T = 100 K) as compared to the model of Chmeliov et al.Footnote 2
Quantum chemical calculations
The TD–DFT calculations for the coupled chromophore system (Chls 610, 611 and 612 in the PCF of the environment, see Fig. 4 for a structure) yielded a large variety of excited states, of which we show only the Q band state energies and the energies of some additional states which are energetically close (Fig. 5). The energies of the Q band states appear slightly blue-shifted compared to the experimental bands due to the calculations providing vertical excitation energies, i.e., not energies corresponding to actual ground state to Q transitions with large Franck–Condon factors.

Molecular structure of the proposed quenching center in LHCII showing the arrangement of the relevant molecular groups for quenching in surface presentation. The protein is shown in dark gray. The highly surface-exposed and strongly exciton coupled Chl-triple (Chl610, Chl611, Chl612, shown in different shades of green) is indicated with blue labels, along with the associated amino acids (K175/E179 pair) and lipid/amino acid pair (PG/K182). Luteins are shown in light transparent yellow and the protruding neoxanthin in transparent orange. Other Chl a and Chl b molecules are shown in transparent light blue

Energy level diagram for locally excited/exciton states (green lines) and CT states (red and black lines for the two different CT states formed) of the quenching center formed by the excitonically coupled cluster containing three Chls. The energies are calculated for the four different protein protonation states of the surrounding protein as indicated in the boxes above and on the right hand side. Left: unquenched situation; middle: quenched situation; Right: gas phase situation for comparison (no stromal charge, no low-energy CT states present)
For the crucial CT state energies, the effect of the protonation changes in the triple Chl cluster environment is very strong as expected. Without any protein environment for the Chl cluster (gas phase, Fig. 5, right side, top), we find only local Q band excitations, e.g., Qy excitations in the lowest energy range at 579, 567 and 563 nm with oscillator strengths of 0.62, 0.17 and 0.05, respectively. Such a result can be expected when coupling three chromophores without any environmental disturbance. No CT state is found in the vicinity of the Qy states in the absence of the protein. Upon introduction of the protein environment and PCFs, the calculations yield a large number of CT states within and below the Qy band region (Fig. 5, left and center). Most importantly the energies of these CT states are strongly dependent on the specific protonation state configuration at the two protonation/deprotonation centers considered here.
The various low-lying CT states differ both in terms of their electronic character and their dependence on the environment. One group of CT states reflects electron transfers from Chl 610 to Chl 612 (states marked red in Fig. 5). These states are very sensitive to the protonation state of the E175/K179 aa pair. The lowest Chl610 → 612 CT state is strongly shifted to lower energies (> 2000 cm−1) upon protonation of K179. It gets even more shifted to lower energies upon switching the protonation state of the PG/K182 pair, albeit the latter effect is significantly smaller. This can be attributed to the larger distance of the PG/K182 pair to Chls 610 and 612.
The second group of CT states (marked black in Fig. 5) represents Chl610 → 611 CT states. The energy of these states is strongly dependent on the PG/K182 protonation state, which is explained by the spatial proximity of PG/K182 to Chl 611. The energies of the lowest CT states shift by about 1500 cm−1 to lower energies upon PG−/H-K182+ to PG-H/K182 reprotonation.
The effect of E175/K179 reprotonation on these states is only a red-shift of no more than 500 cm−1, e.g., when going from column A on the left side of Fig. 5 to column A on the right hand side, one finds only a minor shift in energy for the states marked in black.
Other CT states were not observed, likely due to the presence of D162 close to Chl 610, enforcing Chl 610 to act as an electron donor in all the different protonation states tested. Therefore, we can conclude that the two observed classes of CT states exhibit distinctively different energy response upon changes in the protein environment. While being slightly affected by both (protonation) residue pairs, each class has its dominant control element. For the 610 → 612 CT states that element is the E175/K179 aa pair, for the 610 → 611 CT states it is the PG/K182 pair. The orbital presentations of the involved CT transitions are shown in Fig. S17 (see also Fig. 5 for the energies). These presentations clearly show the CT character of the assigned transitions.
Discussion
New FR-emitting states in LHCII aggregates and crystals
As a general summary of the experimental data one can conclude that both aggregates and crystals show strongly quenched excited state decays around 680 nm, the main emission band of trimers. In that range the lifetime is shorter by a factor ≥ 20 as compared to unquenched trimers (Figs. S3 and S4). Invariably related to the drastically shortened lifetimes around 680 nm is the appearance of strongly temperature-dependent FR-emitting species with lifetimes ranging from − 100 ps to more than 4 ns (note that the emission spectrum of the long-lived component in the quenched case is very different from that of LHCII trimers). The total fluorescence yield increases pronouncedly with decreasing temperature, as has already been noticed earlier (Mullineaux et al. 1993; Ruban et al. 1995a, b) and the temperature dependence of the spectra shown in Fig. 1 is very similar to the one previously reported. At all temperatures, the fluorescence spectrum of both quenched LHCII forms is drastically different from those of unquenched trimeric LHCII at the same temperature (see also Mullineaux et al. 1993; Ruban et al. 1995a, b). A very similar temperature behavior is observed for crystals although the spectra at very low temperatures differ slightly from those of the aggregates. The dominant reason for this is the presence of pronounced self-absorption in the crystal at the short emission wavelengths. The most striking observation for both types of quenched LHCII is that several well-distinguishable FR-emitting states appear at lower temperatures: F684, F − 690, F − 700, F − 720, and F − 743 (Figs. 2 and 3, the numbers reflect the approximate wavelength of the spectral maximum) whose amplitudes show a pronounced temperature dependence. For the states with fluorescence peaks in the range 690–700 nm, the temperature behavior has been reported earlier (Ruban and Horton 1992; Ruban et al. 1995a, b). The states emitting further to the red range have also been resolved previously in time-resolved experiments at low temperatures although their exact origin had not been understood at the time (Mullineaux et al. 1993). The FR-emitting states show a highly unusual temperature dependence (Figs. 2 and 3). The longer-wavelength emitting states are more populated at higher temperatures (Fig. S7), while the shorter wavelength emitting FR states get relatively more populated at the lower temperatures in both aggregates and crystals. The F − 679 state also shows a pronounced temperature dependence. Its relative contribution to total fluorescence increases with temperature, i.e., opposite to the fluorescence intensity from the long-wavelength emitting states (Figs. 2 and 3). This different behavior points to a different origin of the F − 679 state from all the others. The F − 679 state represents the equilibrated excited states of the LHCII complex prior to quenching. It is the same state that is also observed for the trimeric solubilized LHCII (c.f. Figure 1) (Pieper et al. 1999a, b, 2001), while at the low temperatures the aggregates and crystals show only very weak emission around 679 nm, i.e., the lowest energy excited states are very rapidly depopulated to the FR-emitting states. These FR states (c.f. Figure 2) clearly represent a group of fluorescent states that are not present at all in solubilized trimers but only in the quenched LHCII aggregates and crystals.
The most red-shifted components undergo the slowest relaxation (Figs. 2, 3, branched model). Their lifetimes actually do change in the 5–170 K temperature range only by a factor of about 2, but the total intensity of their fluorescence changes by a factor of 7–8. Thus it is clear that the origin of the large temperature dependence of the steady-state fluorescence as well as the differences in the spectral shapes between steady-state fluorescence (Fig. 1) and average lifetimes (Figs. S3, S4 and S5) does not simply reflect a change in lifetimes but has a more complex origin related primarily to pronounced changes in the relative populations of these different states (Fig. S7). As we can see from the rate constants in the target models (Figs. 2 and 3), both the rise and the decay rates of these intermediates are temperature-dependent. The combination of all these changes then gives rise to the observed very complex overall temperature dependence of the steady-state spectra (Fig. 1). Note that this observed temperature dependence is indeed highly unusual. In almost all fluorescing systems, whether simple molecules in solution or, e.g., complex antenna systems, lowering the temperature typically leads to a bathochromic shift of the overall fluorescence (Beechem and Brand 1985; Schulman 1985; Murata and Satoh 1986). However, in the case of quenched LHCII aggregates and crystals lowering the temperature leads to a pronounced hypsochromic shift of the fluorescence. Thus, a very special situation with regard to the properties of the FR-emitting excited states and their kinetics must be realized in that case (vide infra for more detailed discussion of the complex temperature dependence of the reaction rates and the CT state spectra). From qualitative analysis of the temperature dependence of spectra and lifetimes, it can be concluded that in both aggregates and crystals there appear to exist two groups of temperature-activated processes that have their approximate activation energies in a range corresponding to thermal energies of about 30 K and around 80–100 K, respectively.
The lowest energy emitting state in solubilized trimers at low temperature has been found at − 679 nm with an inhomogeneous width of 70–80 cm−1 with several emitting states located at slightly shorter wavelength (Pieper et al. 1999a, b, 2001). This is in good agreement with the properties of the first excited state(s) found in the target models for the two lowest temperatures (Figs. 2 and 3) for both aggregates and crystals (for proper comparison note that the spectral resolution in our time-resolved studies ranges from 2–4 nm, i.e., much lower than that of the above-mentioned detailed studies of low-temperature hole-burning and fluorescence (Pieper et al. 2001)). We assign those states—at least for 5 K and 100 K data—to the same Chl excited states that are also fluorescing in unquenched solubilized trimers. The energy differences of the maximum of the trimers’ lowest energy emitting state to the maxima of the FR-emitting states are in the range of ∼ 200–1000 cm−1. Note that the most red-shifted FR states are significantly populated only at the higher temperature (c.f. Figs. 2 and 3). If this pronounced spectral shift should be caused by exciton coupling between Chls or between Chls and Cars, it would require a drastic increase in the respective excitonic coupling energies, at least 500 cm−1 and up to 1000 cm−1 (depending on the interaction model), when going from the unquenched solubilized to the quenched aggregated or crystalline states. This could only be brought about by rather large conformational rearrangements changing distances and orientations of pigments. We note here that the largest exciton coupling energies in the trimeric LHCII state—for the lowest energy Chl pairs that are located at very close distance —are ≤ 120 cm−1 (Novoderezhkin et al. 2005). Any larger excitonic coupling would have to show up as large correlated changes in the absorption and CD spectra, concomitant with the very large red-shifts in the fluorescence. Several groups have measured absorption and CD spectra in various environments and for both solubilized and aggregated LHCII (Javorfi et al. 1996; Naqvi et al. 1997; Lambrev et al. 2007; Magdaong et al. 2013; Akhtar et al. 2015, 2019a, b). Only some small increase in the absorption at the red tail of the Qy absorption band and at most small changes in the shorter wavelength Qy range as well as large specific CD changes in the Car and Chl Qx (436 nm) absorption range have been reported for non-solubilized aggregates. Moreover, these CD changes were found to be unrelated to the induction of fluorescence quenching (Akhtar et al. 2015). Also anisotropic CD spectra, which are in general more sensitive to isolate exciton interactions, failed to detect significant changes in the FR region upon aggregation (Akhtar et al. 2019a). Quite interestingly, and in direct contrast to all the proposed Chl–Car exciton quenching models, actually an increase, rather than a decrease, of Chl–Lut exciton coupling has been found when going from the non-solubilized aggregated LHCII to the unquenched detergent-solubilized LHCII trimers (Naqvi et al. 1999) along with the expected changes in the CD spectra (Lambrev et al. 2007; Akhtar et al. 2015).
In an early hole-burning study comparing solubilized trimers and aggregates it was found that there occurred at best minor red-shifts in the main Chl Qy absorption band (670–685 nm) of less than 2 nm in aggregates vs. trimers (Pieper et al. 1999a, b). In contrast, clear evidence for strongly red-shifted Chl–Chl CT states has been found by hole-burning in the red tail of LHCII aggregates (Kell et al. 2014) in addition to CT state sensitization by LHCII aggregates in photovoltaic solar cells (Yang et al. 2014) which is only possible if the sensitizer is able to transfer an electron. It follows from all these observations that we can exclude that the strongly red-shifted (200–1000 cm−1) emitting states whose appearance is closely associated with quenching—both in aggregates and in the crystals—are caused by increases in the exciton coupling strength between Chls or between Chls and Cars, thus requiring major conformational rearrangements. It is revealing in this context that authors who actually do propose a Chl–Car quenching model (Fox et al. 2017), were unable to propose any structural changes in LHCII large enough such that an energy transfer quenching by Cars would be switched off (i.e., the long searched for “unquenched conformation”). Taken together these observations essentially exclude already the hitherto proposed Car quenching mechanisms as well as any Chl–Car CT quenching (see more detailed discussion below). Rather, to explain these very large observed fluorescence red-shifts and the observed switching, quite a different mechanism of quenching must be invoked. At this point it is essential to note that despite some small differences in the detailed parameters, the overall high similarity of the fluorescence spectral properties, the rate constants and lifetimes and their temperature dependence in aggregates vs. crystals strongly suggests that the same phenomena give rise to the FR fluorescence bands and their peculiar temperature dependence in both LHCII forms. In fact all our data indicate that (i) these FR-emitting states are Chl–Chl CT states and (ii) that they represent intermediates in the quenching process of the excited Chl* state, as has been proposed earlier both on the basis of fluorescence as well as femtosecond transient absorption kinetics (Miloslavina et al. 2008, 2011; Müller et al. 2010).
Both FR-emitting states (Figs. 2 and 3) show drastically wider bandwidths than the initially excited Chl state(s) and also much wider than the fluorescence emissions of solubilized unquenched trimers, i.e., bandwidths of several hundred cm−1 for the FR states vs. 70–80 cm−1 in trimers are observed (see also Pieper et al. 1999a, b, 2001). Thus either the electron–phonon coupling or the inhomogeneous broadening, or both, of the FR-emitting states are pronouncedly larger than for the typical Chl excited states, indicating drastically different electronic properties. The recent hole-burning experiments in the weak FR absorption tail of LHCII aggregates indeed revealed states with very large electron–phonon coupling (Huang-Rhys factors S = 3–4) (Kell et al. 2014) which could only be interpreted as Chl–Chl CT states that have a weak transition moment to the ground state.
The area under the SAES of an emitting state is proportional to the radiative rate (Holzwarth 1986, 1996). According to this criterium the FR-emitting states also differ drastically from those of the directly excited “normal” Chl excited states. For the FR states in aggregates and crystals the radiative rates are quite low, i.e., in the range of 0.05–0.15 of the radiative rate of a typical Chl excited state. This situation is expected for Chl–Chl CT states.
We have excluded above that major changes of the exciton coupling within the trimers (either between Chls or between Chls and Cars) could explain the FR-emitting states. This excludes also any major increases in inter-trimer coupling. The relative arrangement of LHCII trimers in the crystals is known from the X-ray structure (Liu et al. 2004; Standfuss et al. 2005). For LHCII aggregates several pieces of evidence exist that similar trimer-trimer arrangements might be realized (Li 1985; Garab et al. 1988) as are present in two-dimensional crystals (Kühlbrandt 1984). We may also expect a significant degree of inhomogeneity in the inter-trimer contacts of aggregates, even if they were in principle following a similar structural arrangement as two-dimensional crystals. Such an inhomogeneity is not present in the crystal, which is one of the key reasons why we include the quenching comparison of aggregates and crystals in this study. The comparison leads us to conclude that close trimer–trimer contacts, as appearing undoubtedly in the aggregates, can be excluded as the cause of the quenching. For LHCII crystals, the exciton coupling due to the strongest possible inter-trimer contacts has been calculated for both type of crystals (Barros et al. 2009). This interaction for type II crystals (Standfuss et al. 2005) corresponds to the coupling energy between Chls 612 in two adjacent trimers which is 11 cm−1. For type I crystals (Liu et al. 2004) this coupling energy is maximally 60 cm−1 corresponding to coupling of Chls 605/614 in two adjacent trimers. Thus the largest inter-trimer interaction energy of 60 cm−1 could at best shift the lowest energy emitting state to ca. 683 nm from 680 nm even in type I crystals. For the type II crystals used in the present study the expected shift due to inter-trimer coupling would be even less. Hole-burning experiments on LHCII aggregates have indeed localized the low-energy Chl excited state in the aggregates at the same low-energy Chls as in the trimer at 683 nm (Pieper et al. 1999a, b). Thus the inter-trimer couplings and their associated spectral shifts are far too small in order to explain the appearance of the FR-emitting states but they explain the minor red-shifts of up to 2 nm observed in the spectra. One may argue that for aggregates various inter-trimer arrangements may in principle be possible. However inspection of the crystal structure of LHCII reveals that the closest possible inter-trimer Chl pair would always have to be either the Chl 612–Chl 612′ pair, since Chl 612 is the Chl that is located closest to the protein surface, or the Chl 605/614 pair, depending on relative arrangement. Since in type I crystals these inter-trimer Chls are in a rather favorable position already, we conclude that any other inter-trimer arrangement in aggregates would not lead to a much larger inter-trimer exciton coupling than the value of 60 cm−1 calculated for the type I crystals (Barros et al. 2009). Thus also for aggregates we can exclude that the reason for the emergence of the FR-emitting states could be excitonic inter-trimer contacts. We thus come to the conclusion that the quenching in aggregates and in crystals of LHCII and the associated appearance of similar FR-emitting states in both systems can neither be related to inter-trimer contacts causing inter-trimer exciton coupling nor to conformational distortions caused by inter-trimer interactions. This is so far—even though a definite mechanism was not proposed—the underlying implied assumption (in connection with the conformational change hypothesis and the related search for the so far elusive “unquenched conformation”) for the quenching observed in LHCII aggregates. We can now clearly exclude this hypothesis. This leaves us with the notion that the quenching process must be explained by mechanism(s) that are entirely limited to the size of a single trimer, or possibly even a single monomer within a trimer. It is thus interesting to refer to the study of Ilioaia et al. (2008) who reported strong excited state quenching similar to aggregates in a non-aggregated trimeric form of LHCII embedded in a gel. Quenching was switched on when the detergent micelle in the gel was removed and could be switched off reversibly when the detergent micelle was reconstituted. A more recent study has also shown that the CD changes associated with trimer-trimer contacts and quenching are fully separable and independent from each other (Akhtar et al. 2015). Our conclusions appear to be in full agreement with these works. Consequently quenching in aggregates (and likewise in crystals) cannot be explained by aggregation-induced trimer-trimer contacts. It must be rather explained by the removal of the detergent layer and the ensuing modified properties around the surface-exposed Chls, in particular by the change from an apolar environment present in a lipid micelle (or a lipid membrane) to a polar environment (i.e., LHCII surface exposed to water and salts) in aggregated LHCII and/or crystals ( i.e., a “polarity switch” model).
The molecular model
We will in the following develop a LHCII quenching model that is consistent with the occurrence of the FR fluorescence components in the quenched state taking into account the results of experimental as well a Chl exciton and CT state calculations which locate the lowest energy excited state at the Chl-triple 610/611/612 (Novoderezhkin et al. 2005; Mozzo et al. 2008; Ramanan et al. 2015) and identify this group as the quenching center in LHCII (Ruban et al. 2007; Mozzo et al. 2008; Bode et al. 2009; Müh et al. 2010). In all of these discussions the clear distinction between the implied “quenched state” (which we define as the lowest energy excited state of LHCII) and the “quenching state” (or intermediates formed out of that state) is of crucial importance. Inspection of that region in the crystal structure reveals several groups as candidates for a “polarity switch” model. The easiest way to switch from a non-polar to a polar environment in a protein is a proton transfer between neighboring groups (protonation/deprotonation reaction). Such a localized proton transfer on two aa or cofactor pairs would not have any major consequence for the overall conformation of the protein. We identified two sites to be straightforward proton switching sites in LHCII: The aa pair formed by E175/K179, located between Chls 612 and 611, and a pair formed by K182 and a PG molecule, which coordinates the Mg ion of Chl 611. These two sites have a decisive influence on the energies of the Chl CT states according to the quantum chemical calculations (c.f. Fig. 5). Thus our theoretical results strongly support the idea of a polarity switch controlling the energies of the CT states of the system. Opening the “quenching gate” by a switch to a polar environment in addition requires the surface accessibility of the relevant group(s). This is the case only for the E175/K179 pair which is strongly surface-exposed (Fig. 4). We thus propose this pair as the ideal candidate acting as the initial switch or sensor for controlling quenching. We propose that reprotonation of the initially neutral E175/K179 pair (in the unquenched stated) opens the gate to quenching by lowering drastically the CT state energies of the Chl610/Chl612 pair by about 2000 cm−1 thus allowing the formation of the first CT pair, Chl610+/Chl612−. The pK values of the E/K pair in strongly different polar environments are explained theoretically by the “Depolarization Born effect” (Pace 2009; Pace et al. 2009). Our calculations also suggest that the initially generated CT state is likely not the final state, but it will undergo further relaxation to lower lying CT states. A PG−/K182-H+ reprotonation forming the neutral PG-H/K182 state and lowering the energy of the initial Chl–Chl CT state by another ca. 1500 cm−1 becomes a favorable reaction step once the first CT state (610 → 612 CT) has been formed (c.f. Fig. 5). In such a protonation environment the formation of a Chl610+/Chl611− state by a second electron transfer step is energetically favored (Fig. 5). The overall sequence of events (Fig. 6) described here for the quenching process is a classical two-step proton-coupled electron transfer process (PCET) which occurs in many chemical and biological processes and can control a variety of different functions (Hammes-Schiffer et al. 2008; Hammes-Schiffer 2009).

Energetic and kinetic scheme explaining the suggested sequence of reaction steps for quenching in aggregates/crystal of LHCII. Electron transfer processes are indicated with bent arrows labeled e− and the numbers shown indicate the Chl molecules involved. A polar environment induces a reprotonation in the initially neutral E175/K179 aa pair, which lowers the energy of the Chl CT state(s) below the excited state energy (gate open), thus switching on Chl–Chl CT formation from Chl610 to Chl 612 producing the CT1 state. This electron transfer step triggers a proton transfer step from K182-H+ to PG−, which subsequently reduces the energy of the CT states further and thus induces a second electron transfer step from Chl612 to Chl611 to form the CT2 state. The latter finally recombines to the ground state to complete the quenching process
We have thus identified two groups of CT states, one of which can be controlled directly by an external factor (via the protonation control of the E172/K175 pair), while the other allows for a further drop in overall energy and a second electron transfer step due to an internal CT-driven proton transfer (from K182-H+ to PG−). It is straightforward to correlate these two CT states with the sequentially formed CT1 and CT2 state kinetics (compartments, c.f. Figs. 2 and 3) observed in fluorescence. In the absence of further relaxation possibilities the lowest energy CT state(s) could then recombine to the ground state, either directly or via the rapid formation (on the several nsec time scale) of a Chl triplet state due to spin dephasing of the radical pair in the lowest CT state (Bixon et al. 1988; Ogrodnik et al. 1988; Volk et al. 1993), thus completing the quenching process. This reaction scheme is shown in detail in Fig. 6 and the corresponding potential energy diagram, explaining also the peculiar temperature dependence of the FR fluorescence states is provided in Fig. 7.

Top: Schematic potential energy diagram showing the processes of formation, interconversion and recombination to ground state of the Chl–Chl CT states in LHCII. This scheme is a simplified sequential reaction scheme that reflects only one branch in the kinetic schemes shown in Figs. 2 and 3. GS, ground state (black); ‘Chl*’—lowest excitonic Chl state (green) with fluorescence at − 680 nm; ‘CT1′ (blue) and ‘CT2′ (red)—FR fluorescing Chl–Chl CT states; b1 and b2—temperature-dependent potential energy barriers for CT state formation and recombination; dashed arrows—fluorescence (fl) of Chl–Chl exciton and CT states with fluorescence maxima in the 685–760 nm region; curved arrows—CT reaction steps for CT state formation (green and blue) and finally recombination to ground state (red). Bottom: Cartoon illustrating the “polarity-switching” model for quenching. In a non-polar/symmetric environment the Chl–Chl CT states are higher in energy than the locally excited states (Chl* and exciton states). CT states can not be formed corresponding to the unquenched situation (left). In a polar environment the CT states are energetically below the excited states and the quenching process is possible (right)
The crucial step for the excited state quenching is the very rapid formation of the first CT state out of the Qy excitonic state (with nearly temperature-independent time constants in the range of 50 ps, c.f. Figs. 2 and 3). The quantum chemical calculations result in the local Qy band excitation energies being fairly unaffected by the different protonation states. This agrees with the experimental finding that the absorption spectrum of quenched aggregates and crystals, as compared to unquenched trimeric LHCII, does not differ substantially in the main absorption bands, but only shows a weak FR tailing in the quenched LHCII reflecting absorption of low-lying mixed CT states (Magdaong et al. 2013; Kell et al. 2014).
Controlling the quenching switch
According to this proposition the quenching form of LHCII is the E175−/K179-H+ protonated form and the surface-accessible E175/K179 site plays the key role of the initial switch or “gate-keeper” which decides whether quenching by CT state formation can occur or not. This E/K pair senses the immediate environment of LHCII. As for the neutral, non-quenching H-E/K variety, there exist several arguments to postulate its preference in unquenched LHCII in vivo, especially when bound to the PSII supercomplex. In vivo in the PSII supercomplex (PDB entry 3JCU (Wei et al. 2016)) the immediate environment enforces the neutral state: The E175/K179 pair directly interacts with a cluster of negatively charged residues (e.g., E128/E129/D130 in the “center W protein” of PSII and similar groups in the minor antenna complexes). Thus a charge on E175 in LHCII is energetically unfavorable at the binding site of LHCII in the PSII supercomplex. The monomers not bound to other proteins in the supercomplex are located in the non-polar environment of the hydrophobic membrane lipid tails thus also favoring the neutral form. The same holds true for isolated LHCII trimers in a proteoliposome membrane, or if surrounded by the hydrophobic tails of detergents in a micelle. Such non-polar hydrophobic environments enforce the non-quenching form as a result of the depolarization Born effect, which is known to result in uncharged aa residues in the absence of a stabilizing polar solvent (Pace 2009; Pace et al. 2009). In contrast, the charged form of the E/K pair is realized in aggregates, where the removal of the detergent affords a polar/charged environment around this pair thus switching to the quenched form (Mullineaux et al. 1993; Pascal et al. 2005; Miloslavina et al. 2008; Müller et al. 2010) (c.f. Fig. 7). The mechanism of salt bridge reprotonation by environmental polarization change is also well established through high level computations (Nagy and Erhardt 2010). Essentially the same situation is realized in the crystal which does not contain a sufficient amount of detergent to build a protecting micelle around the trimers and allows access of water and salts to the surface of LHCII, thus inducing also a quenching situation. Interestingly, the direct switching between polar (inducing quenching) and non-polar (non-quenching) environment has been demonstrated with trimeric LHCII in a gel which excludes aggregation (Ilioaia et al. 2008). Our model thus provides a consistent and transparent explanation for the different situations under which quenching is either switched on or is disabled.
Why are the CT states fluorescent?
Chl–Chl CT states, which can be considered Chl+/Chl− radical pairs similar to a primary Chl–Chl radical pair formed in photosynthetic reaction centers, would be non-fluorescent in first approximation since such states do not carry oscillator strength to the ground state. However it has been demonstrated both theoretically as well as experimentally that such Chl–Chl CT states gain oscillator strength by electronic coupling to the parent exciton states (Bixon and Jortner 1969). The exact radiative rates of CT states depend pronouncedly on the relative orientation of the molecules forming the CT states, the specific properties of the environment, and the energy difference to their parent exciton states. Documented cases are the “red fluorescing Chls” in Lhca4 (Novoderezhkin et al. 2016, 2018) and the P-band of the bacterial RC. The P-band has been studied extensively by various spectroscopic methods and it has been concluded that it represents a strongly mixed state between the P-exciton state and the intradimer CT state (Lathrop and Friesner 1994; Zhou and Boxer 1997, 1998; Moore et al. 1999). The FR states in LHCII bear very pronounced similarities to the “red Chl fluorescence” states in the Lhca4 antenna complexes of PSI which have been shown by Stark spectroscopy to represent CT states that are strongly coupled to the respective exciton state(s). They are also fluorescent and show a similar temperature dependence as the LHCII aggregates (Ihalainen et al. 2000; Croce et al. 2002; Frese et al. 2002; Ihalainen et al. 2003; Morosinotto et al. 2003, 2005; Gibasiewicz et al. 2005; Croce et al. 2006, 2007). Another example is the RC of PSII (Konermann, Gatzen et al. 1997; Konermann, Yruela et al. 1997; Hughes et al. 2004; Krausz et al. 2005; Hughes et al. 2006a, b; Novoderezhkin et al. 2007; Thapper et al. 2009). All of these Chl–Chl CT states exhibit large electron–phonon coupling, leading to a very large homogeneous bandwidth, as well as large inhomogeneous broadening (Hayes et al. 2000; Rätsep et al. 2000; Croce et al. 2007; Romero et al. 2009). We thus propose that the FR states observed in the LHCII aggregates and crystal (Figs. 2 and 3) represent Chl–Chl CT states gaining their (weak) optical transition moment to the ground state by intensity borrowing from their parent excitonic Chl states. A strong Stark effect on this fluorescence can thus be expected and has indeed been found experimentally (Wahadoszamen et al. 2012). The CT states observed in this work carry an oscillator strength that is in the range of 15–20% (CT1 and CT1′ states), and ca. 10% (CT2 and CT2′ states), resp. of the lowest excited state of LHCII which is taken as a reference.
The need for a branched kinetic model can be attributed to the pronounced energetic disorder and inhomogeneous broadening of the CT states which are much more sensitive in their energies to small conformational changes (within the usual inhomogeneous linewidth) than ordinary excited or exciton states. Furthermore formation of the CT states is associated with very large reorganization energies, demanding major dielectric and structural relaxations after their formation (Heitele and Michel-Beyerle 1987; Heitele et al. 1989, 1990). Conformational relaxations are, however, increasingly hampered by lowering the temperature which makes it then possible to distinguish different states, representative of broad CT state energy distributions. Consequently, the branched kinetic model describes a bimodal distribution of the reaction rates and energies of the CT states (Heitele and Michel-Beyerle 1987; Heitele et al. 1989, 1990). This model is of course an approximation to the actual distribution functions, albeit a very good one as exemplified by the excellent fits of the model for the experimental data. Overall these properties explain the highly unusual temperature dependence of the spectra (spectral blue-shifts upon lowering the temperature), the unusual temperature dependence of the reaction rates (i.e., non-monotonous rates upon lowering the temperature, Figs. 2 and 3), and the apparently decreasing energy gaps between the CT and exciton states with decreasing temperature.Footnote 3 At low temperatures there exist more or less static distributions allowing only limited and slowed-down relaxation (cooling). At higher temperatures, these static distributions are replaced by rapidly relaxing CT states and dynamic distributions, as exemplified by the strong red-shifts and broadening of the CT fluorescence spectra. This leads to a pronounced overlap of the different CT state spectra and of their kinetics, thus not allowing further separation. Finally, at r.t. a single sequential kinetics describes the experimental data very well (Miloslavina et al. 2008; Müller et al. 2010). Overall LHCII excited state quenching has high similarity to many aspects of the dynamics of the electron transfer and CT state relaxation dynamics in photosynthetic reaction centers. This has been studied extensively by a range of methods both for PSII reaction centers (Roelofs et al. 1991; Roelofs et al. 1993; Gatzen et al. 1996; Konermann and Holzwarth 1996; Konermann, Gatzen et al. 1997; Konermann, Yruela et al. 1997; Holzwarth et al. 2006; Szczepaniak et al. 2008; 2009) as well as for bacterial reaction centers (Müller et al. 1995; Holzwarth and Müller 1996; Wang et al. 2007; Guo et al. 2012; Pan et al. 2012, 2013).
Quenching models that require changes in Chl-carotenoid and Chl–Chl interactions
The still prevailing quenching models for LHCII involve a modification in the electronic coupling between a Chl and an adjacent Car activated by a major conformational change of the protein core backbone and the relative orientations of Chls and Cars. To switch on a quenching process in an unquenched LHCII trimer Chl–Car coupling would have to be modified strongly either by a modified trimer–trimer interaction or intra-trimer in the crystal form. One possibility is the so-called CIC quenching model proposed theoretically by Naqvi (1998) and more recently taken up for explaining in vivo quenching (Bode et al. 2009). Alternatively an increased Chl–Car exciton coupling could increase the energy transfer rate to a Car S1 state, e.g., of Lut1, as the quenching center, which is located next to Chls 610 and 611 (Ruban et al. 2007; Fox et al. 2018). The two models are actually quite similar and both require a pronounced conformational change that would allow for a drastic change in Chl–Car coupling strength (van Amerongen and van Grondelle 2001; Ruban et al. 2007; Fox et al. 2018). Krueger et al. (1998) have calculated the Coulomb couplings between BChl Qy states and the adjacent Car in the bacterial antenna complexes to be in the order of a few cm−1 up to max. 100 cm−1. However the Chl–Car couplings in LHCII are typically weaker by an order of magnitude (Fox et al. 2018) and it has been estimated that the (calculated theoretical) coupling strengths might in fact be largely overestimated. These Coulomb couplings can in principle induce an excited state mixing resulting in an increase in non-radiative decay of the excited state (CIC model) (van Amerongen and van Grondelle 2001). Alternatively they could also induce an energy transfer from Chl to the Car S1 state, given favorable energetic positions. In both cases these small couplings would induce quite a small spectral shift of the involved Chl excited state (and also the Car state) in the same order of magnitude. Thus these relatively small coupling energies would lead to minor energy shifts only in the modified fluorescing states of at most a few nm as compared to the unquenched trimer. Such small shifts would be consistent with the relatively minor band-shifts in the absorption and hole-burning spectra that have been reported upon aggregation of LHCII (Pieper et al. 1999a, b, 2001). They would, however, not be able to explain energetic shifts up to 1000 cm−1, as is required to explain the strongly shifted FR-emitting states observed in this work for quenched aggregates and crystals of LHCII. To induce energetic shifts of that magnitude, very large conformational changes would have to occur. We consider the possibility to achieve, by a mere conformational change, such a large increase in coupling strength between Chl and Car and corresponding spectral shifts as highly unlikely in view of the fact that the LHCII protein is very rigid in its hydrophobic core where the Chls and Cars are located (Barros et al. 2009). The decisive question is, however, not whether some energy transfer to Car or some non-radiative decay of the Chl excited state is possible due to electronic mixing. Rather the decisive question is whether the required changes would be large enough to allow a controlled switching on and off of the deactivation rate by a factor of 30–100 (Ruban et al. 2007; Miloslavina et al. 2008; Holzwarth et al. 2009; Müller et al. 2010), a magnitude required to explain the observed in vitro as well as the in vivo quenching effects in LHCII. To switch on and off quenching—going for example from LHCII trimers in detergent to aggregates—this conformational change would have to be not only very drastic but also non-random and reversible in a controlled manner to allow a controlled increase in the quenching rate by nearly 2 orders of magnitude relative to the unquenched state. Notably the authors who do propose a conformational switching model and a Car-induced Chl excited state quenching (Krüger et al. 2014; Chmeliov et al. 2015; Duffy and Ruban 2015; Chmeliov et al. 2016) were not able—despite many efforts—to come up with any defined conformational change model that would allow the required large changes in quenching rates by Cars (Fox et al. 2017).
Thus the small changes in quenching rates in various conformation states suggested by the theoretical calculations mentioned above and the large changes in quenching rates induced upon aggregation (see Horton et al. 1991; Mullineaux et al. 1993; Miloslavina et al. 2008; Magdaong et al. 2013; Chmeliov et al. 2016 and data presented in this work) as well as in the crystal are in pronounced disagreement. Clearly to explain the fluorescence yield changes, quenching rate changes, and the huge spectral shifts to the FR region by a change in Chl–Car coupling one would require to observe correspondingly large changes in the absorption, and in particular shifts in the CD spectra which is not the case (vide supra). In principle the CIC hypothesis might be consistent with the observed small absorption changes, but it is, however, neither consistent with the documented weak hole-burning spectra accompanied by very large Huang-Rhys factors (Kell et al. 2014) nor with the pronouncedly shifted FR fluorescence bands.
As an alternative scenario, the formation of a Chl–Car CT state, which could also be brought about by a changed electronic Chl–Car coupling and which might in principle explain the observed FR fluorescence bands, has been considered. Such a mechanism has been proposed for example recently on the basis of theoretical calculations (Cupellini et al. 2020). This model causes, however, several contradictions with established experimental data. To explain the lack of experimental evidence for such a mechanism from ultrafast transient absorption measurements (Ruban et al. 2007; Müller et al. 2010), the authors proposed that the formed Car+Chl− CT state might be extremely short-lived. This is, however, quite unlikely since the recombination of CT states to the ground state typically occurs on much slower time scales than their formation for fundamental reasons related to the large reorganization energies involved in their formation which then either involves singlet-to-triplet dephasing or thermally activated recombination (Ogrodnik et al. 1988a, b; Heitele et al. 1989; Bixon et al. 1994). Furthermore, a Chl–Car CT state would be easily observable even in the case of very short lifetimes in the NIR around 800–1000 nm due to the extremely strong difference absorption band of the associated Car cation with difference extinction coefficients of 140 000 to 200 000 M−1 cm−1 and the absence of other strong signals in that region (Amarie et al. 2007, 2008). Furthermore, an extremely short-lived Chl–Car CT state, such that it cannot be detected anymore by femtosecond transient absorption, would then also fail to explain the key observation of the FR fluorescence which has lifetimes in the range from several 100 ps to many ns. In contrast clear evidence for a long-lived (ns), Chl–Chl CT state was observed in the transient absorption data (Müller et al. 2010) as well as the hole-burning data (Kell et al. 2014). We can thus exclude both the Chl–Car CT hypothesis as well as the Chl–Car CIC or excitonic coupling hypothesis as viable quenching mechanisms for LHCII. Furthermore, no viable mechanism was proposed in the work of Cupellini et al. (2020) that would allow the controlled switching between quenched and unquenched conformations.
Chmeliov et al. (2016) have recently also measured the temperature dependence of the fluorescence kinetics of LHCII aggregates (but not of crystals), albeit at much lower signal-to-noise ratio due to the employed streak camera detection. Formally their data are similar to ours. The authors have, however, only been able to resolve the kinetics into two spectral components and three kinetic components. Our measurements show that the kinetics of both aggregated LHCII as well as of crystals is, however, much more complex (c.f. Figs. S3, S5, and Fig. S12 ff.), showing up to 6 lifetime components. This discrepancy can be explained to the most part by the lower signal-to-noise ratio of the streak camera detection vs. the single-photon counting detection employed in the present measurements. Chmeliov et al. (2016) consequently suggested a three-component kinetic model, comprising the (lowest) excited state of LHCII, one “red emitting form”, which is interpreted as a Chl–Chl CT state, but according to their interpretation is explicitly not involved in quenching. Interestingly that “red emitting form”, present always in the aggregates in low amounts, is populated in their model by long-distance, possibly multi-step, energy transfer from the LHCII excited state(s), from energy donors residing to a large part in other LHCII trimers than the “red state” (Gelzinis et al. 2018).Footnote 4 The third state in their model is a non-fluorescent “dark quenched state” populated by the quenching process. In such a case their kinetic data should be fitted equally well with a mere two-component model (see footnote 2 above). The authors, however, explicitly excluded such a two-component as not describing their data (Chmeliov et al. 2016) which creates an internal contradiction. Testing similar simplified models (Figs. S12 ff.) on our data clearly demonstrates that there must exist more than two fluorescing species differing in both their spectra and in their kinetics.
Ignoring the different levels of complexity in the analysis, the major differences of the model of Chmeliov et al. (2016) to our kinetic model are two-fold. As suggested by detailed target analysis performed here the appearance kinetics of the FR fluorescent state(s) corresponds exactly with the decay kinetics of the excited state of LHCII. This shows that the FR state(s) are the immediate reaction products of the excited state quenching process and thus represent direct intermediates in the quenching reaction, in contrast to the interpretation of Chmeliov et al. The fast electron transfer reaction is only possible if it is occurring from the excitonically coupled Chl dimer/trimer which then also carries the CT state. This also excludes the possibility that the FR-emitting state(s) are representing merely some heterogeneous conformational states of the LHCII excited state or a Chl–Chl CT state being i) populated independent of the quenching process to a small extent and ii) are not involved in quenching, as proposed in the model of Chmeliov et al. Quite in contrast to that interpretation our data show unequivocally that the actual excited state quenching of both aggregates as well as crystals is very fast (decay times of − 30–50 ps, depending on temperature), while Chmeliov et al. state that the “NPQ trap is rather slow”, a conclusion which also appears to be in contradiction to their proposed kinetic scheme. The second major difference of our model to that of Chmeliov et al. consists in the fact that in our case the excited state decay of LHCII is homogeneous and that virtually all LHCII complexes undergo quenching. The proposal of a high percentage of “unquenched” LHCII complexes and the occurrence of fast inter-complex energy transfer processes in the aggregates (between different trimers) (Chmeliov et al. 2016) is inconsistent with the essentially identical quenching processes in the crystal, which not only possesses a homogeneous environment for each LHCII trimer but also where such fast energy transfer processes do not occur due to low inter-trimer coupling (Barros et al. 2009). The assumption of fast inter-complex energy transfer (to the “red states”) and highly inhomogeneous quenching of only parts of the trimeric complexes is, however, a central element in the quenching model of Chmeliov et al. (2016). In summary, the quenching data obtained here on crystals and the close similarity of quenching processes in both types of samples—despite their extreme differences in relative arrangement and environment of LHCII trimers—do prove that the quenching model of Chmeliov et al. does not represent a valid description of the quenching kinetics in LHCII.
Conclusion
On the basis of the reported kinetic and spectral properties of the FR-emitting states, we propose a Chl–Chl CT state model for quenching. All experimental data, including the previously published r.t. fluorescence and femtosecond TA data are in excellent agreement with this model and it is supported also clearly by the quantum chemical calculations. The proposed mechanism is a sequence of two proton-coupled electron transfer steps. This model involving two emissive Chl–Chl CT states as quenching intermediates is also in excellent agreement with the results of fluorescence Stark spectroscopy in aggregated LHCII which demonstrated the presence of two fluorescent CT states, both directly related to quenching (Wahadoszamen et al. 2012). Finally, the proposed quenching model is also in excellent agreement with single-molecule fluorescence studies on LHCII trimers which revealed the relatively slow switching between unquenched complexes emitting around 680 nm and several quenched states emitting in the FR region (Krüger et al. 2010, 2011a, b). Of particular interest is also the finding that a change of the local environment of the complexes to more polar and/or more acidic conditions caused more frequent switching to the quenched state(s) (Krüger et al. 2011a), although internal aa pair reprotonation was not considered in these papers as the switch controlling quenching.
More work is required to understand the detailed molecular interactions and changes in the vicinity of the quenched Chls for the different cases of in vivo quenching, which requires the interaction with PsbS, a small protein present in the thylakoid membrane (Li et al. 2004). It is interesting in this connection that we have been able very recently to demonstrate strong PsbS-induced quenching of LHCII in proteoliposomes co-reconstituted with PsbS. These samples showed far-red CT state fluorescence exclusively in the quenched state very similar to the one reported here (Pawlak et al. 2020), and very similar to the LHCII quenching detected in intact leaves (Holzwarth et al. 2009; Miloslavina et al. 2011). For the discussed cases of in vitro quenching, it is remarkable that such seemingly different situations like depleting the LHCII from its detergent layer, or putting it into a crystal, is able to switch on essentially identical quenching processes in a reversible manner. Furthermore all available data are fully consistent with the idea that the quenched and unquenched states of LHCII do not differ significantly in their protein backbone and cofactor conformations and their relative orientations in the relevant hydrophobic core part, as has been proposed earlier (Barros et al. 2009). This interpretation is strongly supported by the “…near identity of the cryo-EM structure of LHCII within the supercomplex… to the previous crystal structure of isolated LHCII (Liu et al. 2004)…” (Wei et al. 2016). In summary all these observations and correlations should help to put an end to the intense and long-standing—but so far elusive—search for the “unquenched conformation” of the LHCII trimer complex.
Notes
The crystal structure of LHCII (Standfuss et al. 2005) as deposited in file ID 2BHW in the protein database has been used as structural basis.
We note here that the three-state model used by Chmeliov et al. (two fluorescing components, plus one non-fluorescent “dark state”) (Chmeliov et al. 2016) is kinetically indistinguishable in fluorescence data from a simpler two-state model with two fluorescent components since presence or absence of the “dark state” has no influence on the fluorescence kinetics. The proposal of the “dark state” in the kinetic model is thus an unproven assumption lacking experimental evidence.
Due to the pronounced CT state relaxation at higher temperatures this system does not show the temperature dependence of reaction rates expected for a normal Boltzmann equilibrium.
The authors (Gelzinis et al. 2018) claim that they have tested our Chl–Chl CT state kinetic model (Miloslavina et al. 2008; Müller et al. 2010) and found it to be inconsistent with their fluorescence kinetic data on LHCII aggregates. We note that their interpretation is a gross misinterpretation of our CT state model, since they modeled an energy transfer process as populating the “red state”. In contrast in our Chl–Chl CT state kinetic model an electron transfer process from the excited LHCII state to the Chl–Chl CT state, residing in the same LHCII monomer as the formed CT state, populates the “red state”. state. Thus the interpretation used in their modeling (Chmeliov et al. 2016; Gelzinis et al. 2019) has no resemblance to and deviates fundamentally from our proposed kinetic model.
Abbreviations
- LHCII:
-
Major light-harvesting complex of photosystem II
- PSII:
-
Photosystem II
- RC:
-
Reaction center
- Chl:
-
Chlorophyll
- Car:
-
Carotenoid
- Lut:
-
Lutein
- Vx:
-
Violaxanthin
- Zx:
-
Zeaxanthin
- Nx:
-
Neoxanthin
- DAES:
-
Decay-associated emission spectrum
- r.t:
-
Room temperature
- NPQ:
-
Non-photochemical quenching
- qE:
-
Energy-dependent quenching
- CT (state):
-
Charge-transfer (state)
- RP:
-
Radical pair
- FR:
-
Far-red
- HL:
-
High light
References
Akhtar P, Dorogi M, Pawlak K, Kovacs L, Bota A, Kiss T, Garab G, Lambrev PH (2015) Pigment interactions in light-harvesting complex II in different molecular environments. J Biol Chem 290:4877–4886
Akhtar P, Görföl F, Garab G, Lambrev PH (2019a) Dependence of chlorophyll fluorescence quenching on the lipid-to-protein ratio in reconstituted light-harvesting complex II membranes containing lipid labels. Chem Phys 522:242–248
Akhtar P, Lindorfer D, Lingvay M, Pawlak K, Zsiros O, Siligardi G, Javorfi T, Dorogi M, Ughy B, Garab G, Renger T, Lambrev PH (2019b) Anisotropic circular dichroism of light-harvesting complex ii in oriented lipid bilayers: theory meets experiment. J Phys Chem B 123:1090–1098
Amarie S, Arefe K, Starcke JH, Dreuw A, Wachtveitl J (2008) Identification of an additional low-lying excited state of carotenoid radical cations. J Phys Chem B 112:14011–14017
Amarie S, Standfuss J, Barros T, Kühlbrandt W, Dreuw A, Wachtveitl J (2007) Carotenoid radical cations as a probe for the molecular mechanism of nonphotochemical quenching in oxygenic photosynthesis. J Phys Chem B 111:3481–3487
Amarie S, Wilk L, Barros T, Kühlbrandt W, Dreuw A, Wachtveitl J (2009) Properties of zeaxanthin and its radical cation bound to the minor light-harvesting complexes CP24, CP26 and CP29. Biochim Biophys Acta-Bioenergetics 1787:747–752
Avenson TJ, Ahn TK, Zigmantas D, Niyogi KK, Li Z, Ballottari M, Bassi R, Fleming GR (2008) Zeaxanthin radical cation formation in minor light-harvesting complexes of higher plant antenna. J Biol Chem 283:3550–3558
Balevicius V Jr, Fox KF, Bricker WP, Jurinovich S, Prandi IG, Mennucci B, Duffy CDP (2017) Fine control of chlorophyll-carotenoid interactions defines the functionality of light-harvesting proteins in plants. Sci Rep 7:13956
Barros T, Royant A, Standfuss J, Dreuw A, Kühlbrandt W (2009) Crystal structure of plant light-harvesting complex shows the active, energy-transmitting state. EMBO J 28:298–306
Beechem JM, Brand L (1985) Time-resolved fluorescence of proteins. Ann Rev Biochem 54:43–71
Bennett DIG, Amarnath K, Park S, Steen CJ, Morris JM, Fleming GR (2019) Models and mechanisms of the rapidly reversible regulation of photosynthetic light harvesting. Open Biol 9:190043
Betterle N, Ballottari M, Zorzan S, de Bianchi S, Cazzaniga S, Dall'Osto L, Morosinotto T, Bassi R (2009) Light-induced dissociation of an antenna hetero-oligomer is needed for non-photochemical quenching induction. J Biol Chem 284:15255–15266
Bixon M, Jortner J (1969) Electronic relaxation in large molecules. J Chem Phys 50:4061–4070
Bixon M, Jortner J, Cortes J, Heitele H, Michel-Beyerle ME (1994) Energy gap law for nonradiative and radiative charge transfer in isolated and in solvated supermolecules. J Phys Chem 98:7289–7729
Bixon M, Michel-Beyerle ME, Jortner J (1988) Formation dynamics, decay kinetics, and singlet-triplet splitting of the (bacteriochlorophyll dimer)+ (bacteriopheophytin)- radical pair in bacterial photosynthesis. Isr J Chem 28:155–168
Bode S, Quentmeier CC, Liao PN, Barros T, Walla PJ (2008) Xanthophyll-cycle dependence of the energy transfer between carotenoid dark states and chlorophylls in NPQ mutants of living plants and in LHC II. Chem Phys Lett 450:379–385
Bode S, Quentmeier CC, Liao PN, Hafi N, Barros T, Wilk L, Bittner F, Walla PJ (2009) On the regulation of photosynthesis by excitonic interactions between carotenoids and chlorophylls. Proc Natl Acad Sci USA 106:12311–12316
Chai JD, Head-Gordon M (2008) Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys Chem Chem Phys 10:6615–6620
Chmeliov J, Bricker WP, Lo C, Jouin E, Valkunas L, Ruban AV, Duffy CD (2015) An 'all pigment' model of excitation quenching in LHCII. Phys Chem Chem Phys 17:15857–15867
Chmeliov J, Gelzinis A, Songaila E, Augulis R, Duffy CD, Ruban AV, Valkunas L (2016) The nature of self-regulation in photosynthetic light-harvesting antenna. Nat Plants 2:16045
Crisafi E, Krishnan M, Pandit A (2018) Time-resolved fluorescence analysis of LHCII in the presence of PsbS at neutral and low pH. bioRxiv 456046
Croce R, Chojnicka A, Morosinotto T, Ihalainen JA, van Mourik F, Dekker JP, Bassi R, van Grondelle R (2007) The low-energy forms of photosystem I light-harvesting complexes: Spectroscopic properties and pigment-pigment interaction characteristics. Biophys J 93:2418–2428
Croce R, Morosinotto T, Bassi R (2006) LHCI: The antenna complex of photosystem I in plants and green algae. In: Golbeck JH (ed) Photosytem I: The Light-Driven Plastocyanin: Ferredoxin Oxidoreductase. Springer, Dordrecht, pp 119–137
Croce R, Morosinotto T, Castelletti S, Breton J, Bassi R (2002) The Lhca antenna complexes of higher plants photosystem I. Biochim Biophys Acta 1556:29–40
Cupellini L, Calvani D, Jacquemin D, Mennucci B (2020) Charge transfer from the carotenoid can quench chlorophyll excitation in antenna complexes of plants. Nature Communications 11:662
Cupellini L, Corbella M, Mennucci B, Curutchet C (2019) Electronic energy transfer in biomacromolecules. Wiley Interdisciplinary Reviews: Computational Molecular Science 9:e1392
Dall'Osto L, Cazzaniga S, Bressan M, Palecek D, Zidek K, Niyogi KK, Fleming GR, Zigmantas D, Bassi R (2017) Two mechanisms for dissipation of excess light in monomeric and trimeric light-harvesting complexes. Nat Plants 3:17033
Dapprich S, Komarómi I, Byun KS, Morokuma K, Frisch MJ (1999) A new ONIOM implementation in Gaussian98. Part I. the calculation of energies, gradients, vibrational frequencies and electric field derivatives. J Mol Struct 461–462:1–21
Demmig-Adams B, Cohu CM, Muller O, Adams WW III (2012) Modulation of photosynthetic energy conversion efficiency in nature: from seconds to seasons. Photosynth Res 113:75–88
Dreuw A, Fleming GR, Head-Gordon M (2003) Chlorophyll fluorescence quenching by xanthophylls. Phys Chem Chem Phys 5:3247–3256
Duffy CD, Ruban AV (2015) Dissipative pathways in the photosystem-II antenna in plants. J Photochem Photobiol B 152:215–226
Fox KF, Balevicius V, Chmeliov J, Valkunas L, Ruban AV, Duffy CDP (2017) The carotenoid pathway: what is important for excitation quenching in plant antenna complexes? Phys Chem Chem Phys 19:22957–22968
Fox KF, Unlu C, Balevicius V Jr, Ramdour BN, Kern C, Pan X, Li M, van Amerongen H, Duffy CDP (2018) A possible molecular basis for photoprotection in the minor antenna proteins of plants. Biochim Biophys Acta 1859:471–481
Frank HA, Cua A, Chynwat V, Young A, Gosztola D, Wasielewski MR (1994) Photophysics of the carotenoids associated with the xanthophyll cycle in photosynthesis. Photosynth Res 41:389–395
Frese RN, Palacios MA, Azzizi A, van Stokkum IHM, Kruip J, Rögner M, Karapetyan NV, Schlodder E, van Grondelle R, Dekker JP (2002) Electric field effects on red chlorophylls, beta-carotenes and P700 in cyanobacterial Photosystem I complexes. Biochim Biophys Acta 1554:180–191
Garab G, Faludi-Daniel A, Sutherland JC, Hind G (1988) Macroorganization of chlorophyll a/b light-harvesting complex in thylakoids and aggregates: Information from circular differential scattering. Biochemistry 27:2425–2430
Gatzen G, Müller MG, Griebenow K, Holzwarth AR (1996) Primary processes and structure of the photosystem II reaction center: 3. Kinetic analysis of picosecond energy transfer and charge separation processes in the D1-D2-cyt-b559 complex measured by time-resolved fluorescence. J Phys Chem 100:7269–7278
Gelzinis A, Augulis R, Butkus V, Robert B, Valkunas L (2019) Two-dimensional spectroscopy for non-specialists. Biochim Biophys Acta Bioenerg 1860:271–285
Gelzinis A, Chmeliov J, Ruban AV, Valkunas L (2018) Can red-emitting state be responsible for fluorescence quenching in LHCII aggregates? Photosynth Res 135:275–284
Gibasiewicz K, Croce R, Morosinotto T, Ihalainen JA, van Stokkum IHM, Dekker JP, Bassi R, van Grondelle R (2005) Excitation energy transfer pathways in Lhca4. Biophys J 88:1959–1969
Grimme S (1996) Density functional calculations with configuration interaction for the excited states of molecules. Chem Phys Lett 259:128–137
Guo Z, Woodbury NW, Pan J, Lin S (2012) Protein dielectric environment modulates the electron-transfer pathway in photosynthetic reaction centers. Biophys J 103:1979–1988
Hammes-Schiffer S (2009) Theory of proton-coupled electron transfer in energy conversion processes. Acc Chem Res 42:1881–1889
Hammes-Schiffer S, Hatcher E, Ishikita H, Skone JH, Soudackov AV (2008) Theoretical studies of proton-coupled electron transfer: models and concepts relevant to bioenergetics. Coord Chem Rev 252:384–394
Hayes JM, Matsuzaki S, Rätsep M, Small GJ (2000) Red chlorophyll a antenna states of photosystem I of the cyanobacterium Synechocystis sp PCC 6803. J Phys Chem B 104:5625–5633
Heitele H, Finckh P, Weeren S, Pöllinger F, Michel-Beyerle ME (1989) Solvent polarity effects of intramolecular electron transfer. 1. Energetic aspects J Phys Chem 93:5173–5179
Heitele H, Michel-Beyerle ME (1987) The influence of dielectric relaxation on intramolecular electron transfer. Chem Phys Lett 138:237–243
Heitele H, Pöllinger F, Weeren S, Michel-Beyerle ME (1990) Influence of solvent polarity on intramolecular electron transfer: a consistency test of free energies of reaction and solvent reorganization with experimental rates. Chem Phys 143:325–332
Holt NE, Zigmantas D, Valkunas L, Li X-P, Niyogi KK, Fleming GR (2005) Carotenoid cation formation and the regulation of photosynthetic light harvesting. Science 307:433–436
Holzwarth AR (1986) Excited state kinetics of chlorophyll antenna pigments. In: Staehelin LA, Arntzen CJ (eds) Encyclopedia of plant physiology: photosynthesis III new serie. 19. Springer, Berlin, pp 299–309
Holzwarth AR (1988) Time resolved chlorophyll fluorescence:. what kind of information on photosynthetic systems does it provide? In: Lichtenthaler HK (ed) Applications of Chlorophyll Fluorescence. Kluwer Academic, Dordrecht, pp 21–31
Holzwarth AR (1991) Excited-state kinetics in chlorophyll systems and its relationship to the functional organization of the photosystems. In: Scheer H (ed) Chlorophylls. CRC Press, Boca Raton, pp 1125–1151
Holzwarth AR (1995) Time-resolved fluorescence spectroscopy. In: Sauer K (ed) Methods in Enzymology, vol 246. Academic Press, San Diego, pp 334–362
Holzwarth AR (1996) Data analysis of time-resolved measurements. In: Amesz J, Hoff AJ (eds) Biophysical Techniques in Photosynthesis. Kluwer Academic Publishers, Dordrecht, Advances in Photosynthesis Research, pp 75–92
Holzwarth AR, Jahns P (2014) Non-photochemical quenching mechanisms in intact organisms as derived from ultrafast-fluorescence kinetic studies. In: Demmig-Adams B, Garab G, Adams WWI, Govindjee (eds) Non-photochemical quenching and thermal energy dissipation in plants, algae and cyanobacteria. Springer, Dordrecht, pp 129–156
Holzwarth AR, Miloslavina Y, Nilkens M, Jahns P (2009) Identification of two quenching sites active in the regulation of photosynthetic light-harvesting studied by time-resolved fluorescence. Chem Phys Lett 483:262–267
Holzwarth AR, Müller MG (1996) Energetics and kinetics of radical pairs in reaction centers from Rhodobacter sphaeroides. A femtosecond transient absorption study Biochemistry 35:11820–11831
Holzwarth AR, Müller MG, Reus M, Nowaczyk M, Sander J, Rögner M (2006) Kinetics and mechanism of electron transfer in intact photosystem II and in the isolated reaction center: Pheophytin is the primary electron acceptor. Proc Natl Acad Sci USA 103:6895–6900
Horton P, Ruban AV, Rees D, Pascal AA, Noctor G, Young AJ (1991) Control of the light-harvesting function of chloroplast membranes by aggregation of the LHCII chlorophyll-protein complex. FEBS Lett 292:1–4
Horton P, Ruban AV, Walters RG (1996) Regulation of light harvesting in green plants. Annu Rev Plant Physiol Plant Mol Biol 47:655–684
Horton P, Wentworth M, Ruban AV (2005) Control of the light harvesting function of chloroplast membranes: The LHCII-aggregation model for non-photochemical quenching. FEBS Lett 579:4201–4206
Hughes JL, Prince BJ, Arsköld SP, Smith PJ, Pace RJ, Riesen H, Krausz E (2004) The native reaction centre of photosystem II: a new paradigm for P680. Aust J Chem 57:1179–1183
Hughes JL, Smith P, Pace R, Krausz E (2006a) Charge separation in photosystem II core complexes induced by 690–730 nm excitation at 1.7K. Biochim Biophys Acta 1757:841–851
Hughes JL, Smith PJ, Pace RJ, Krausz E (2006b) Spectral hole burning at the low-energy absorption edge of photosystem II core complexes. J Luminesc 119–120:298–303
Ihalainen JA, Gobets B, Sznee K, Brazzoli M, Croce R, Bassi R, van Grondelle R, Korppi-Tommola JEI, Dekker JP (2000) Evidence for two spectroscopically different dimers of light- harvesting complex I from green plants. Biochemistry 39:8625–8631
Ihalainen JA, Rätsep M, Jensen PE, Scheller HV, Croce R, Bassi R, Korppi-Tommola JEI, Freiberg A (2003) Red spectral forms of chlorophylls in green plant PSI —a site-selective and high-pressure spectroscopy study. J Phys Chem B 107:9086–9093
Ilioaia C, Johnson MP, Horton P, Ruban AV (2008) Induction of efficient energy dissipation in the isolated light-harvesting complex of photosystem II in the absence of protein aggregation. J Biol Chem 283:29505–29512
Ilioaia C, Johnson MP, Liao PN, Pascal AA, van Grondelle R, Walla PJ, Ruban AV, Robert B (2011) Photoprotection in plants involves a change in lutein 1 binding domain in the major light-harvesting complex of Photosystem II. J Biol Chem 286:27247–27254
Jahns P, Holzwarth AR (2012) The role of the xanthophyll cycle and of lutein in photoprotection of photosystem II. Biochim Biophys Acta, Bioenerg 1817:182–193
Javorfi T, Naqvi KR, Melo TB, Bangar BR, Garab G (1996) Effect of scattering and sieving in the absorption and circular dichroism spectra of trimers and macro-aggregates of the CHL A/B light-harvesting complex, LHCII, and in thylakoid membranes. Prog Biophys & Mol Biol 65:E225
Johnson MP, Goral TK, Duffy CD, Brain AP, Mullineaux CW, Ruban AV (2011) Photoprotective energy dissipation involves the reorganization of photosystem II light-harvesting complexes in the grana membranes of spinach chloroplasts. Plant Cell 23:1468–1479
Kell A, Feng X, Lin C, Reus M, Holzwarth AR, Jankowiak R (2014) On the charge-transfer character of the low-energy Chl a Qy absorption band in aggregated LHCII pigment protein complexes. J Phys Chem B 118:6086–6091
Konermann L, Gatzen G, Holzwarth AR (1997a) Primary processes and structure of the photosystem II reaction center. 5. Modeling of the fluorescence kinetics of the D1-D2-cyt-b559 complex at 77K. J Phys Chem B 101:2933–2944
Konermann L, Holzwarth AR (1996) Analysis of the absorption spectrum of photosystem II reaction centers: Temperature dependence, pigment assignment and inhomogeneous broadening. Biochemistry 35:829–842
Konermann L, Yruela I, Holzwarth AR (1997b) Pigment assignment in the absorption spectrum of the photosystem II reaction center by site selection fluorescence spectroscopy. Biochemistry 36:7498–7502
Konig C, Neugebauer J (2011) First-principles calculation of electronic spectra of light-harvesting complex II. Phys Chem Chem Phys 13:10475–10490
Krausz E, Hughes JL, Smith P, Pace R, Arsköld SP (2005) Oxygen-evolving Photosystem II core complexes: a new paradigm based on the spectral identification of the charge-separating state, the primary acceptor and assignment of low-temperature fluorescence. Photochem Photobiol Sci 4:744–753
Kröner D, Götze JP (2012) Modeling of a violaxanthin-chlorophyll b chromophore pair in its LHCII environment using CAM-B3LYP. J Photochem Photobiol, B 109:12–19
Krueger BP, Scholes GD, Fleming GR (1998) Calculation of couplings and energy-transfer pathways between the pigments of LH2 by the ab-inito transition density cube method. J Phys Chem B 102:5378–5386
Krüger TP, Ilioaia C, Johnson MP, Ruban AV, van Grondelle R (2014) Disentangling the low-energy states of the major light-harvesting complex of plants and their role in photoprotection. Biochim Biophys Acta-Bioenergetics 1837:1027–1038
Krüger TPJ, Ilioaia C, Valkunas L, van Grondelle R (2011a) Fluorescence intermittency from the main plant light-harvesting complex: sensitivity to the local environment. J Phys Chem B 115:5083–5095
Krüger TPJ, Novoderezhkin V, Ilioaia C, van Grondelle R (2010) Fluorescence spectral dynamics of single LHCII trimers. Biophys J 98:3093–3101
Krüger TPJ, Wientjes E, Croce R, van Grondelle R (2011b) Conformational switching explains the intrinsic multifunctionality of plant light-harvesting complexes. Proc Natl Acad Sci USA 108:13516–13521
Kühlbrandt W (1984) Three-dimensional structure of the light-harvesting chlorophyll a/b-protein complex. Nature 307:478–480
Lambrev P, Várkonyi Z, Krumova S, Kovács L, Miloslavina Y, Holzwarth AR, Garab G (2007) Importance of trimer-trimer interactions for the native state of the plant light-harvesting complex II. Biochim Biophys Acta 1767:847–853
Lambrev PH, Nilkens M, Miloslavina Y, Jahns P, Holzwarth AR (2010) Kinetic and spectral resolution of multiple nonphotochemical quenching components in Arabidopsis leaves. Plant Physiol 152:1611–1624
Lathrop EJP, Friesner RA (1994) Simulation of optical spectra from the reaction center of Rb. sphaeroides. Effects of an internal charge-separated state of the special pair. J Phys Chem 98:3056–3066
Leuenberger M, Morris JM, Chan AM, Leonelli L, Niyogi KK, Fleming GR (2017) Dissecting and modeling zeaxanthin- and lutein-dependent nonphotochemical quenching in Arabidopsis thaliana. Proc Natl Acad Sci USA 114:E7009–E7017
Li J (1985) Light-harvesting chlorophyll a/b protein: three- dimensional structure of a reconstituted membrane lattice in negative stain. Proc Natl Acad Sci USA 82:386–390
Li X-P, Gilmore AM, Caffarri S, Bassi R, Golan T, Kramer D, Niyogi KK (2004) Regulation of photosynthetic light harvesting involves intrathylakoid lumen pH sensing by the PsbS protein. J Biol Chem 279:22866–22874
Li Z, Wakao S, Fischer BB, Niyogi KK (2009) Sensing and responding to excess light. Ann Rev Plant Biol 60:239–260
Liao PN, Holleboom CP, Wilk L, Kühlbrandt W, Walla PJ (2010) Correlation of Car S1 -%3eChl with Chl -%3eCar S1 energy transfer supports the excitonic model in quenched light harvesting complex II. J Physi Chem B 114:15650–15655
Liguori N, Periole X, Marrink SJ, Croce R (2015) From light-harvesting to photoprotection: structural basis of the dynamic switch of the major antenna complex of plants (LHCII). Sci Rep 5:15661
Liu Z, Yan H, Wang K, Kuang T, Zhang J, Gui L, An X, Chang W (2004) Crystal structure of spinach major light-harvesting complex at 2.72 angstrom resolution. Nature 428:287–292
Magdaong NM, Enriquez MM, LaFountain AM, Rafka L, Frank HA (2013) Effect of protein aggregation on the spectroscopic properties and excited state kinetics of the LHCII pigment-protein complex from green plants. Photosynth Res 118:259–276
Mascoli V, Liguori N, Xu P, Roy LM, van Stokkum IHM, Croce R (2019) Capturing the quenching mechanism of light-harvesting complexes of plants by zooming in on the ensemble. Chem 5:2900–2912
Matsubara S, Förster B, Waterman M, Robinson SA, Pogson BJ, Gunning B, Osmond B (2012) From ecophysiology to phenomics: some implications of photoprotection and shade-sun acclimation in situ for dynamics of thylakoids in vitro. Philos Trans R Soc Lond B Biol Sci 367:3503–3514
Miloslavina Y, DeBianchi S, Dall'Osto L, Bassi R, Holzwarth AR (2011) Quenching in Arabidopsis thaliana mutants lacking monomeric antenna proteins of photosystem II. J Biol Chem 286:36830–36840
Miloslavina Y, Wehner A, Wientjes E, Reus M, Lambrev P, Garab G, Croce R, Holzwarth AR (2008) Far-red fluorescence: a direct spectroscopic marker for LHCII oligomers forming in non-photochemical quenching. FEBS Lett 582:3625–3631
Moore LJ, Zhou H, Boxer SG (1999) Excited-state electronic asymmetry of the special pair in photosynthetic reaction center mutants: Absorption and stark spectroscopy. Biochemistry 38:11949–11960
Morosinotto T, Breton J, Bassi R, Croce R (2003) The nature of a chlorophyll ligand in Lhca proteins determines the far red fluorescence emission typical of photosystem I. J Biol Chem 278:49223–49229
Morosinotto T, Mozzo M, Bassi R, Croce R (2005) Pigment-pigment interactions in Lhca4 antenna complex of higher plants photosystem I. J Biol Chem 280:20612–20619
Mozzo M, Passarini F, Bassi R, van Amerongen H, Croce R (2008) Photoprotection in higher plants: the putative quenching site is conserved in all outer light-harvesting complexes of photosystem II. Biochim Biophys Acta 1777:1263–1267
Müh F, Madjet M, Renger T (2010) Structure-based identification of energy sinks in plant light-harvesting complex II. J Phys Chem B 114:13517–13535
Müh F, Renger T (2012) Refined structure-based simulation of plant light-harvesting complex II: linear optical spectra of trimers and aggregates. Biochim Biophys Acta 1817:1446–1460
Müller MG, Dorra D, Holzwarth AR, Gad'on N, Drews G (1995) Time-dependent radical pair relaxation in chromatophores of an antenna-free mutant from Rhodobacter capsulatus. In: Mathis P (ed) Photosynthesis: From Light to Biosphere, vol 1. Kluwer Academic Publishers, Dordrecht, pp 595–598
Müller MG, Drews G, Holzwarth AR (1996) Primary charge separation processes in reaction centers of an antenna-free mutant of Rhodobacter capsulatus. Chem Phys Lett 258:194–202
Müller MG, Griebenow K, Holzwarth AR (1991) Primary processes in isolated photosynthetic bacterial reaction centers from Chloroflexus aurantiacus studied by picosecond fluorescence spectroscopy. Biochim Biophys Acta 1098:1–12
Müller MG, Griebenow K, Holzwarth AR (1992) Primary processes in isolated bacterial reaction centers from Rhodobacter sphaeroides studied by picosecond fluorescence kinetics. Chem Phys Lett 199:465–469
Müller MG, Lambrev P, Reus M, Wientjes E, Croce R, Holzwarth AR (2010) Singlet energy dissipation in photosystem II light-harvesting complex does not involve energy transfer to carotenoids. ChemPhysChem 11:1289–1296
Mullineaux CW, Pascal AA, Horton P, Holzwarth AR (1993) Excitation energy quenching in aggregates of the LHC II chlorophyll-protein complex: a time-resolved fluorescence study. Biochim Biophys Acta 1141:23–28
Murata N, Satoh K (1986) Absorption and fluorescence emission by intact cells, chloroplasts, and chlorophyll-protein complexes. In: Amesz J, Fork DC (eds) Govindjee. Light Emission by Plants and Bacteria. Academic Press, New York, pp 137–160
Nagy PI, Erhardt PW (2010) Theoretical studies of salt-bridge formation by amino acid side chains in low and medium polarity environments. J Phys Chem B 114:16436–16442
Naqvi KR (1998) Carotenoid-induced electronic relaxation of the first excited state of antenna chlorophylls. In: Garab G (ed) Photosynthesis: mechanisms and effects, vol 1. Kluver Academic Publishers, Netherlands, pp 265–270
Naqvi KR, Javorfi T, Melo TB, Garab G (1999) More on the catalysis of internal conversion in chlorophyll a by an adjacent carotenoid in light-harvesting complex (Chla/b LHCII) of higher plants: time-resolved triplet-minus-singlet spectra of detergent-perturbed complexes. Spectrochim Acta A 55:193–204
Naqvi KR, Melo TB, Bangar Raju B, Javorfi T, Garab G (1997) Comparison of the absorption spectra of trimers and aggregates of chlorophyll a/b light-harvesting complex LHC II. Spectrochim Acta A 53:1925–1936
Nicol L, Nawrocki WJ, Croce R (2019) Disentangling the sites of non-photochemical quenching in vascular plants. Nat Plants 5:1177–1183
Novoderezhkin V, Marin A, van Grondelle R (2011) Intra- and inter-monomeric transfers in the light harvesting LHCII complex: the Redfield-Forster picture. Phys Chem Chem Phys 13:17093–17103
Novoderezhkin VI, Croce R, van GR, (2018) Dynamics of the mixed exciton and charge-transfer states in light-harvesting complex Lhca4: Hierarchical equation approach. Biochim Biophys Acta Bioenerg 1859:655–665
Novoderezhkin VI, Croce R, Wahadoszamen M, Polukhina I, Romero E, van Grondelle R (2016) Mixing of exciton and charge-transfer states in light-harvesting complex Lhca4. Phys Chem Chem Phys 18:19368–19377
Novoderezhkin VI, Dekker JP, van Grondelle R (2007) Mixing of exciton and charge-transfer states in Photosystem II reaction centers: Modeling of stark spectra with modified redfield theory. Biophys J 93:1293–1311
Novoderezhkin VI, Palacios MA, van Amerongen H, van Grondelle R (2005) Excitation dynamics in the LHCII complex of higher plants: Modeling based on the 2.72 angstrom crystal structure. J Phys Chem B 109:10493–10504
Ogrodnik A, Volk M, Letterer R, Feick R, Michel-Beyerle ME (1988a) Determination of free energies in reaction centers of Rb. sphaeroides. Biochim Biophys Acta 936:361–371
Ogrodnik A, Volk M, Michel-Beyerle ME (1988b) On the energetics of the states 1P,3P and P+H- in reaction centers of Rb. sphaeroides. In: Breton J, Vermeglio A (eds) The photosynthetic bacterial reaction center. Plenum Press, New York, pp 177–183
Ostroumov EE, Fadeev VV, Khristin MS, Pashchenko VZ, Tusov VB (2007) Fluorescence characteristics and photophysical parameters of light-harvesting chlorophyll a/b complex aggregates. Biophysics 52:462–467
Pace CN (2009) Energetics of protein hydrogen bonds. Nat Struct Mol Biol 16:681–682
Pace CN, Grimsley GR, Scholtz JM (2009) Protein ionizable groups: pK values and their contribution to protein stability and solubility. J Biol Chem 284:13285–13289
Pan J, Lin S, Woodbury NW (2012) Bacteriochlorophyll excited-state quenching pathways in bacterial reaction centers with the primary donor oxidized. J Phys Chem B 116:2014–2022
Pan J, Saer RG, Lin S, Guo Z, Beatty JT, Woodbury NW (2013) The protein environment of the bacteriopheophytin anion modulates charge separation and charge recombination in bacterial reaction centers. J Phys Chem B 117:7179–7189
Pascal AA, Liu ZF, Broess K, van Oort B, van Amerongen H, Wang C, Horton P, Robert B, Chang WR, Ruban AV (2005) Molecular basis of photoprotection and control of photosynthetic light-harvesting. Nature 436:134–137
Pawlak K, Paul S, Liu C, Reus M, Yang C, Holzwarth AR (2020) On the PsbS-induced quenching in the major light-harvesting complex LHCII studied inproteoliposomes. Photosyn Res. https://doi.org/10.1007/s11120-020-00740-z
Pieper J, Irrgang K-D, Rätsep M, Schrötter T, Voigt J, Small GJ, Renk G, Jankowiak R (1999a) Effects of aggregation on trimeric light-harvesting complex II of green plants: A hole-burning study. J Phys Chem A 103:2422–2428
Pieper J, Rätsep M, Jankowiak R, Irrgang K-D, Voigt J, Renger G, Small GJ (1999b) Q(Y)-level structure and dynamics of solubilized light- harvesting complex II of green plants: Pressure and hole burning studies. J Phys Chem A 103:2412–2421
Pieper J, Schödel R, Irrgang K-D, Voigt J, Renger G (2001) Electron-phonon coupling in solubilized LHC II complexes of green plants investigated by line-narrowing and temperature-dependent fluorescence spectroscopy. J Phys Chem B 105:7115–7124
Ramanan C, Gruber JM, Maly P, Negretti M, Novoderezhkin V, Kruger TP, Mancal T, Croce R, van Grondelle R (2015) The role of exciton delocalization in the major photosynthetic light-harvesting antenna of plants. Biophys J 108:1047–1056
Rätsep M, Johnson TW, Chitnis PR, Small GJ (2000) The red-absorbing chlorophyll a antenna states of photosystem I: a hole-burning study of Synechocystis sp PCC 6803 and its mutants. J Phys Chem B 104:836–847
Robert B, Horton P, Pascal AA, Ruban AV (2004) Insights into the molecular dynamics of plant light-harvesting proteins in vivo. Trends Plant Sci 9:385–390
Roelofs TA, Gilbert M, Shuvalov VA, Holzwarth AR (1991) Picosecond fluorescence kinetics of the D1-D2-cyt-b559 photosystem II reaction center complex. Energy Transfer and primary charge separation processes. Biochim Biophys Acta 1060:237–244
Roelofs TA, Kwa SLS, van Grondelle R, Dekker JP, Holzwarth AR (1993) Primary processes and structure of the photosystem II reaction center: II. Low-temperature picosecond fluorescence kinetics of a D1-D2-cyt-b-559 reaction center complex isolated by short Triton exposure. Biochim Biophys Acta 1143:147–157
Romero E, Mozzo M, van Stokkum IHM, Dekker JP, van Grondelle R, Croce R (2009) The origin of the low-energy form of photosystem I light-harvesting complex Lhca4: mixing of the lowest exciton with a charge-transfer state. Biophys J 96:L35–L37
Ruban AV (2015) Evolution under the sun: optimizing light harvesting in photosynthesis. J Exp Bot 66:7–23
Ruban AV (2016) Nonphotochemical Chlorophyll Fluorescence Quenching: Mechanism and Effectiveness in Protecting Plants from Photodamage. Plant Physiol 170:1903–1916
Ruban AV (2019) The mechanism of nonphotochemical quenching: the end of the ongoing debate. Plant Physiol 181:383–384
Ruban AV, Berera R, Ilioaia C, van Stokkum IHM, Kennis JTM, Pascal AA, van Amerongen H, Robert B, Horton P, van Grondelle R (2007) Identification of a mechanism of photoprotective energy dissipation in higher plants. Nature 450:575–578
Ruban AV, Dekker JP, Horton P, van Grondelle R (1995a) Temperature dependence of chlorophyll fluorescence from the light harvesting complex II of higher plants. Photochem Photobiol 61:216–221
Ruban AV, Horton P (1992) Mechanism of DpH-dependent dissipation of absorbed excitation energy by photosynthetic membranes. 1. Spectroscopic analysis of isolated light-harvesting complexes. Biochim Biophys Acta-Bioenergetics 1102:30–38
Ruban AV, Horton P, Robert B (1995b) Resonance raman spectroscopy of the photosystem II light- harvesting complex of green plants: A comparison of trimeric and aggregated. Biochemistry 34:2333–2337
Ruban AV, Rees D, Noctor GD, Young A, Horton P (1991) Long-wavelength chlorophyll species are associated with amplification of high-energy-state excitation quenching in higher plants. Biochim Biophys Acta 1059:355–360
Schulman SG (1985) Molecular luminescence spectroscopy. In: Elving PJ, Winefordner JP (eds) Chemical Analysis, vol 77. Wiley, New York, pp 1–27
Segatta F, Cupellini L, Garavelli M, Mennucci B (2019) Quantum Chemical Modeling of the Photoinduced Activity of Multichromophoric Biosystems: Focus Review. Chem Rev 119:9361–9380
Standfuss J, van Scheltinga ACT, Lamborghini M, Kühlbrandt W (2005) Mechanisms of photoprotection and nonphotochemical quenching in pea light-harvesting complex at 2.5Å resolution. EMBO J 24:919–928
Su X, Ma J, Wei X, Cao P, Zhu D, Chang W, Liu Z, Zhang X, Li M (2017) Structure and assembly mechanism of plant C2S2M2-type PSII-LHCII supercomplex. Science 357:815–820
Szczepaniak M, Sander J, Nowaczyk M, Müller MG, Rögner M, Holzwarth AR (2008) Influence of the protein environment on the regulation of the Photosystem II activity—a time-resolved fluorescence study. In: Allen JF, Gantt E, Golbeck JH, Osmond B (eds) Photosynthesis energy from the sun. Springer, Dordrecht, pp 211–214
Szczepaniak M, Sander J, Nowaczyk M, Müller MG, Rögner M, Holzwarth AR (2009) Charge separation, stabilization, and protein relaxation in photosystem II core particles with closed reaction center. Biophys J 96:621–631
Thapper A, Mamedov F, Mokvist F, Hammarström L, Styring S (2009) Defining the far-red limit of photosystem II in spinach. Plant Cell 21:2391–2401
Tutkus M, Akhtar P, Chmeliov J, Görföl F, Trinkunas G, Lambrev PH, Valkunas L (2018) Fluorescence microscopy of single liposomes with incorporated pigment-proteins. Langmuir 34:14410–14418
van Amerongen H, Croce R (2013) Light harvesting in photosystem II. Photosynth Res 116:251–263
van Amerongen H, van Grondelle R (2001) Understanding the energy transfer function of LHCII, the major light-harvesting complex of green plants. J Phys Chem B 105:604–617
van Leeuwen PJ, Nieveen MC, van de Meent EJ, Dekker JP, van Gorkom HJ (1991) Rapid and simple isolation of pure photosystem II core and reaction center particles from spinach. Photosynth Res 28:149–153
van Oort B, Roy LM, Xu P, Lu Y, Karcher D, Bock R, Croce R (2018) Revisiting the role of xanthophylls in nonphotochemical quenching. J Phys Chem Lett 9:346–352
Volk M, Häberle T, Feick R, Ogrodnik A, Michel-Beyerle ME (1993) What can be learned from the singlet-triplet splitting of the radical pair P+H- in the photosynthetic reaction center—conclusions from electric field effects on the P+H- recombination dynamics. J Phys Chem 97:9831–9836
Wahadoszamen M, Berera R, Ara AM, Romero E, van Grondelle R (2012) Identification of two emitting sites in the dissipative state of the major light harvesting antenna. Phys Chem Chem Phys 14:759–766
Wang HY, Lin S, Allen JP, Williams JC, Blankert S, Laser C, Woodbury NW (2007) Protein dynamics control the kinetics of initial electron transfer in photosynthesis. Science 316:747–750
Wang J, Cieplak P, Kollman PA (2000) How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J Comput Chem 21:1049–1074
Wei X, Su X, Cao P, Liu X, Chang W, Li M, Zhang X, Liu Z (2016) Structure of spinach photosystem II-LHCII supercomplex at 3.2 A resolution. Nature 534:69–74
Wormit M, Harbach PHP, Mewes JM, Amarie S, Wachtveitl J, Dreuw A (2009) Excitation energy transfer and carotenoid radical cation formation in light harvesting complexes; a theoretical perspective. Biochim Biophys Acta-Bioenergetics 1787:738–746
Yakushevska AE, Keegstra W, Boekema EJ, Dekker JP, Andersson J, Jansson S, Ruban AV, Horton P (2003) The structure of photosystem II in Arabidopsis: Localization of the CP26 and CP29 antenna complexes. Biochemistry 42:608–613
Yanai T, Tew DP, Handy NC (2004) A new hybrid exchange-correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem Phys Lett 393:51–57
Yang Y, Jankowiak R, Lin C, Pawlak K, Reus M, Holzwarth AR, Li J (2014) Effect of the LHCII pigment-protein complex aggregation on photovoltaic properties of sensitized TiO2 solar cells. PCCP 16:20856–20865
Zhou H, Boxer SG (1997) Charge resonance effects on electronic absorption line shapes: application to the heterodimer absorption of bacterial photosynthetic reaction centers. J Phys Chem B 101:5759–5766
Zhou H, Boxer SG (1998) Probing excited-state electron transfer by resonance Stark spectroscopy. 2. Theory and application. J Phys Chem B 102:9148–9160
Acknowledgements
This research was supported by grants to ARH from the Deutsche Forschungsgemeinschaft (DFG HO-924/3–1 and in part by SFB 663), and by the EU Training and Research Network “Harvest” of the European Union. PHL acknowledges support from the Hungarian National Research, Development and Innovation Fund (grants NN 124904, 2018–1.2.1-NKP-2018–00009). JPG is currently funded by the DFG, project no. 393271229. We thank Prof. W. Kühlbrandt and Dr. Tiago Barros (Max-Planck-Institute for Biophysics, Frankfurt a. Main, Germany) for providing the LHCII crystals. We also thank the Max-Planck-Institute for Chemical Energy Conversion for generous support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ostroumov, E.E., Götze, J.P., Reus, M. et al. Characterization of fluorescent chlorophyll charge-transfer states as intermediates in the excited state quenching of light-harvesting complex II. Photosynth Res 144, 171–193 (2020). https://doi.org/10.1007/s11120-020-00745-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11120-020-00745-8